
Abstract
DyP-type peroxidases comprise a novel superfamily of heme-containing peroxidases which is unrelated to the superfamilies of known peroxidases and of which only a few members have been characterized in some detail. Here, we report the identification and characterization of a DyP-type peroxidase (TfuDyP) from the thermophilic actinomycete Thermobifida fusca. Biochemical characterization of the recombinant enzyme showed that it is a monomeric, heme-containing, thermostable, and Tat-dependently exported peroxidase. TfuDyP is not only active as dye-decolorizing peroxidase as it also accepts phenolic compounds and aromatic sulfides. In fact, it is able to catalyze enantioselective sulfoxidations, a type of reaction that has not been reported before for DyP-type peroxidases. Site-directed mutagenesis was used to determine the role of two conserved residues. D242 is crucial for catalysis while H338 represents the proximal heme ligand and is essential for heme incorporation. A genome database analysis revealed that DyP-type peroxidases are frequently found in bacterial genomes while they are extremely rare in other organisms. Most of the bacterial homologs are potential cytosolic enzymes, suggesting metabolic roles different from dye degradation. In conclusion, the detailed biochemical characterization reported here contributes significantly to our understanding of these enzymes and further emphasizes their biotechnological potential.
Similar content being viewed by others


Introduction
Peroxidases (EC 1.11.1.x) represent a large group of oxidoreductases that catalyze the oxidation of substrate molecules using hydrogen peroxide as electron acceptor. The vast majority of peroxidases contain heme as cofactor (Banci 1997). These enzymes are consistently found in eukaryotic and prokaryotic cells and play a key role in important biological processes, such as biosynthesis of lignin, degradation pathways, and host-defense mechanisms (Passardi et al. 2005; Davies et al. 2008). Additionally, the chemical nature of peroxidase-catalyzed reactions, the oxidation of a wide variety of compounds with the help of hydrogen peroxide, has resulted in a wide range of peroxidase-based biotechnological applications. For example, peroxidases are utilized in biobleaching, wastewater treatment, and various analytical biosensors (Regalado et al. 2004).
Peroxidases are commonly subdivided into two superfamilies: the plant peroxidases and the animal peroxidases. The plant peroxidase superfamily also includes evolutionarily related, heme-containing peroxidases from fungi and bacteria and has been further subdivided into three classes based on cellular localization and function (Welinder et al. 1992). Class I plant peroxidases are intracellular peroxidases, including yeast cytochrome c peroxidase and chloroplast ascorbate peroxidase. Class II plant peroxidases are extracellular fungal peroxidases, like the lignin peroxidase and manganese peroxidase. Class III plant peroxidases represent extracellular plant peroxidases, such as horseradish peroxidase. The superfamily of animal peroxidases is not sequence-related to the group of plant peroxidases. Evolutionary relationships between different mammalian heme-containing peroxidase subclasses are just beginning to emerge (Loughran et al. 2008).
Recently, a novel superfamily of heme-containing peroxidases has been identified: the so-called dye-decolorizing peroxidase (DyP-type) superfamily (Sugano 2009). DyP-type peroxidases are not related in primary sequence, structure, and reaction characteristics to peroxidases belonging to the plant and animal peroxidase superfamilies. DyP-type peroxidases have been included as a separate superfamily in databases, such as PeroxiBase and Pfam (Passardi et al. 2007; Finn et al. 2008). DyP-type peroxidases were first discovered in fungi and were shown to be capable to degrade a wide range of dyes (Kim and Shoda 1999). This discovery was subsequently followed by the identification of some homologs in bacteria. For BtDyP from Bacteroides thetaiotaomicron and TyrA from Shewanella oneidensis the crystal structure has been determined but other biochemical data are limited (Zubieta et al. 2007a, b). Escherichia coli YcdB represents the best characterized bacterial DyP-type peroxidase thus far. It has been established that YcdB is a hemoprotein, exhibits modest peroxidase activity with guaiacol as substrate and is most active under acidic conditions (Sturm et al. 2006). The physiological function of all reported DyP-type peroxidases is as yet unknown.
In the present study, we report the identification and characterization of a novel DyP-type peroxidase (TfuDyP; accession number Q47KB1) from the thermophilic actinomycete Thermobifida fusca. Heterologous expression of TfuDyP in E. coli confirmed periplasmic export by the Tat system. The monomeric and robust enzyme contains non-covalently bound heme as cofactor, is most active at pH 3.5, is able to convert a large number of compounds, and catalyzes enantioselective sulfoxidations. Furthermore, we have been able to show that the conserved residues D242 and H338 are crucial for proper functioning of this type of peroxidases. In conclusion, the data show that TfuDyP is indeed a bona fide DyP-type peroxidase and also represents the first characterized substrate protein of the T. fusca Tat system. Moreover, the detailed biochemical characterization reported here contributes significantly to our understanding of these enzymes.
Materials and methods
Reagents, enzymes, and sera
Restriction enzymes were from Roche Applied Science and New England Biolabs. GC-rich PCR system and Pfu DNA polymerase were from Roche Applied Science and Invitrogen. Enhanced chemiluminescence (ECL) Western-blotting detection reagent was from Amersham Biosciences. Reactive Blue 19 was from Acros Organics. All other chemicals were supplied by Sigma and were of analytical grade. Ni2+–NTA agarose was obtained from Qiagen. Antiserum against the Myc sequence was from Abcam. DsbA and DnaK antisera were kind gifts of H. D. Bernstein and A. Mogk, respectively.
Strains, plasmids, and growth conditions
E. coli strain MC1061 (Casadaban and Cohen 1980) was used for routine cloning and maintenance of all plasmid constructs. This strain was also used for overexpression and purification of TfuDyP and its derivatives. E. coli strain B1LK0 (MC4100 ΔtatC) has been described previously (Bogsch et al. 1998) and was kindly provided by F. Sargent. BL1K0 and its isogenic control strain MC4100 (Casadaban 1976) was used for subcellular localization experiments. Cultures were grown to saturation at 37°C overnight. The following day, overnight cultures were back-diluted 1:100 into fresh media containing 0.2% l-arabinose to induce the expression of TfuDyP or its derivatives and grown to saturation at 37°C. All strains were routinely grown in Luria Bertani medium (LB; per liter, 10 g tryptone, 5 g yeast extract, 5 g NaCl) under aerobic conditions unless indicated otherwise. Where appropriate, ampicillin (100 μg/ml) was added to the culture medium.
