
Abstract
In the present study, EMA (ethidium monoazide) treatment was applied to a silty-sand reference soil prior to DNA extraction to enable a differentiation between dead and living cells. For this purpose, a reference soil was spiked with Listeria monocytogenes cells or cell equivalents, respectively. With the purpose of evaluating optimum treatment conditions, different EMA concentrations have been tested. However, the results remained largely inconclusive. Furthermore, varied dark incubation periods allowing EMA to penetrate dead cells did not allow the selective removal of DNA from membrane-compromised cells in downstream analyses. In contrast to undiluted soil, an effect of EMA treatment during DNA extraction could be observed when using a 1:10 dilution of the reference soil; however, the effect has not been sufficiently selective to act on heat-treated cells only. Although the application of EMA to soil requires further evaluation, the procedure harbors future potential for improving DNA-based approaches in microbial ecology studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Polymerase chain reaction (PCR) has progressively developed to become an indispensable technique in medical and biological applications as it allows the amplification of any DNA fragment present in a certain sample. Despite the many advantages, data interpretation of PCR-based approaches in microbial ecology is still hampered by lacking discrimination between DNA originating from intact, alive cells and dead, fragmented cell deposits. In particular, the extraction of the requested intracellular DNA from environmental samples is biased by co-extraction of extracellular nucleic acids that may considerably contribute to the total DNA yield, and thus overestimate the number of viable cells in a certain habitat [1,2,3]. However, for a reliable (and exact) determination of microbial communities—regarding both, diversity and abundance—it is of particular interest to specifically target the living population when employing molecular biological, DNA-based methods in microbial ecology studies. Methodical progress remains essential to optimize DNA extraction and amplification efficiencies to avoid false-positive signals [4]. Otherwise, RNA-based approaches, which would target the active part of a microbial community, can be problematic due to the fast RNA-decay rates after loss of cell viability, are more expensive, laborious, and error-prone with respect to DNA-based analysis, and experience difficulties as well as often need extensive protocol adaptation when extracted from biosolids [5,6,7,8].
A rather novel technique employs the addition of intercalating dyes including ethidium monoazide (EMA) or propidium monoazide (PMA) during DNA extraction to enable the differentiation between live and dead cells in subsequent molecular biological analyses. This technique has been extensively used for qPCR applications to mask DNA from dead, membrane-compromised cells [9,10,11,12,13,14,15,16]. It has proven to be successful with pathogen-related samples [17,18,19,20,21,22,23] and is promising to study environmental samples [24,25,26,27,28,29,30,31,32,33,34]. The procedure involves the application of EMA or PMA, followed by the extraction of genomic DNA and subsequent PCR amplification. Prior to conventional DNA extraction, the intercalating dye is added to the desired extraction matrix that covalently binds to genomic DNA by conversion of the azide group into a nitrene radical upon photolysis [35]. As a consequence, DNA extraction and PCR amplification of these fragments is restricted [12, 27, 36]. Since EMA and PMA are hypothesized not to be able to penetrate cytoplasmic membranes [37], free DNA or DNA derived from microorganisms not maintaining cell wall integrity is selectively bound during extraction. Hence, the active (viable) part of the microbial community of a certain habitat can be targeted. Although both dyes share similar characteristics as intercalating dyes, they differ in respect of their permeation behavior. Due to its chemical composition, EMA is more efficient in signal suppression and equally effective on Gram-positive and Gram-negative bacteria [38]; however, compared with PMA it is less selective and could underestimate the viable population [32, 39]. Recent studies also investigated a mixture of both dyes [40, 41]. With a few exceptions, this technique has rarely been used for microbial diversity and/or abundance characterization of environmental samples and an investigation of the possible scope of application is still missing for many habitats including soil.
The present study therefore aimed at evaluating the overall applicability of EMA (Phenanthridium, 3-amino-8-azido-5-ethyl-6-phenyl, bromide) treatment to selectively target and mask DNA from cell-compromised Listeria monocytogenes in an Eutric Flavisol reference soil.
Material and Methods
Reference Soil
The present study used a reference soil derived from Landwirtschaftliche Untersuchungs- und Forschungsanstalt Speyer, Germany (https://www.lufa-speyer.de/). It is defined as “Reference Soil 2.3” for soil type silty sand (uS) (after German DIN) or sandy loam (after U.S. Department of Agriculture (USDA)). Based on the “World Reference Base for Soil Resources 2014” (FAO, 2015), it is classified as Eutric Flavisol and was also used for previous investigations [36]. Basic soil characteristics can be found in Table 1.
Experimental Design and Setup
In order to investigate the applicability of EMA pretreatment for DNA extraction of vital cells only in soil, a series of experiments was conducted. These had in common that DNA derived from Listeria monocytogenes was added in known concentrations to soil either as intact or heat-treated cells (cell equivalents, heat-treated cells). Subsequently, DNA extraction was performed with (EMA-DNA extraction) and without EMA pretreatment (refer to 2.5 and 2.6) followed by the quantification of the DNA extracts and/or listeriolysin O gene copy numbers via qPCR assays (refer to 2.7). All experiments using soil as a matrix were carried out at least in triplicate.
As an initial step, and before using soil as starting material, the successful removal of DNA derived from damaged cells using EMA was evaluated in a non-soil environment. Serial dilutions in PBS buffer from 1 × 109 down to 1 × 103 cells ml−1 were subjected to DNA extraction with and without an additional EMA pretreatment step. Equivalent amounts of heat-treated cells served as controls. EMA treatment was carried out using a concentration of 50 µg ml−1 according to previously published recommendations [13, 36] under identical dark incubation and light activation conditions as described in [27] (please also refer to 2.5). This analysis was carried out in duplicate for EMA- and non-EMA-treated samples, respectively.
In a second step, parameter variation studies including EMA concentrations and dark incubation periods aimed at finding a protocol for the EMA-DNA extraction from soil. To evaluate the impact of initial EMA concentrations (0, 66, 100, 250, and 500 µg EMA g−1 soil) the reference soil was spiked with 1 × 108 ml−1 intact (C) or heat-inactivated, disrupted cells (D), respectively (n = 70).
Optimum dark incubation conditions during EMA treatment were investigated by adding 100 µg EMA g−1 soil (n = 28) under varied time periods spanning 20, 60, and 90 min. As a control, DNA extracts from soil containing 1 × 108 cells or cell equivalents per gram, respectively, were evaluated without EMA treatment.
Furthermore, in sterile distilled water a 1:10 diluted solution of the reference soil (0.5 g fresh weight) was prepared in triplicates and spiked with intact and heat-treated cells, respectively. Thus, different mixtures of cells and cell equivalents in one sample (n = 24) (Table 2) were resulting and the effect of EMA pretreatment on heat-treated cells in the presence of intact cells was evaluated. An aliquot of diluted soil served as a control; spiked soil dilutions as well as controls were treated in the same way. The prepared suspension was shaken for 15 min at 125 rpm and 25 °C.
In a last experiment, a soil dilution was prepared in PBS buffer supplemented with 5 mM EDTA (PBS-EDTA) in order to minimize possibly interacting of the soil matrix with EMA treatment (n = 18). For this purpose, three 50-ml tubes were filled with 1.0 g soil; 1 × 108 cells were added (suspended in PBS-EDTA), as well as 1 × 109, 1 × 108, and 1 × 107 cell equivalents, respectively, and filled up with PBS-EDTA to a final volume of 10 ml in order to achieve a mixture of 1 × 107 cells plus 1 × 108, 1 × 107, and 1 × 106 cell equivalents per ml, respectively. The prepared suspensions were shaken for 15 min at 125 rpm and 25 °C, with 1.0 ml of the liquid phase being subsequently used for DNA extraction in three replicates.
Cultivation of L. monocytogenes
Listeria monocytogenes (DSM 15675) was grown in DSM medium 92 containing 30.0 g Trypticase soy broth, 3.0 g yeast extract, and 1000 ml of distilled water (pH 7.0). Cells were harvested after 24–30 h of incubation by centrifugation (7000×g, 10 min), washed twice (¼ Ringer solution or PBS-EDTA), and re-suspended in ¼ Ringer solution or PBS-EDTA. Freshly prepared suspensions were either used directly for spiking or underwent a heat treatment (refer to 2.4). Cell counting was performed using a Thoma-chamber (0.0025 mm2 × 0.01 mm) after dilution in Ringer solution. CFU numbers were determined by preparing serial dilutions and plating 50 µl on DSM medium 92 agar dishes.
Heat Treatment
Cell suspensions (0.75 ml, in ¼ Ringer solution) were filled into micro-centrifuge tubes and heated for 5 min at 95 °C in a water bath as described before [33]. Aliquots were then plated on DSM medium 92, incubated for 72 h at 37 °C, and checked for microbial growth. Remaining samples were stored in low DNA binding tubes (Biozym, Germany) at − 20 °C until further processing.
EMA Treatment
EMA pretreatment for soil samples was started by mixing 250 mg soil with 250 µl of molecular grade water to the extraction tubes containing the soil samples. After the addition of EMA (Biotium), the tubes were vortexed for 10 s to secure a homogenous distribution of EMA. Subsequently, the samples were incubated in the dark for 10 min (non-soil samples) and 45 min (soil samples) allowing EMA to penetrate membrane-compromised cells. Then, the samples were exposed to light by placing them 10 cm away from a 650-W halogen device. In previous investigations using non-environmental samples, light activation periods ranged from 60 s [9, 34], to 5 min [17], and up to 20 min [19]. In the present study, light activation in light transparent tubes (Biozym, Germany) was performed for 20 min to secure the best possible covalent binding of EMA to DNA, since turbidity can impede the efficiency of EMA treatment using environmental samples [42]. To avoid heating of the samples, light activation was carried out on ice, which had to be exchanged in 10-min intervals. Samples were mixed very gently every 5 min. Following the treatment with EMA, DNA extraction was conducted according to “DNA Extraction and Quantification” section starting with the addition of buffer and the beat-beating step. Control samples (without EMA treatment) were handled analogously (dark incubation and light activation) but without EMA addition.
For EMA pretreatment of 1:10 soil extracts 0.5 ml (extract in water) or 1.0 ml (extract in PBS-EDTA) was transferred into DNA extraction kit’s beat-beating tubes. Subsequently, EMA (100 µg EMA ml−1 extract) was added and a dark incubation conducted for 10 min followed by 15-min light activation time. Samples were centrifuged for 5 min at 12,000×g. For the PBS-EDTA extracts, 250 µl of supernatant were discarded and taken into account when calculating copy numbers. Posterior DNA extraction steps were conducted as described in “DNA Extraction and Quantification.”