The gene encoding TfuDyP was polymerase chain reaction (PCR) amplified from T. fusca genomic DNA thereby removing the original stop codon. The PCR fragment was cloned between the SacI and HindIII restriction sites of pBAD/Myc-His A (Invitrogen), resulting in pBAD/Myc-His A-TfuDyP. TfuDyPD242A and TfuDyPH338A were obtained by site-directed mutagenesis, using the QuikChange kit (Stratagene) and pBAD/Myc-His A-TfuDyP as template. Nucleotide sequences were verified by DNA sequencing (GATC, Konstanz). Primer sequences are available upon request.
Ni2+–NTA agarose purification of TfuDyP
MC1061 cells expressing TfuDyP or its derivatives were grown in 0.5 L LB medium at 37°C to saturation. Cells were collected by centrifugation (5000×g for 10 min at 4°C) and resuspended in phosphate-buffered saline (PBS). Following addition of lysozyme (0.5 mg/ml), cells were disrupted by sonication. The cellular debris were removed by a short clarifying spin after which the supernatant was subjected to an ultracentrifugation step (100,000×g for 40 min at 4°C) to obtain the soluble fraction (containing the cytoplasmic and periplasmic fraction). The NaCl concentration was adjusted to 0.5 M, imidazole (15 mM) was added, and the samples were incubated with Ni2+–NTA agarose for 120 min at 4°C. The slurry was loaded into a column and washed with buffer A (PBS with 0.5 M NaCl) supplemented with 15 mM imidazole followed by buffer A plus 30 mM imidazole. Samples were eluted with buffer A supplemented with 150 mM imidazole. To remove the imidazole and NaCl, the collected enzyme fraction was applied to a pre-equilibrated desalting column (Biorad). To monitor the purification procedure, samples were taken of each fraction and analyzed by sodium dodecyl sulfate-poly acrylamide gel electroforese (SDS-PAGE) and protein staining.
Cell fractionations
Cells expressing TfuDyP were grown as described above. Twenty OD660 units of cells were harvested and fractionated into a spheroplast and periplasmic fraction as described previously (Huber et al. 2005). The cytoplasmic fraction was obtained as follows. After disruption of the spheroplasts by sonication and a brief, clarifying spin, the clarified lysate was centrifuged (100,000×g for 40 min at 4°C), and the supernatant was taken as the cytoplasmic fraction. Subsequently, proteins were precipitated by trichloroacetic acid and analyzed by SDS-PAGE and immunoblotting.
SDS-PAGE and immunoblotting
Cellular fractions were normalized on the basis of the OD660, and samples of these fractions, containing equal OD660 units, were analyzed on standard 12% SDS-PAGE gels. Proteins were transferred to nitrocellulose membrane (Amersham Biosciences) using a semidry apparatus from Biorad. Immunodetection was performed using the primary antisera described above, a secondary horseradish peroxidase-coupled antiserum (Rockland) and the ECL system from Amersham Biosciences (according to the instructions of the manufacturer). Proteins were visualized using the Fujifilm LAS-3000 imaging system. For native poly acrylamide gel electroforese (PAGE) gel electrophoresis, 3.0 μg of purified enzyme was analyzed on a 7.5% PAGE gel; SDS was omitted from all buffers, and samples were not reduced. Proteins were visualized by protein staining or stained for peroxidase activity, using 1.0 mM 3,3-diaminobenzidine (DAB) and 0.03% H2O2 in 25 mM citrate buffer pH 3.5.
Enzyme assay and steady-state kinetic parameters
TfuDyP activity was measured spectrophotometrically (Perkin–Elmer Lambda Bio40) at ambient temperature in 25 mM citrate buffer pH 3.5, containing 35 nM of purified enzyme and 100 μM H2O2. The oxidation of the following substrates was tested at the indicated wavelength: Reactive Blue 19 (100 μM) at 595 nm (ε = 10 mM−1 × cm−1), Reactive Blue 4 (100 μM) at 595 nm (ε = 4.2 mM−1 × cm−1), Reactive Black 5 (20 μM) at 597 nm (ε = 37 mM−1 × cm−1), guaiacol (100 μM) at 465 nm (ε = 26.6 mM−1 × cm−1), 2,6-dimethoxyphenol (100 μM) at 470 nm (ε = 49.6 mM−1 × cm−1), veratryl alcohol (100 μM) at 340 nm (ε = 93 mM−1 × cm−1) and o-phenylenediamine (100 μM) at 420 nm (ε = 31.3 mM−1 × cm−1). Control reactions were included without enzyme, H2O2, or both. Conversion of substrates was only observed when both TfuDyP and H2O2 were present. The steady-state kinetic parameters of TfuDyP were determined by analyzing the reactivity of the enzyme at different Reactive Blue 19 or H2O2 concentrations. Data were fitted with Sigmaplot 10 software, using the Michaelis–Menten equation. Catalase activity of TfuDyP in 25 mM citrate buffer pH 3.5 was analyzed spectrophotometrically by following the consumption of H2O2 at 240 nm as described (Yumoto et al. 2000). All experiments were performed in duplicates, and the values obtained were within 10% of each other.
Influence of pH and temperature on enzyme activity and stability
The pH optimum of TfuDyP was determined by measuring the Reactive Blue 19-decolorizing activity of the enzyme as described above in 25 mM citrate buffer adjusted to different pH values. To establish the optimum temperature of TfuDyP, the activity towards Reactive Blue 19 was tested at temperatures in the range of 25–65°C. Before adding the enzyme, the assay mixture was equilibrated for 20 min at the appropriate temperature. The influence of the temperature on TfuDyP stability was investigated by incubating 40-μl aliquots of the purified enzyme (19 μM) at ambient temperature, 30°C, 40°C, 50°C, and 60°C. Samples were taken, placed on ice, after which enzyme activity was analyzed as described above.