DNA Extraction and Quantification
As the extraction of DNA from soil can be critical, the procedure adopted here was optimized in a previous investigation [36]. DNA was extracted using the Soil Extract II Kit (Macherey & Nagel, Germany) following the recommendation of the manufacturer, except that the beat-beating step was done twice on a FastPrep 24-5G (MPbio) with an interval of 300 s. Subsequently, the quantity and quality of the extracted DNA was evaluated fluorimetrically with PicoGreen dsDNA quantification reagent (Invitrogen, Carlsbad, USA, Anthos-Zenyth Multimode Detector) and spectrophotometrically with NanoDrop 2000c™ (PEQLAB, Germany) (Abs@260 nm = 1.0 referred to a concentration of 50 ng µl−1), respectively.
qPCR
qPCR was performed to quantify the listeriolysin O gene (hly A) in DNA extracts, which further allowed for the indirect estimation of Listeria monocytogenes abundance in spiked soils. A method proposed by [43] was used and qPCR performed on a Corbett Life Science (Qiagen) Rotor-Gene 6000 system. Prior to experiments, the primers and reaction conditions were validated with DNA extracts of a known number of L. monocytogenes cells. All PCR reactions and approaches were performed at least in duplicates, with “non-template controls” (NTCs) and positive controls using 2 µl of template DNA in a final volume of 20 µl being applied. Template DNA was diluted to achieve total DNA concentrations of 0.5–5 ng µl−1 prior addition to the reaction mix (at least 1:10) and taken into account in the calculation of copy numbers. As a standard, the purified PCR product targeting the hly A gene associated with DNA from a pure culture of Listeria monocytogenes was used in known concentrations as described before for the quantification of Archaea [44]. For quantification and efficiency calculations, diluted standards were used and the CT (cycle threshold) values were plotted against the log of given templates to obtain standard graphs as described in [45]. qPCR data were used when an efficiency value of at least 0.9 (90%) was reached. Standard curves resulted in R2 between 0.999 and 0.947. The efficiency analysis of the qPCR reactions was calculated by the Rotor-Gene software.
Statistical Analysis
Statistical analysis was performed by using the Software package Statistica 9.0 (StatSoft®) and SigmaPlot 12.0 (Systat Software Inc.). Results are given as means ± standard deviation. Significant differences were ascertained by one-way or multifactorial ANOVA. A significance level of 0.05 was used to assess differences between treatments. Bonferroni Test was used to discriminate between single variants.
Results and Discussion
EMA Application in a Non-soil Environment
To validate the previously shown potential of EMA to selectively bind to DNA from cells not maintaining cell wall integrity [9, 13, 46], serial dilutions of Listeria monocytogenes cultures or equivalent amounts of heat-treated cells (1 × 109 to 1 × 103 ml−1) were used in a non-soil environment. Prepared dilutions were either subjected to EMA treatment or not, followed by DNA extraction and subsequent determination of the copy number of hyl A gene by qPCR. Although previous studies used lower concentrations of intercalating dyes [47] or demonstrated a positive effect of EMA if applied in much lower concentrations [17, 33], we used 50 µg EMA ml−1. This was taking into account the given high number of desired 1 × 109 cells ml−1, which corresponds to an approximate amount of 5 µg of total DNA when calculating 5 fg DNA per cell [48].
qPCR results for the desired amount of 1 × 109 to 1 × 103 copies ml−1 showed higher copy numbers (14–38%) for samples with intact cells than for those with equivalent amounts of heat-inactivated cells. While the number for intact cell samples remained in the expected range, the copy number obtained for heat-treated cells significantly ceased when EMA treatment was applied. From an initial concentration of 2.84 × 109 (as measured by qPCR in the sample containing intact cells), a reduction to 5.92 × 105 copies ml−1 could be achieved. Although the complete portion of cracked cell’s DNA could not be eliminated by EMA treatment, at least a reduction of approx. 4 log10 could be attained. For samples containing a spike of 1.08 × 107, 7.66 × 104, and 3.09 × 103 heat-treated cells (copy numbers obtained from samples without EMA treatment), at least one of two replicates resulted in a qPCR result below the detection limit (no template control, NTC), while the remaining ones were very close to NTC. Thus, efficient binding of EMA to the DNA present in the samples for samples containing < 1.08 × 107 heat-treated cells was assumed as also observed in other studies [13]. In conclusion, preliminary results in a non-soil matrix were promising and clearly indicated a high potential of EMA treatment. Subsequent experiments aimed at the investigation of EMA treatment and the removal of DNA from non-vital cells in a reference soil.
EMA Application in Soil Environment
To evaluate the potential of EMA treatment prior DNA extraction from soil, the same parameters as for the non-soil environment were applied; however, instead of using 1 × 109 cells or cell equivalents (heat-treated cells) 1 × 108 cells were used in order to decrease the total amount of DNA that is addressed to EMA binding. However, it became obvious that the treatment parameters (EMA concentration, dark incubation, light activation) were not appropriate to see a reduction in hyl A copy numbers via qPCR. Melt curve analysis did not reveal any differences in melting peaks between EMA-treated and untreated samples, respectively, so the poor effect was attributable to the insufficient action of EMA.
So in a next step different EMA concentrations were tested. Results are depicted in Fig. 1. Even though concentrations of up to 500 µg EMA g−1 soil were applied during EMA treatment, a significant effect could not be verified. Although the results indicated sufficient resolution of qPCR and demonstrated that the comparability of results for intact and heat-treated cell was given even in the soil environment (Fig. 1, samples lacking EMA treatment C0 and D0), EMA treatment did not result in the expected decrease in target DNA from samples containing heat-treated cells. Possible explanations include a too high number of target molecules [49] as either the concentration of EMA was still too low or the binding of EMA was insufficient during dark incubation. Moreover, the interaction of EMA with soil particles might have been responsible for a reduced effectivity of EMA since the high content of silt and sand together with a substantial amount of clay reflect very large surface areas. This possibly led to an absorbtion of EMA as well as to problems with the light activation due to the matrix hampering the covalent binding of EMA [27]. However, also various other environmental factors are known to negatively influence the binding of EMA to DNA [17].

Copy number (qPCR) for different EMA concentrations (0, 65.5, 100, 250, 500) applied prior DNA extraction from a reference soil spiked with 1 × 108 viable cells (C) or an equivalent amount of heat-treated, disrupted (D) cells. Inset: box plot calculated from all EMA-treated C and D samples showing no significant difference (ANOVA, p < 0.05). Numbers reflect the applied EMA concentrations (µg g−1 soil); C0 and D0 the co-processed controls without EMA addition
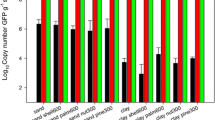
The number of target DNA from cells not maintaining cell wall integrity was varied by adding 1 × 106, 1 × 107, and 1 × 108 cells or equivalents g−1 soil (FW) (Fig. 2); however, a significant impact of EMA treatment on total DNA yield between soil spiked with intact and heat-treated cells, respectively, was missing (Fig. 2a). On the contrary, the addition of viable cells became clearly apparent by classical cultivation, although CFU counts were slightly lower than the calculated spike. Furthermore, in samples containing heat-treated cells, no living cells could be verified (Fig. 2b). While data for total DNA lacked significant differences between the spiking variations, qPCR amplification of the listeriolysin gene allowed a clear differentiation but underestimated the amount of added cells and cell equivalents considerably. A significant effect of EMA treatment to mask DNA from heat-treated cells and protect them from being amplified during qPCR could not be observed, although a general trend of higher Ct values of EMA-treated samples was found irrespective of the addition of cells or heat-treated cell equivalents (Fig. 2c). In conclusion, an increase of EMA concentration up to 100 µg g−1 soil (FW) and a concurrent reduction of target DNA from 1 × 109 to max. 1 × 108 did not result in the desired reduction of extractable DNA from heat-treated cells in the applied soil as determined by qPCR measurement and led to clearly false-positive signals in EMA-treated samples.

Total DNA concentration (a), CFU count (b), and hly A gene copy number (c) for a reference soil spiked with 1 × 106, 1 × 107, or 1 × 108 cells (C) or an equivalent amount of heat-treated, disrupted (D) cells. Samples were either subjected to an EMA treatment (+) (100 µg g−1 soil FW) or not. Significant differences are indicated by different characters (p < 0.05, Bonferroni post hoc test)
We thus hypothesized that the soil matrix or its composition reduced the effect of EMA treatment. To test the impact of the amount of extracted soil on DNA extraction in combination with EMA, the initial weight of soil used for DNA extraction was reduced. For this experiment, 50 or 100 mg of soil (instead of 250 mg) spiked with 1 × 108 cells or equivalents of L. monocytogenes was treated with 25 µg EMA and used for DNA extraction. The effect, however, was statistically negligible and resulted in 4.55 × 107 and 8.37 × 107 copies g−1 soil after qPCR for EMA-treated and non-treated samples, respectively.
Besides testing different EMA concentrations, the impact of dark incubation periods was also evaluated. Previously described procedures applied time periods ranging from 5 [9, 34], to 10 [17, 50], and up to 60 min [19]. In the present study, dark incubation periods extending over 20, 60, and 90 min were applied allowing EMA to penetrate membranes of cells not maintaining cell wall integrity prior to light activation. While the controls without EMA treatment showed copy numbers in the expected range of 1 × 108 copies g−1 soil for intact as well as for heat-treated cells, copy numbers for samples with intact cells were slightly higher. However, EMA treatment in combination with 20 min of dark incubation did not result in significant differences between samples containing intact and heat-treated cells, respectively. Even a prolongation of dark incubation during EMA treatment did not result in a significant reduction of copy numbers obtained from samples containing heat-treated cells and samples with intact cells. For a dark incubation time during EMA treatment of 60 min, 1.34 × 107 and for 90 min 3.83 × 107 copies were found.
In a last step, 1:10 soil extracts were prepared, first in distilled water and then in PBS-EDTA. The water soil extract was spiked with different concentrations of L. monocytogenes cells or equivalents (5 × 108, 1 × 107, 1 × 105) and EMA treatment was tested. For this experiment, a combination of intact cells and heat-treated cells was used according to Table 2. Negative controls were tested several times before using DNA extracts from soil without adding any target DNA that resulted in Ct values within the range of the no template control during qPCR analysis (data not shown). Then, the effect of EMA treatment for DNA from heat-treated cells was evaluated via qPCR in the presence of target DNA from intact cells.