Spectral assays
Absorption spectra of purified TfuDyP or its mutants were recorded on a Perkin–Elmer Lambda Bio40 spectrophotometer at ambient temperature. In reduction experiments, solutions were made anaerobic by flushing the cuvette with argon. Enzyme reduction was achieved by sodium dithionite or H2O2. The protoheme content was determined by the pyridine ferro-hemochrome method as described previously (Yumoto et al. 2000), and the heme content was calculated on the basis of the extinction coefficient (34.5 mM−1 × cm−1) of pyridine hemochrome b (Berry and Trumpower 1987).
TfuDyP catalyzed sulfoxidations
The enantioselectivity of TfuDyP in sulfoxidations was determined as follows. Reactions were performed in Pyrex tubes in a total volume of 1 ml. The reaction mixture typically contained 25 mM citrate buffer pH 3.5, 1 mM H2O2, 2.5 mM substrate, and 8 μM of TfuDyP. Reactions were incubated for 36 h at 30°C at 200 rpm and were subsequently analyzed as previously described (van Hellemond et al. 2007; Torres Pazmiño et al. 2008).
Analytical methods
Protein concentrations were determined using the Bradford method with BSA as standard. For enzymatic assays, the protein content was analyzed by Waddell’s method (Wolf 1983). The oligomeric form of TfuDyP was investigated by gel permeation chromatography, using a Workbeads 40 SEC column (Bio-Works). The column was equilibrated with PBS and subsequently calibrated with a set of protein standards (6.5, 13.7, 29, 43, and 75 kDa).
Sequence analysis
TfuDyP was identified by PSI-BLAST searches of the NCBI bacterial genome sequence database. The presence of a potential Tat-dependent signal sequence was verified, using the TatP server (Bendtsen et al. 2005). Sequence alignment was performed using ClustalX version 2.0 (Larkin et al. 2007) with subsequent manual refinements.
Results
Identification, expression, and verification of Tat-dependent periplasmic export of TfuDyP
Using the protein sequence of Thanatephorus cucumeris Dec1 DyP, a prototype fungal DyP-type peroxidase (Kim and Shoda 1999), PSI-BLAST searches were performed to identify open reading frames (ORFs), encoding putative DyP-type peroxidases. This revealed a large number of ORFs encoding bacterial DyP-type peroxidases while relatively few homologs were found in sequenced genomes of fungi, other eukaryotes, or archeae. The bacterial hits were subsequently analyzed for the presence of signal sequences, and several were found to contain an N-terminal twin-arginine sequence motif which was indicative for a Tat-signal sequence. We chose to focus on a gene that encoded a potential Tat-signal sequence containing DyP-type peroxidase from T. fusca (TfuDyP; Fig. 1a). We have shown before that this actinomycete can be a good source for robust enzymes (Fraaije et al. 2005). TfuDyP consists of 430 amino acids with a calculated mass of 46 kDa for the unprocessed precursor protein. Sequence alignment showed that TfuDyP is homologous to the Tat-dependently exported DyP-type peroxidases YwbN from Bacillus subtilis and YcdB from E. coli with a sequence identity of ∼30% (Jongbloed et al. 2004; Sturm et al. 2006). The known fungal DyP-type peroxidases display sequence similarities of <20%. The so-called GXXDG motif, which represents a highly conserved cluster of residues in the heme-binding site of DyP-type peroxidases, was readily identified in the sequence of TfuDyP (Sugano 2009) (Fig. 1a).

Export of TfuDyP to the periplasm by the Tat system in E. coli. a Alignment of TfuDyP and the sequences of YwbN and YcdB from B. subtilis and E. coli, respectively. Solid bars under the sequences indicate the Tat-signal sequence in which the two arginines of the twin-arginine motif are highlighted by two asterisks. For clarity, only residues conserved in all proteins are shaded. The conserved GXXDG motif is boxed, and residues typically conserved amongst DyP-type peroxidases are displayed by asterisks. b Tat-dependent periplasmic transport of TfuDyP was investigated in wild-type (WT) MC4100 cells and strain B1LK0 (ΔTatC). E. coli cells expressing TfuDyP were fractionated into total cells (T), cytoplasm (C), and periplasm (P). Samples were normalized on the basis of OD660 and analyzed by immunoblotting with the indicated antisera. Black lines indicate that intervening lanes have been spliced out. The precursor (black dot) and mature form (arrowhead) are indicated
Next, the gene encoding TfuDyP (including the signal sequence) was PCR-amplified from T. fusca genomic DNA and cloned into an arabinose-inducible expression plasmid. To aid in the detection and purification of the recombinant protein, a Myc–His6 bipartite tag preceded by a flexible linker, was attached to the C-terminus of the protein. We first tested whether the E. coli Tat system is required for periplasmic export of heterologously expressed TfuDyP. In E. coli, the membrane-bound Tat translocase is formed by TatABC subunits and is dedicated to the periplasmic transport of fully folded and often cofactor-containing substrate proteins (Lee et al. 2006). The TatBC subcomplex binds substrate proteins, which is believed to trigger subsequent recruitment of TatA to form the TatABC translocation complex (Lee et al. 2006). Consequently, the absence of TatC results in a block of Tat-dependent export as protein substrates are unable to associate with the receptor complex. The cellular localization of recombinant TfuDyP was studied in wild-type E. coli cells and cells of a TatC null strain (ΔTatC). Cells expressing TfuDyP were fractionated into a cytoplasmic and periplasmic fraction. The different subcellular fractions were analyzed by immunoblotting, using the indicated antisera. Figure 1b shows that in wild-type cells TfuDyP is present as two forms (indicated by a closed circle and arrowhead, respectively). The upper band is found predominantly in the cytoplasmic fraction (C), unlike the lower form, which is mainly observed in the periplasmic fraction (P). Therefore, the upper band represents most likely the unprocessed precursor, whereas the lower band corresponds to the mature form. These data clearly show that, in wild-type cells, the precursor form has been exported to the periplasm and processed to the mature form. In the absence of TatC, periplasmic export of TfuDyP is almost completely blocked as the vast majority of the protein is observed in the cytoplasmic fraction. Remarkably, under these conditions, TfuDyP is mainly present as a cytoplasmic, mature-sized species, which may result from degradation of the signal sequence by cytoplasmic proteases as observed previously by us and others (Thomas et al. 2001; Sturm et al. 2006; van Bloois et al. 2009).