In contrast to the results obtained from DNA extracted directly from soil, qPCR results from 1:10 diluted soil samples showed an impact of EMA treatment. However, the desired effect to selectively and exclusively mask DNA from heat-treated cells could further not be verified (Table 2, Fig. 3). While CFU counts represented spiked cell concentrations fairly well, the effectiveness of EMA failed and did not eliminate DNA from compromised cells sufficiently (Fig. 3). Obviously, the action of EMA was unspecific also targeting intact cells and ineffective when using high numbers of heat-treated cells in diluted soil samples. Besides, applying EMA treatment to both cells and cell equivalents resulted in similar log10 copy numbers between 6.36 and 6.83, although a difference of 3 log10 between samples containing the highest and lowest amount of intact cells was expected (Table 2). Therefore, an underestimation of the real copy numbers was the consequence, an effect that has also been described earlier [37, 38]. On the other hand, an overestimation of copy number could be observed when using diluted soil samples without EMA treatment. This could be related to the additional incubation time during preparation of dilutions, which was further reflected in a slight increase in the number of CFU counts.

CFU and copy number per ml of soil extract (Mean, Box: Mean ± SE; Whisker: Mean ± 2*SD). The extract was spiked with A: 5 × 108 cells and 1 × 105 heat-treated cells, B: 1 × 107 cells and 1 × 107 heat-treated cells, C: 1 × 105 cells and 5 × 108 heat-treated cells, and D: 1 × 105 cells and 1 × 105 heat-treated cells simultaneously. Please also refer to Table 2
These results implied a still insufficient action (specificity and effectivity) of EMA possibly due to interaction of EMA with the soil matrix. Thus, an additional dilution was prepared in PBS-EDTA comprised of 1 × 107 intact cells and 1 × 108, 1 × 107, and 1 × 106 cell equivalents, respectively. Results are depicted in Fig. 4. Via the utilization of an EDTA containing buffer, the effectivity of EMA could be increased and the non-desired effect on intact cells was reduced. Although for example the copy number of 1 × 107 intact cells was overestimated by a factor of 2.28 when 1 × 108 heat-treated cells were present, this indicates that DNA from approx. 7.7 × 107 cell equivalents were masked by EMA and excluded from qPCR amplification (Fig. 4). However, an impact of EMA on vital cells was still observed and led to underestimation of the real number of vital cells (Fig. 4).

Copy number per ml of PBS-EDTA soil extract (Mean, Box: Mean ± SE; Whisker: Mean ± 2*SD, green lines refer to calculated copy numbers). The extract was spiked with 1 × 107 cells and 1 × 108 (7 + 8), 1 × 107 (7 + 7) and 1 × 106 (7 + 6) heat-treated cells per ml, respectively. The samples were treated with (EMA) and without EMA, arrows indicate the reduction of copy numbers by EMA
Generally speaking, the application of EMA directly to a silty-sand reference soil did not enable a clear differentiation between DNA from cell wall-compromised and living cells, respectively. Although DNA extraction from spiked soil is routine now it remains a challenging technique per se [51,52,53]. In the present investigation, qPCR resulted in copy numbers reflecting the dimension of the spiking and thus indicated proper DNA extraction (efficiency) and accurate qPCR amplification. Nevertheless, the application of different EMA concentrations led to inconclusive results. Varying periods of dark incubation to permit the passage of EMA into the compromised cells did not allow the complete exclusion of DNA from cells not maintaining cell wall integrity in downstream analysis. Lacking effectivity of EMA treatment can be associated with the matrix it was applied to, since complex matrices can adversely influence the efficiency of EMA treatment [49, 54]. The high content of silt and sand together with a substantial amount of approx. 10% clay reflect very large surface areas, which might have been able to absorb concentrations of up to 500 mg EMA g−1 soil. Other parameters known to limit the success of EMA treatment include high salt concentrations, pH, presence of high amounts of dead cells, and turbid samples [27, 49].
In contrast to direct application, an effect of EMA treatment during DNA extraction could be observed when using a 1:10 dilution of the reference soil in water or PBS-EDTA. By applying soil dilutions (instead of direct application), the effect of EMA on high target numbers (1 × 108 cell equivalents) could be strengthened und resulted in a copy number close to the calculated ones. However, by the applied EMA-DNA extraction procedure still a portion of non-target molecules was masked by EMA.
Insights gained in the present study, especially the use of soil dilutions applying PBS-EDTA buffer prior to DNA extraction, might serve as a basis for subsequent investigations. Suggested improvements further include the application of primers generating longer fragments as a solution to avoid false-positive signals [18, 55, 56]. However, promising results of removing DNA from non-viable cells via intercalating dyes in other habitats [29, 40, 41, 57,58,59,60] give courage for the applicability of this methodology for soil habitats in the future.
References
Pietramellara G, Ascher J, Borgogni F, Ceccherini MT, Guerri G, Nannipieri P (2009) Extracellular DNA in soil and sediment: fate and ecological relevance. Biol Fert Soils 45:219–235. https://doi.org/10.1007/s00374-008-0345-8
Ascher J, Ceccherini MT, Pantani OL, Agnelli A, Borgogni F, Guerri G, Nannipieri P, Pietramellara G (2009) Sequential extraction and genetic fingerprinting of a forest soil metagenome. Appl Soil Ecol 42:176–181. https://doi.org/10.1016/j.apsoil.2009.03.005
Ceccherini MT, Ascher J, Agnelli A, Borgogni F, Pantani OL, Pietramellara G (2009) Experimental discrimination and molecular characterization of the extracellular soil DNA fraction. Antonie Van Leeuwenhoek 96:653–657. https://doi.org/10.1007/s10482-009-9354-3
Wintzingerode FV (1997) Determination of microbial diversity in environmental samples: pitfalls of PCR-based rRNA analysis. FEMS Microbiol Rev 21:213–229. https://doi.org/10.1016/S0168-6445(97)00057-0
Belasco J (1993) mRNA degradation in prokaryotic cells: an overview. In: Belasco J, Brawerman G (eds) Control of messenger RNA stability. Academic Press Inc, San Diego, pp 3–12
Dunaev T, Alanya S, Duran M (2008) Use of RNA-based genotypic approaches for quantification of viable but non-culturable Salmonella sp. in biosolids. Water Sci Technol 58:1823–1828. https://doi.org/10.2166/wst.2008.555
Lim NYN, Roco CA, Frostegård Å (2016) Transparent DNA/RNA co-extraction workflow protocol suitable for inhibitor-rich environmental samples that focuses on complete DNA removal for transcriptomic analyses. Front Microbiol 7:1588. https://doi.org/10.3389/fmicb.2016.01588
Lever MA, Rogers KL, Lloyd KG, Overmann J, Schink B, Thauer RK, Hoehler TM, Jørgensen BB (2015) Life under extreme energy limitation: a synthesis of laboratory- and field-based investigations. FEMS Microbiol Rev 39:688–728. https://doi.org/10.1093/femsre/fuv020
Rudi K, Moen B, Dromtorp M, Holck AL (2005) Use of ethidium monoazide and PCR in combination for quantification of viable and dead cells in complex samples. Appl Environ Microbiol 71:1018–1024
Nocker A, Sossa KE, Camper AK (2007) Molecular monitoring of disinfection efficacy using propidium monoazide in combination with quantitative PCR. J Microbiol Methods 70:252–260
Nocker A, Sossa-Fernandez P, Burr MD, Camper AK (2007) Use of propidium monoazide for live/dead distinction in microbial ecology. Appl Environ Microbiol 73:5111–5117
Nogva HK, Dromtorp SM, Nissen H, Rudi K (2003) Ethidium monoazide for DNA-based differentiation of viable and dead bacteria by 5′-nuclease PCR. Biotechniques 34:804–808
Nocker A, Camper AK (2006) Selective removal of DNA from dead cells of mixed bacterial communities by use of ethidium monoazide. Appl Environ Microbiol 72:1997–2004. https://doi.org/10.1128/AEM.72.3.1997-2004.2006
Agulló-Barceló M, Moss JA, Green J, Gillespie S, Codony F, Lucena F, Nocker A (2014) Quantification of relative proportions of intact cells in microbiological samples using the example of Cryptosporidium parvum oocysts. Lett Appl Microbiol 58:70–78
Desneux J, Chemaly M, Pourcher A-M (2015) Experimental design for the optimization of propidium monoazide treatment to quantify viable and non-viable bacteria in piggery effluents. BMC Microbiol 15:164. https://doi.org/10.1186/s12866-015-0505-6
Minami J, Yoshida K, Soejima T, Yaeshima T, Iwatsuki K (2010) New approach to use ethidium bromide monoazide as an analytical tool. J Appl Microbiol 109:900–909
Shi H, Xu W, Luo Y, Chen L, Liang Z, Zhou X, Huang K (2011) The effect of various environmental factors on the ethidium monazite and quantitative PCR method to detect viable bacteria. J Appl Microbiol 111:1194–1204
Soejima T, Iida K-I, Qin T, Taniai H, Seki M, Yoshida S-I (2008) Method to detect only live bacteria during PCR amplification. J Clin Microbiol 46:2305–2313. https://doi.org/10.1128/JCM.02171-07
Soejima T, Iida K-I, Qin T, Taniai H, Seki M, Takade A, Yoshida S-I (2007) Photoactivated ethidium monoazide directly cleaves bacterial DNA and is applied to PCR for discrimination of live and dead bacteria. Microbiol Immunol 51:763–775