As controls to monitor the efficiency of the fractionation procedure, the levels of DnaK and DsbA, which serve as cytoplasmic or periplasmic marker, respectively, were analyzed in the same samples by immunoblotting. The data show that DnaK is restricted to the cytoplasmic fraction and DsbA is mainly detected in the periplasmic fraction, demonstrating the efficiency of the fractionation procedure. These data show that TfuDyP requires the Tat system for periplasmic export as expected.
Purification of recombinant TfuDyP and spectral characterization of heme cofactor
To enable further biochemical characterization, TfuDyP was heterologously expressed in wild-type E. coli cells and subsequently purified from the soluble fraction (containing the cytoplasmic and periplasmic fraction) by one-step Ni2+–NTA agarose chromatography under native conditions. This procedure yielded approximately 3 mg of purified enzyme from 1 L of culture broth. Samples were taken of the cell extract (CE), soluble fraction, containing cytoplasmic and periplasmic proteins, flowthrough (FT), wash steps (W1 and W2), and eluate and subjected to SDS-PAGE analysis followed by protein staining of the gel (Fig. 2a). As observed previously (compare Fig. 1a), TfuDyP is present as two forms in the eluate (indicated by a closed circle and arrowhead, respectively) of which the unprocessed precursor migrates in the gel at a position corresponding to 45 kDa. This corresponds nicely to the calculated mass of the unprocessed precursor of 46 kDa.

TfuDyP contains non-covalently bound heme. a MC1061 cells expressing TfuDyP were fractionated into a cell extract (CE) and soluble fraction (Sol), containing cytoplasmic and periplasmic proteins. TfuDyP was isolated from the soluble fraction by Ni2+–NTA agarose purification as described in “Materials and methods”. To monitor the purification procedure, samples were taken and analyzed by SDS-PAGE followed by protein staining of the gel. FT flow through, W1 and W2 first and second wash fractions, E eluate. The precursor (black dot) and mature form (arrowhead) are indicated. b UV-visible spectra of oxidized (ox) and reduced (red) TfuDyP. Spectra were recorded with the purified enzyme in PBS at ambient temperature. TfuDyP was reduced by addition of sodium dithionite. The inset shows the pyridine hemochrome spectrum of TfuDyP after reduction with dithionite in pyridine as described in “Materials and methods”
Several oligomeric states of DyP-type peroxidases have been reported, ranging from monomers to hexamers (Ebihara et al. 2005; Sugano et al. 2007; Zubieta et al. 2007b). Gel permeation experiments with purified TfuDyP revealed that this enzyme exists as a monomer in solution (data not shown).
All hitherto characterized members of the DyP-type peroxidase family contain non-covalently bound heme as cofactor (Sturm et al. 2006; Sugano et al. 2007; Zubieta et al. 2007a, b). Therefore, we analyzed the spectral properties of recombinant TfuDyP to assess whether this enzyme also contains a heme cofactor. Notably, during purification of TfuDyP, a brown / reddish color of the soluble fraction was observed, which was more pronounced in the eluate, indicating that a chromogenic cofactor is associated with the enzyme. Figure 2b shows the spectral characteristics of purified TfuDyP. A large Soret band was observed at 409 nm together with two small absorbance maxima at 540 and 575 nm. The Reinheitzahl value (the ratio of A 409/A 280) for the purified enzyme was 0.90, which compares favorably with those reported for other DyP-type peroxidases (Kim and Shoda 1999; Sturm et al. 2006; Zubieta et al. 2007b). These data indicate that TfuDyP indeed contains a heme cofactor, which is in an oxidized state. A more detailed analysis of the heme cofactor was performed by determining the spectral properties of the enzyme after pyridine/NaOH treatment. This revealed a pyridine hemochrome spectrum (Fig. 2b inset) which is identical to that of protoheme IX (non-covalently bound heme b) (Berry and Trumpower 1987). From this spectrum, the heme content per mole of TfuDyP was estimated at 0.6, indicating that TfuDyP contains a single heme cofactor. Thus similar to other DyP-type peroxidases, TfuDyP protomers possess a non-covalently bound heme (protoheme IX) as cofactor and appear to be partly apo upon purification (Sturm et al. 2006; Sugano et al. 2007; Zubieta et al. 2007a, b).
Determination of enzymatic activity and substrate specificity
After having established that TfuDyP is a hemoprotein, we further delineated the biochemical characteristics of this enzyme. First, we tested whether TfuDyP can be reduced by dithionite. Anaerobic reduction of the enzyme with sodium dithionite altered the spectral shape significantly. The Soret band decreased and shifted towards 431 nm, and the two peaks at 540 and 575 nm condensed, resulting in a broad peak with a maximum at 560 nm (Fig. 2b). Also, the reactivity of the enzyme with H2O2 was tested, resulting in a major decrease of the Soret band (data not shown). The reactivity of TfuDyP with dithionite and H2O2 and the related observed spectral characteristics are fully in line with typical features of a heme-containing peroxidase.
To probe whether the purified enzyme displays (DyP-type) peroxidase activity, its reactivity towards a subset of well-known peroxidase substrates and anthraquinone and azo dyes was tested (Table 1). The oxidation of the indicated compounds was assayed spectrophotometrically at the appropriate wavelengths in 25 mM citrate buffer (pH 3.5) containing 100 μM H2O2. TfuDyP shows a modest activity towards substrates that are typical substrates for plant peroxidases, such as guaiacol and 2,6-dimethoxyphenol. Additionally, we found that TfuDyP is able to act on veratryl alcohol, o-phenylenediamine, and 3,3-diaminobenzidine but not very efficiently (Table 1 and data not shown). The enzyme showed high reactivity towards the anthraquinone dyes, Reactive Blue 19, and Reactive Blue 4, whereas the azo dye Reactive Black 5 was poorly decolorized. The data also show that TfuDyP displays dye-decolorizing activity similar to related fungal and bacterial proteins (Kim and Shoda 1999; Zubieta et al. 2007a). Moreover, no catalase activity was detected under these experimental conditions, indicating that TfuDyP does not function as a catalase–peroxidase (data not shown).
Analysis of catalytic properties and steady-state kinetic parameters
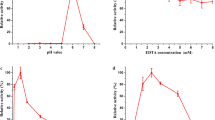
Because the physiological substrate of TfuDyP is not known, we used Reactive Blue 19 as a representative substrate in subsequent experiments because of the high reactivity of the enzyme towards this dye. All DyP-type peroxidases characterized so far exhibit significant peroxidase activity at low pH. To determine the pH profile of TfuDyP, the decolorizing activity of the enzyme was analyzed spectrophotometrically in 25 mM citrate buffer adjusted at different pHs and containing 100 μM H2O2. We found that TfuDyP displayed the best Reactive Blue 19 decolorizing activity at pH 3.5 (Fig. 3a). A similar pH profile has been observed for fungal and bacterial DyP-type peroxidases (Kim and Shoda 1999; Sturm et al. 2006).

TfuDyP is a bona fide DyP-type peroxidase. a Influence of pH on Reactive Blue 19-decolorizing activity of TfuDyP. The activity of TfuDyP with Reactive Blue 19 was determined in 25 mM citrate buffer adjusted to different pH values and containing 100 μM H2O2. b Identification of enzymatically active forms of TfuDyP. Purified TfuDyP was analyzed on a 7.5% native PAGE gel, and one part of the gel was stained with Coomassie Brilliant Blue (CBB), and the other part was stained for peroxidase activity, using 3,3-diaminobenzidine (DAB). The apo-precursor (asterisk), precursor (black dot), and mature form (greater than symbol) are indicated. c Role of D242 and H338 in enzymatic activity and heme binding. UV-visible spectra of purified TfuDyPD242A (D242A), TfuDyPH338A (H338A), and the wild-type enzyme (WT). Spectra were recorded with the enzyme in PBS at ambient temperature
In order to assess the optimum temperature, the activity of TfuDyP towards Reactive Blue 19 was assayed spectrophotometrically at various temperatures in 25 mM citrate buffer (pH 3.5), containing 100 μM H2O2. It was found that the optimum temperature for enzymatic activity was approximately 25°C. The thermostability of TfuDyP was examined by testing the reactivity towards Reactive Blue 19 after heat treatment of the enzyme at different temperatures. These studies revealed that TfuDyP became more active at higher temperatures. Specifically, upon incubation at 60°C, an almost two-fold increase in activity was observed within 10 min. A similar activation effect at elevated temperatures has been observed for other thermostable enzymes (Stutzenberger and Lupo 1986; Antoine et al. 1999; Fraaije et al. 2005). After incubation for 2 h at 60°C, the enzyme lost 50% of its original activity, while at 30°C and 40°C, TfuDyP retained its activity after this period. This indicates that TfuDyP is a reasonable thermostable peroxidase.
For Reactive Blue 19 and H2O2, the steady-state kinetic parameters were determined. This revealed that TfuDyP displays a comparable affinity and turnover number for both substrates as K m values of 29 and 27 μM and k cat values of 10 and 9 s−1 were found. The catalytic efficiency (k cat/ K m ) for Reactive Blue 19 and H2O2 was 345 and 333 s−1 × mM−1, respectively.
Enantioselectivity of TfuDyP
So far, no DyP-type peroxidase has ever been reported to be active with sulfides while (enantioselective) oxidations of sulfides have been extensively studied with plant and animal peroxidases. The enantioselective sulfoxidation of aromatic sulfides by plant peroxidases, such as HRP and LiP, is well established (van Rantwijk and Sheldon 2000; Klibanov 2003; Veitch 2004). To investigate whether TfuDyP is also able to catalyze this type of reaction enantioselectively, the sulfoxidation of several aromatic sulfides was tested followed by analysis of the products on a chiral GC column. All tested sulfides were enantioselectively converted into the corresponding sulfoxides by TfuDyP (Table 2). The best enantioselectivity was obtained with methyl phenyl sulfide, which yielded the (R)-sulfoxide with 61% ee. Notably, this value represents a lower limit as a non-enantioselective background oxidation was also observed. Despite the enantioselective oxidation of the tested aromatic sulfides, the overall conversion was relatively poor, consistent with the observed modest activity of TfuDyP towards relatively small aromatic substrates (Table 1). Nonetheless, the enantioselectivity of TfuDyP also confirms that the observed oxidations are truly enzyme-catalyzed by enantioselective binding of the substrate near the oxidizing heme cofactor.
Identification of enzymatically active forms
E. coli YcdB is able to incorporate heme in the cytoplasm prior to translocation across the inner membrane. This indicates that the unprocessed precursor and the periplasmic mature protein are enzymatically active as both contain the heme cofactor (Sturm et al. 2006). To investigate whether the unprocessed precursor and mature form of TfuDyP are enzymatically active, we subjected purified TfuDyP to native gel electrophoresis. Staining of the gel with Coomassie Brilliant Blue (CBB) revealed three protein bands (Fig. 3b). When staining for peroxidase activity using DAB only two bands appeared (Fig. 3b). The upper band, which only appeared after CBB staining, was not observed after staining for peroxidase activity. Most likely, this band represents the apo-precursor which may have a different conformation due to the absence of heme, explaining its different migration behavior in native gel electrophoresis. DAB staining identified two bands that could be assigned to the unprocessed precursor and mature form after comparison with the CBB stained gel. The peroxidase activity displayed by the unprocessed precursor and mature form of TfuDyP indicate that both forms contain the heme cofactor like YcdB (Sturm et al. 2006).
Role of D242 and H338
The heme-binding site of DyP-type peroxidases contains a cluster of highly conserved residues, which includes the so-called GXXDG motif (Sugano 2009). Structural and biochemical evidence suggests that the aspartate of this motif (D242 in TfuDyP) dictates that DyP-type peroxidases are most active at low pH (Sugano et al. 2007). Furthermore, all DyP-type peroxidases contain a highly conserved histidine residue in the C-terminal domain of the protein (H338 in TfuDyP), which is part of the active site (Sugano 2009). The available structures of DyP, BtDyP, and TyrA show that this histidine residue is an important heme ligand (Sugano et al. 2007; Zubieta et al. 2007a, b). To verify that D242 and H338 are essential for peroxidase activity and/or cofactor binding, site-directed mutagenesis was employed to replace these residues by alanine. The mutant enzymes were purified, and their UV-visible spectra were recorded. Figure 3c clearly shows that the H338A mutant lacks the heme cofactor as the diagnostic Soret band is not observed in contrast to the D242A variant and the wild-type protein. Interestingly, the spectral properties of the D242A mutant are significantly different when compared with the wild-type protein. These spectral changes may reflect structural changes in the heme-binding region of the D242A mutant or altered ligation of the heme iron atom. The mutant enzymes displayed 0.7% (D242A) and 3% (H338A) of the Reactive Blue 19-decolorizing activity of the wild-type protein. These data clearly show that D242 is important for enzymatic activity, and H338 is crucial for proper cofactor assembly.