Soejima T, Minami J-I, Yaeshima T, Iwatsuki K (2012) An advanced PCR method for the specific detection of viable total coliform bacteria in pasteurized milk. Appl Microbiol Biotechnol 95:485–497. https://doi.org/10.1007/s00253-012-4086-0
Rawsthorne H, Dock CN, Jaykus LA (2009) PCR-based method using propidium monoazide to distinguish viable from nonviable Bacillus subtilis spores. Appl Environ Microbiol 75:2936–2939. https://doi.org/10.1128/AEM.02524-08
Pan Y, Breidt F Jr (2007) Enumeration of viable Listeria monocytogenes cells by real-time PCR with propidium monoazide and ethidium monoazide in the presence of dead cells. Appl Environ Microbiol 73:8028–8031
Bae S, Wuertz S (2012) Survival of host-associated bacteroidales cells and their relationship with Enterococcus spp, Campylobacter jejuni, Salmonella enterica serovar Typhimurium, and Adenovirus in freshwater microcosms as measured by propidium monoazide-quantitative PCR. Appl Environ Microbiol 78:922–932
Heise J, Nega M, Alawi M, Wagner D (2016) Propidium monoazide treatment to distinguish between live and dead methanogens in pure cultures and environmental samples. J Microbiol Methods 121:11–23. https://doi.org/10.1016/j.mimet.2015.12.002
van Frankenhuyzen JK, Trevors JT, Lee H, Flemming CA, Habash MB (2011) Molecular pathogen detection in biosolids with a focus on quantitative PCR using propidium monoazide for viable cell enumeration. J Microbiol Methods 87:263–272. https://doi.org/10.1016/j.mimet.2011.09.007
van Frankenhuyzen JK, Trevors JT, Flemming CA, Lee H, Habash MB (2013) Optimization, validation, and application of a real-time PCR protocol for quantification of viable bacterial cells in municipal sewage sludge and biosolids using reporter genes and Escherichia coli. J Ind Microbiol Biotechnol 40:1251–1261. https://doi.org/10.1007/s10295-013-1319-x
Wagner AO, Malin C, Knapp BA, Illmer P (2008) Removal of free extracellular DNA from environmental samples by ethidium monoazide and propidium monoazide. Appl Environ Microbiol 74:2537–2539. https://doi.org/10.1128/AEM.02288-07
Luo J-F, Lin W-T, Guo Y (2010) Method to detect only viable cells in microbial ecology. Appl Microbiol Biotechnol 86:377–384. https://doi.org/10.1007/s00253-009-2373-1
Desneux J, Biscuit A, Picard S, Pourcher A-M (2016) Fate of viable but non-culturable listeria monocytogenes in pig manure microcosms. Front Microbiol 7:124. https://doi.org/10.3389/fmicb.2016.00245
Salam KW, El-Fadel M, Barbour EK, Saikaly PE (2014) A propidium monoazide-quantitative PCR method for the detection and quantification of viable Enterococcus faecalis in large-volume samples of marine waters. Appl Microbiol Biotechnol 98:8707–8718. https://doi.org/10.1007/s00253-014-6023-x
Eichmiller JJ, Borchert AJ, Sadowsky MJ, Hicks RE (2014) Decay of genetic markers for fecal bacterial indicators and pathogens in sand from Lake Superior. Water Res 59:99–111. https://doi.org/10.1016/j.watres.2014.04.005
Elizaquivel P, Aznar R, Sanchez G (2013) Recent developments in the use of viability dyes and quantitative PCR in the food microbiology field. J Appl Microbiol 116:1–13
Lee J-L, Levin RE (2007) Quantification of total viable bacteria on fish fillets by using ethidium bromide monoazide real-time polymerase chain reaction. Int J Food Microbiol 118:312–317
Pisz JM, Lawrence JR, Schafer AN, Siciliano SD (2007) Differentiation of genes extracted from non-viable versus viable micro-organisms in environmental samples using ethidium monoazide bromide. J Microbiol Methods 71:312–318
Trevors JT (2012) Can dead bacterial cells be defined and are genes expressed after cell death? J Microbiol Methods 90:25–28
Wagner AO, Praeg N, Reitschuler C, Illmer P (2015) Effect of DNA extraction procedure, repeated extraction and ethidium monoazide (EMA)/propidium monoazide (PMA) treatment on overall DNA yield and impact on microbial fingerprints for bacteria, fungi and archaea in a reference soil. Appl Soil Ecol 93:56–64. https://doi.org/10.1016/j.apsoil.2015.04.005
Nocker A, Cheung C-Y, Camper AK. Comparison of propidium monoazide with ethidium monoazide for differentiation of live vs. dead bacteria by selective removal of DNA from dead cells. J Microbiol Methods 2006; 67: 310–320. doi: 10.1016/j.mimet.2006.04.015.