Discussion
Although DyP-type peroxidases represent a novel superfamily of peroxidases, only a few members have been characterized in some detail (Sugano 2009). Hence, our understanding of these enzymes is limited. In the present study, we have used the available bacterial genome sequence information by searching for proteins with homology to a known fungal DyP-type peroxidase and identified an ORF in the genome of T. fusca encoding a putative DyP-type peroxidase bearing a Tat-signal sequence. Heterologous expression of TfuDyP in E. coli revealed that the protein is transported to the periplasm by the Tat system. Similar to other DyP-type peroxidases, TfuDyP contains non-covalently bound heme (protoheme IX) as cofactor; it is most active at low pH and shows high reactivity towards anthraquinone dyes and a moderate activity towards standard peroxidase substrates, aromatic sulfides and azo dyes (Kim and Shoda 1999; Sugano et al. 2000, 2007; Johjima et al. 2003; Sturm et al. 2006; Zubieta et al. 2007a, b). These data suggest that TfuDyP is indeed a bona fide DyP-type peroxidase. Similar to plant peroxidases, TfuDyP is able to oxidize aromatic sulfides enantioselectively, resulting in the corresponding (R)-sulfoxides. Notably, we show for the first time that a DyP-type peroxidase is able to catalyze this type of reaction. The displayed enantioselectivity by TfuDyP in sulfoxidations suggest a selective binding of the substrate within the active site. Moreover, TfuDyP is quite robust as the enzyme lost 50% of its original activity after 2 h incubation at 60°C.
The most distinguishing features of DyP-type peroxidases are their unique reaction characteristics and structure (Sugano 2009). The activity of plant peroxidases against standard peroxidase substrates and azo dyes has been well documented in contrast to the degradation of anthraquinone dyes by these enzymes (Burner et al. 2000; Reszka et al. 2001; Stolz 2001; Reszka et al. 2005; Chen 2006). Conversely, the reported fungal DyP-type peroxidases are known to be highly active against anthraquinone dyes and display only modest activity against standard peroxidase substrates and azo dyes (Kim and Shoda 1999; Sugano et al. 2000). For TfuDyP, we have now also established that sulfides can be oxidized by a DyP-type peroxidase but with a poor efficiency. These observations reveal a major difference in substrate acceptance profiles for the different peroxidase superfamilies. Structurally, DyP-type peroxidases comprise two domains that contain α-helices and anti-parallel β-sheets, unlike plant peroxidases that are primarily α-helical proteins. Both domains adopt a unique ferredoxin-like fold and form an active site crevice with the heme cofactor sandwiched in between (Banci 1997; Sugano 2009). The molecular basis for the different and complementing substrates profiles for plant and DyP-type peroxidases most likely stems from their structural differences.
DyP-type peroxidases have been included as a separate superfamily in databases, such as Peroxibase, Pfam, and InterPro. The most comprehensive overview of the DyP-type peroxidase superfamily is offered by the InterPro database. According to this database, which surveys all available genome sequences, the DyP superfamily comprises almost 1,000 members of which 881 members are found in bacteria, 11 in cyanobacteria, 39 in fungi, 19 in higher eukaryotes, and one is unclassified. With regards to the remarkable abundance of DyP-type peroxidases in bacteria, we propose that the superfamily of DyP-type peroxidases should be renamed into the superfamily of bacterial peroxidases in analogy and addition to the superfamilies of plant and animal peroxidases. Furthermore, Dyp-type peroxidases are, according to PeroxiBase, further subdivided into the phylogenetically distinct classes A, B, C, and D.
Intriguingly, many of the putative bacterial DyP-type peroxidases are predicted to be cytoplasmic enzymes (PeroxiBase class B and C), which suggests that they play a role in an intracellular metabolic pathway. The exact role(s) of these cytosolic bacterial peroxidases have to be established. In contrast, a small group of putative bacterial DyP-type peroxidases contain a Tat-signal sequence (PeroxiBase class A), indicating that these enzymes function outside the cytoplasm and in the case of TfuDyP extracellularly. This would fit in a role of this enzyme in dye degradation, as for the sequence-related peroxidases in fungi. Such an activity has not been described before for this actinomycete but is in line with various reports that indicate that actinomycetes have the capacity to degrade to some extent complex molecules, e.g., lignin or lignin-derived compounds (Kirby 2006). Thereby, it would represent a bacterial counterpart of the fungal lignin peroxidases. It also complies with the finding that T. fusca harbors many genes that encode for enzymes involved in degradation of aromatic compounds.
The highly conserved heme-binding motif of plant peroxidases is not present in DyP-type peroxidases. Rather, the heme-binding site of DyP-type peroxidases contains a cluster of highly conserved residues, which includes the so-called GXXDG motif (Fig. 1a; Sugano 2009). Recently, the importance of the conserved aspartate (D171) in the GXXDG motif of a related protein, DyP of T. cucumeris, was investigated. It was found that replacement of aspartate by asparagine abolished enzymatic activity. This is in agreement with the proposed function as an acid–base catalyst in the catalytic mechanism at low pH, as indicated by structural data (Sugano et al. 2007). Consistent with these data, replacement of the aspartate in the GXXDG motif of TfuDyP (D242) by alanine inactivated the enzyme but did not interfere with cofactor assembly, indicating that D242 of TfuDyP plays a similar role in the catalytic mechanism as D171 of T. cucumeris DyP.
Sequence alignments have also identified a conserved histidine residue in the N-terminal domain of fungal DyP-type peroxidases, such as H164 in T. cucumeris DyP. H164 was previously assigned as ligand of the heme but recent structural data demonstrated that this residue does not contribute to heme binding (Sugano et al. 2004, 2007). While this residue is not present in bacterial DyP-type peroxidases, all DyP-type peroxidases contain a highly conserved histidine residue in the C-terminal domain of the protein, which is part of the active site. The available structures show that this histidine residue is an important heme ligand and represents the proximal histidine residue (Sugano et al. 2007; Zubieta et al. 2007a, b). However, the role of this residue in heme binding was never experimentally verified. To confirm whether the proximal histidine is indeed required for proper heme binding, we replaced the corresponding residue in TfuDyP, H338 (Fig. 1a), by an alanine. This abrogated heme binding and inactivated the enzyme, suggesting that the conserved H338 is indeed the proximal histidine of TfuDyP and other DyP-type peroxidases.
Taken together, our data show that TfuDyP is a novel member of the growing superfamily of bacterial peroxidases (previously DyP-type peroxidases) and represents the first characterized substrate protein of the T. fusca Tat system. The detailed biochemical characterization of TfuDyP reported here contributes significantly to our understanding of these enzymes and further underscores the biotechnological potential of TfuDyP because: (1) it was obtained mainly as a holoenzyme in contrast to other heterologously expressed bacterial DyP-type peroxidases (Zubieta et al. 2007a, b), (2) the enzyme appears to be robust, and (3) it accepts a broad range of substrates. Thereby, it is a promising alternative for other peroxidases, such as HRP, which are notoriously difficult to express in, e.g., E. coli. In addition, TfuDyP seems a good candidate for whole-cell biotransformations as it is transported to the E. coli periplasm, which increases its substrate accessibility as most substrates are able to enter the periplasm in contrast to the cytoplasm (Chen 2007).
References
Antoine E, Rolland JL, Raffin JP, Dietrich J (1999) Cloning and over-expression in Escherichia coli of the gene encoding NADPH group III alcohol dehydrogenase from Thermococcus hydrothermalis. Characterization and comparison of the native and the recombinant enzymes. Eur J Biochem 264:880–889
Banci L (1997) Structural properties of peroxidases. J Biotechnol 53:253–263
Bendtsen JD, Nielsen H, Widdick D, Palmer T, Brunak S (2005) Prediction of twin-arginine signal peptides. BMC Bioinformatics 6:167
Berry EA, Trumpower BL (1987) Simultaneous determination of hemes a, b, and c from pyridine hemochrome spectra. Anal Biochem 161:1–15
Bogsch EG, Sargent F, Stanley NR, Berks BC, Robinson C, Palmer T (1998) An essential component of a novel bacterial protein export system with homologues in plastids and mitochondria. J Biol Chem 273:18003–18006
Burner U, Krapfenbauer G, Furtmuller PG, Regelsberger G, Obinger C (2000) Oxidation of hydroquinone, 2,3-dimethylhydroquinone and 2,3,5-trimethylhydroquinone by human myeloperoxidase. Redox Rep 5:185–190
Casadaban MJ (1976) Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage lambda and Mu. J Mol Biol 104:541–555
Casadaban MJ, Cohen SN (1980) Analysis of gene control signals by DNA fusion and cloning in Escherichia coli. J Mol Biol 138:179–207
Chen H (2006) Recent advances in azo dye degrading enzyme research. Curr Protein Pept Sci 7:101–111
Chen RR (2007) Permeability issues in whole-cell bioprocesses and cellular membrane engineering. Appl Microbiol Biotechnol 74:730–738
Davies MJ, Hawkins CL, Pattison DI, Rees MD (2008) Mammalian heme peroxidases: from molecular mechanisms to health implications. Antioxid Redox Signal 10:1199–1234
Ebihara A, Okamoto A, Kousumi MR, Ueyama N, Yokoyama S, Kuramitsu S (2005) Structure-based functional identification of a novel heme-binding protein from Thermus thermophilus HB8. J Struct Funct Genomics 6:21–32
Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer EL, Bateman A (2008) The Pfam protein families database. Nucleic Acids Res 36:D281–288
Fraaije MW, Wu J, Heuts DP, van Hellemond EW, Spelberg JH, Janssen DB (2005) Discovery of a thermostable Baeyer–Villiger monooxygenase by genome mining. Appl Microbiol Biotechnol 66:393–400
Huber D, Boyd D, Xia Y, Olma MH, Gerstein M, Beckwith J (2005) Use of thioredoxin as a reporter to identify a subset of Escherichia coli signal sequences that promote signal recognition particle-dependent translocation. J Bacteriol 187:2983–2991
Johjima T, Ohkuma M, Kudo T (2003) Isolation and cDNA cloning of novel hydrogen peroxide-dependent phenol oxidase from the basidiomycete Termitomyces albuminosus. Appl Microbiol Biotechnol 61:220–225
Jongbloed JD, Grieger U, Antelmann H, Hecker M, Nijland R, Bron S, van Dijl JM (2004) Two minimal Tat translocases in Bacillus. Mol Microbiol 54:1319–1325
Kim SJ, Shoda M (1999) Purification and characterization of a novel peroxidase from Geotrichum candidum dec 1 involved in decolorization of dyes. Appl Environ Microbiol 65:1029–1035
Kirby R (2006) Actinomycetes and lignin degradation. Adv Appl Microbiol 58:125–168
Klibanov AM (2003) Asymmetric enzymatic oxidoreductions in organic solvents. Curr Opin Biotechnol 14:427–431
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948
Lee PA, Tullman-Ercek D, Georgiou G (2006) The bacterial twin-arginine translocation pathway. Annu Rev Microbiol 60:373–395