Flekna G, Polonca S, Wagner M, Smulders FJM, Mozina SS, Hein I (2007) Insufficient differentiation of live and dead Campylobacter jejuni and Listeria monocytogenes cells by ethidium monoazide (EMA) compromises EMA/real-time PCR. Res Microbiol 158:405–412
Pacholewicz E, Swart A, Lipman LJA, Wagenaar JA, Havelaar AH, Duim B (2013) Propidium monoazide does not fully inhibit the detection of dead Campylobacter on broiler chicken carcasses by qPCR. J Microbiol Methods 95:32–38
Dinh Thanh M, Agustí G, Mader A, Appel B, Codony F (2017) Improved sample treatment protocol for accurate detection of live Salmonella spp. in food samples by viability PCR. PLoS ONE 12:e0189302. https://doi.org/10.1371/journal.pone.0189302
Daranas N, Bonaterra A, Francés J, Cabrefiga J, Montesinos E, Badosa E (2018) Monitoring viable cells of the biological control agent Lactobacillus plantarum PM411 in aerial plant surfaces by means of a strain-specific viability quantitative PCR Method. Appl Environ Microbiol. https://doi.org/10.1128/AEM.00107-18
Gedalanga PB, Olson BH (2009) Development of a quantitative PCR method to differentiate between viable and nonviable bacteria in environmental water samples. Appl Microbiol Biotechnol 82:587–596. https://doi.org/10.1007/s00253-008-1846-y
Nogva HK, Rudi K, Naterstad K, Holck A, Lillehaug D (2000) Application of 5′-nuclease PCR for quantitative detection of Listeria monocytogenes in pure cultures, water, skim milk, and unpasteurized whole milk. Appl Environ Microbiol 66:4266–4271. https://doi.org/10.1128/AEM.66.10.4266-4271.2000
Reitschuler C, Lins P, Illmer P (2014) Primer evaluation and adaption for cost-efficient SYBR green-based qPCR and its applicability for specific quantification of methanogens. World J Microbiol Biotechnol. 30:293–304
Bustin S, Benes V, Garson J, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl M, Shipley G et al (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55:611–622
Hein I, Schneeweiss W, Stanek C, Wagner M (2007) Ethidium monoazide and propidium monoazide for elimination of unspecific DNA background in quantitative universal real-time PCR. J Microbiol Methods 71:336–339
Carini P, Marsden PJ, Leff JW, Morgan EE, Strickland MS, Fierer N (2016) Relic DNA is abundant in soil and obscures estimates of soil microbial diversity. Nat Microbiol 2:16242. https://doi.org/10.1038/nmicrobiol.2016.242
Rudd KE, Miller W, Ostell J, Benson DA (1990) Alignment of Escherichia coli K12 DNA sequences to a genomic restriction map. Nucl Acids Res 18:313–321. https://doi.org/10.1093/nar/18.2.313
Fittipaldi M, Nocker A, Codony F (2012) Progress in understanding preferential detection of live cells using viability dyes in combination with DNA amplification. J Microbiol Methods 91:276–289. https://doi.org/10.1016/j.mimet.2012.08.007
Rueckert A, Morgan HW (2007) Removal of contaminating DNA from polymerase chain reaction using ethidium monoazide. J Microbiol Methods 68:596–600
Carrigg C, Rice O, Kavanagh S, Collins G, Olaherty V (2007) DNA extraction method affects microbial community profiles from soils and sediment. Appl Microbiol Biotechnol 77:955–964. https://doi.org/10.1007/s00253-007-1219-y
Feinstein LM, Sul WJ, Blackwood CB (2009) Assessment of bias associated with incomplete extraction of microbial DNA from soil. Appl Environ Microbiol 75:5428–5433. https://doi.org/10.1128/aem.00120-09
Inceoglu O, Hoogwout EF, Hill P, van Elsas JD (2010) Effect of DNA extraction method on the apparent microbial diversity of soil. Appl Environ Microbiol 76:3378–3382
Kramer M, Obermajer N, Bogovic Matijasic B, Rogelj I, Kmetec V (2009) Quantification of live and dead probiotic bacteria in lyophilised product by real-time PCR and by flow cytometry. Appl Microbiol Biotechnol 84:1137–1147. https://doi.org/10.1007/s00253-009-2068-7
Nkuipou-Kenfack E, Engel H, Fakih S, Nocker A (2013) Improving efficiency of viability-PCR for selective detection of live cells. J Microbiol Methods 93:20–24. https://doi.org/10.1016/j.mimet.2013.01.018
Schnetzinger F, Pan Y, Nocker A (2013) Use of propidium monoazide and increased amplicon length reduce false-positive signals in quantitative PCR for bioburden analysis. Appl Microbiol Biotechnol 97:2153–2162. https://doi.org/10.1007/s00253-013-4711-6
Garcia AB, Vigre H, Josefsen MH (2015) Towards the production of reliable quantitative microbiological data for risk assessment: direct quantification of Campylobacter in naturally infected chicken fecal samples using selective culture and real-time PCR. Food Control 55:133–140. https://doi.org/10.1016/j.foodcont.2015.02.044
Gyawali P, Hewitt J (2018) Detection of infectious noroviruses from wastewater and seawater using PEMAXTM treatment combined with RT-qPCR. Water 10:841. https://doi.org/10.3390/w10070841
Ruike W, Higashimori A, Yaguchi J, Li Y-Y (2016) Use of real-time PCR with propidium monoazide for enumeration of viable Escherichia coli in anaerobic digestion. Water Sci Technol 74:1243–1254. https://doi.org/10.2166/wst.2016.327
Kibbee RJ, Örmeci B (2017) Development of a sensitive and false-positive free PMA-qPCR viability assay to quantify VBNC Escherichia coli and evaluate disinfection performance in wastewater effluent. J Microbiol Methods 132:139–147. https://doi.org/10.1016/j.mimet.2016.12.004
Acknowledgments
Open access funding provided by University of Innsbruck and Medical University of Innsbruck.
Funding
This study is funded by Austrian Science Fund (P 22815).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wagner, A.O., Praeg, N. & Illmer, P. Spiking a Silty-Sand Reference Soil with Bacterial DNA: Limits and Pitfalls in the Discrimination of Live and Dead Cells When Applying Ethidium Monoazide (EMA) Treatment. Curr Microbiol 76, 1425–1434 (2019). https://doi.org/10.1007/s00284-019-01772-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-019-01772-y