Loughran NB, O’Connor B, O’Fagain C, O’Connell MJ (2008) The phylogeny of the mammalian heme peroxidases and the evolution of their diverse functions. BMC Evol Biol 8:101
Passardi F, Cosio C, Penel C, Dunand C (2005) Peroxidases have more functions than a Swiss army knife. Plant Cell Rep 24:255–265
Passardi F, Theiler G, Zamocky M, Cosio C, Rouhier N, Teixera F, Margis-Pinheiro M, Loannidis V, Penel C, Falquet L, Dunand C (2007) PeroxiBase: the peroxidase database. Phytochemistry 68:1605–1611
Regalado C, García-Almendárez BE, Duarte-Vázquez MA (2004) Biotechnological applications of peroxidases. Phytochem Rev 3:243–256
Reszka KJ, McCormick ML, Britigan BE (2001) Peroxidase- and nitrite-dependent metabolism of the anthracycline anticancer agents daunorubicin and doxorubicin. Biochemistry 40:15349–15361
Reszka KJ, Wagner BA, Burns CP, Britigan BE (2005) Effects of peroxidase substrates on the Amplex red/peroxidase assay: antioxidant properties of anthracyclines. Anal Biochem 342:327–337
Stolz A (2001) Basic and applied aspects in the microbial degradation of azo dyes. Appl Microbiol Biotechnol 56:69–80
Sturm A, Schierhorn A, Lindenstrauss U, Lilie H, Bruser T (2006) YcdB from Escherichia coli reveals a novel class of Tat-dependently translocated hemoproteins. J Biol Chem 281:13972–13978
Stutzenberger F, Lupo D (1986) Ph-dependent thermal-activation of endo-1, 4-beta-glucanase in Thermomonospora curvata. Enzyme Microb Technol 8:205–208
Sugano Y (2009) DyP-type peroxidases comprise a novel heme peroxidase family. Cell Mol Life Sci 66:1387–1403
Sugano Y, Nakano R, Sasaki K, Shoda M (2000) Efficient heterologous expression in Aspergillus oryzae of a unique dye-decolorizing peroxidase, DyP, of Geotrichum candidum Dec 1. Appl Environ Microbiol 66:1754–1758
Sugano Y, Ishii Y, Shoda M (2004) Role of H164 in a unique dye-decolorizing heme peroxidase DyP. Biochem Biophys Res Commun 322:126–132
Sugano Y, Muramatsu R, Ichiyanagi A, Sato T, Shoda M (2007) DyP, a unique dye-decolorizing peroxidase, represents a novel heme peroxidase family: ASP171 replaces the distal histidine of classical peroxidases. J Biol Chem 282:36652–36658
Thomas JD, Daniel RA, Errington J, Robinson C (2001) Export of active green fluorescent protein to the periplasm by the twin-arginine translocase (Tat) pathway in Escherichia coli. Mol Microbiol 39:47–53
Torres Pazmiño DE, Baas BJ, Janssen DB, Fraaije MW (2008) Kinetic mechanism of phenylacetone monooxygenase from Thermobifida fusca. Biochemistry 47:4082–4093
van Bloois E, Winter RT, Janssen DB, Fraaije MW (2009) Export of functional Streptomyces coelicolor alditol oxidase to the periplasm or cell surface of Escherichia coli and its application in whole-cell biocatalysis. Appl Microbiol Biotechnol 83:679–687
van Hellemond EW, Janssen DB, Fraaije MW (2007) Discovery of a novel styrene monooxygenase originating from the metagenome. Appl Environ Microbiol 73:5832–5839
van Rantwijk F, Sheldon RA (2000) Selective oxygen transfer catalysed by heme peroxidases: synthetic and mechanistic aspects. Curr Opin Biotechnol 11:554–564
Veitch NC (2004) Horseradish peroxidase: a modern view of a classic enzyme. Phytochemistry 65:249–259
Welinder KG, Mauro JM, Norskov-Lauritsen L (1992) Structure of plant and fungal peroxidases. Biochem Soc Trans 20:337–340
Wolf P (1983) A critical reappraisal of Waddell’s technique for ultraviolet spectrophotometric protein estimation. Anal Biochem 129:145–155
Yumoto I, Ichihashi D, Iwata H, Istokovics A, Ichise N, Matsuyama H, Okuyama H, Kawasaki K (2000) Purification and characterization of a catalase from the facultatively psychrophilic bacterium Vibrio rumoiensis S-1(T) exhibiting high catalase activity. J Bacteriol 182:1903–1909
Zubieta C, Joseph R, Krishna SS, McMullan D, Kapoor M, Axelrod HL, Miller MD, Abdubek P, Acosta C, Astakhova T, Carlton D, Chiu HJ, Clayton T, Deller MC, Duan L, Elias Y, Elsliger MA, Feuerhelm J, Grzechnik SK, Hale J, Han GW, Jaroszewski L, Jin KK, Klock HE, Knuth MW, Kozbial P, Kumar A, Marciano D, Morse AT, Murphy KD, Nigoghossian E, Okach L, Oommachen S, Reyes R, Rife CL, Schimmel P, Trout CV, van den Bedem H, Weekes D, White A, Xu Q, Hodgson KO, Wooley J, Deacon AM, Godzik A, Lesley SA, Wilson IA (2007a) Identification and structural characterization of heme binding in a novel dye-decolorizing peroxidase, TyrA. Proteins 69:234–243
Zubieta C, Krishna SS, Kapoor M, Kozbial P, McMullan D, Axelrod HL, Miller MD, Abdubek P, Ambing E, Astakhova T, Carlton D, Chiu HJ, Clayton T, Deller MC, Duan L, Elsliger MA, Feuerhelm J, Grzechnik SK, Hale J, Hampton E, Han GW, Jaroszewski L, Jin KK, Klock HE, Knuth MW, Kumar A, Marciano D, Morse AT, Nigoghossian E, Okach L, Oommachen S, Reyes R, Rife CL, Schimmel P, van den Bedem H, Weekes D, White A, Xu Q, Hodgson KO, Wooley J, Deacon AM, Godzik A, Lesley SA, Wilson IA (2007b) Crystal structures of two novel dye-decolorizing peroxidases reveal a beta-barrel fold with a conserved heme-binding motif. Proteins 69:223–233
Acknowledgements
We thank Tom van den Berg for excellent technical assistance. This research is supported by the Dutch Technology Foundation STW, Applied Science Division of NWO, and the Technology Program of the Ministry of Economic Affairs.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
van Bloois, E., Torres Pazmiño, D.E., Winter, R.T. et al. A robust and extracellular heme-containing peroxidase from Thermobifida fusca as prototype of a bacterial peroxidase superfamily. Appl Microbiol Biotechnol 86, 1419–1430 (2010). https://doi.org/10.1007/s00253-009-2369-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-009-2369-x