
Abstract
Natural products have been developed as an alternative source of novel antibacterials in combating food-borne pathogens such as Staphylococcus aureus (S. aureus), Salmonella, Listeria monocytogenes. The aims of this study were to investigate the antimicrobial effect of chlorogenic acid (CA) on S. aureus and to elucidate its mechanism of action. Minimum inhibitory concentrations of CA were determined using the agar dilution method. Intracellular pH (pHin), membrane potential, intracellular and extracellular ATP concentrations, and the release of cell constituents were measured to elucidate antibacterial mechanism. Morphological changes induced by CA were examined by electron microscopy. CA induced a decrease in the intracellular ATP concentrations, but no proportional increase in the extracellular ATP. And the release of cell constituents increased significantly when treated with CA. A reduction in pHin and cell membrane hyperpolarization were observed in S. aureus after CA treatment. Electron microscopic observations showed that the cell membrane of S. aureus was damaged by CA. It is concluded that CA inhibited the proliferation of S. aureus and destroyed the permeability of the cell membrane. The findings indicated that CA has the potential to be developed as an alternative to control S. aureus and the diseases associated with this microorganism.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Staphylococcus aureus (S. aureus) is one of the important bacterial pathogens and could cause skin infections, endocarditis, septicemia, meningitis, septic arthritis, toxic shock syndrome, medical device-related infections, gastroenteritis and staphylococcal food poisoning [1, 2]. Staphylococcus aureus produces various virulence factors including enterotoxins, exfoliative toxins, toxic shock syndrome toxin-1, Panton-Valentine leucocidin, and other exoproteins [3]. Staphylococcal food poisoning, one of the most common food-borne diseases in the world, is usually caused by staphylococcal enterotoxins (SEs) [4], which include various types such as SEA, SEB, SEC, SED, SEE, SEG, and SEH [5].
With ever-increasing antimicrobial resistant S. aureus strains (especially methicillin-resistant S. aureus), novel substances, other than antibiotics, to treat and prevent infections caused by this pathogen have been explored. Plant-derived compounds have gained tremendous interest in the search for such alternatives. Extracts from plants have been demonstrated to inhibit different microbial pathogens such as S. aureus, Salmonella, and Listeria [6–8]. The natural compounds responsible for antimicrobial effects include tannins, flavonoids, essential oil, organic acid, and so on [9]. Most of those compounds are derived from plants which have a long history of use in folk medicine or culinary use.
Chlorogenic acid (CA) is a secondary plant metabolite and it is commonly found in plant materials such as apples, coffee beans, and kiwi fruit [10]. CA has antiphlogistic, antioxidant, antimutagenic, and other biological activities [11, 12]. Furthermore, CA has been shown to have antibacterial activity against Trichosporon beigelii, Malassezia furfur, Shigella dysenteriae (S. dysenteriae), Escherichia coli (E. coli), and Salmonella Typhimurium (S. Typhimurium) [13, 14]. Sung et al. [14] have reported that CA exhibits antifungal activities by disrupting the structure of the cell membrane. Lou et al. [13] have found that CA was able to inhibit S. dysenteria and Streptococcus pneumoniae (S. pneumoniae) by provoking irreversible permeability changes in cell membrane, causing cells to lose the ability to maintain membrane potential and cytoplasm macromolecules including nucleotide.
Although CA has been reported to inhibit several pathogens, its effect on S. aureus and its mechanism of action have not been fully explored. Therefore, we investigated the effect of CA on the growth of S. aureus and examined its antistapholococcal mechanism by examining membrane structure under electron microscope and by measuring the intracellular and extracellular ATP concentrations, membrane potential, the intracellular pH (pHin), and the release of cell components.
Materials and methods
Reagents
CA [≥95 %, CAS 327-97-9] was bought from the Sigma Chemical Co. (St. Louis, Mo, USA), and solutions were prepared in 0.9 % (w/v) saline solution containing 1 % (v/v) of DMSO and stored at −20 °C. All other chemicals (including DMSO, HEPES, glycine, citric acid, and glutaraldehyde) were of analytical grade and made by Tianjin Kemiou Chemical Reagent Co., Ltd. (Tianjin, China).
Bacterial strains and culture conditions
Staphylococcus aureus ATCC 25923 was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Seven other S. aureus isolates were taken from our laboratory strain collection and originally isolated from various food sources in China. All isolates were used in minimum inhibitory concentration assay and only S. aureus ATCC 25923 was used in antimicrobial mechanism study. Before each experiment, stock cultures were propagated through two consecutive 12 h growth cycles in LB at 37 °C and washed three times in saline solution. The OD600 of working cultures was 0.5 (containing approximately 107 CFU/ml).
Minimum inhibitory concentration (MIC) determination
The MICs of the CA against S. aureus were determined by the agar dilution method recommended by the European Committee for Antimicrobial Susceptibility Testing [15] with a few modifications. Ampicillin was used as a reference antibiotic. The stock solution of the antibiotic was prepared in sterile water and sterilized with 0.2-μm Acrodisc filter (Gelman, USA). LB agar was aseptically added to sterile glass tubes containing CA or ampicillin. The content (a final volume of 4 ml) of each tube was gently mixed and poured in 24-well plates. The final concentration of CA was 0, 1.25, 2.5, 5, and 10 mg/ml, respectively, and the concentration of ampicillin was 0.1 mg/ml. After hardening, the agar media were spotted with 2 μl (approximately 104 CFU/ml) of the tested bacterium. The spots were left to dry and then plates were incubated at 37 °C for 12 h. The MIC was defined as the lowest concentration (mg/ml) of the extract resulting in no growth of bacteria.
Growth curves
The method described by Qiu et al. [16] was followed. Briefly, S. aureus strain ATCC 25923 was grown to an OD600 value of 0.1 in LB. Then, 150 μl of the culture were placed into wells in 96-well microplates. CA was added to the cultures to obtain final concentrations of 1/32MIC, 1/16MIC, 1/8MIC, 1/4MIC, 1/2MIC and MIC. The S. aureus culture containing 1 % DMSO without CA was the control. Bacteria were further cultured at 37 °C, and cell growth was determined at OD600 at 1 h intervals by a SmartSpec™ Plus Spectrophotometer (Bio-Rad, California, USA).
Cell constituents’ release
The release of cell constituents was measured according to the method described by Rhayour et al. [17]. Cells from the working culture (50 ml) of ATCC 25923 was collected by centrifugation at 4,500 rpm/min for 5 min (Model 5804R, Eppendorf, Hamburg, Germany), washed three times, and resuspended in 0.9 % saline solution at OD600 of 0.5. A 20-ml of cell suspension was incubated at 37 °C for 1 h in the presence of CA at three different concentrations (0, 2MIC, and 4MIC). Then, 10 ml of samples were taken and filtered through a 0.2-μm Acrodisc filter, and then diluted with 0.9 % saline solution. The concentration of the constituents released was determined by a UV spectrophotometer (SmartSpec™ Plus Spectrophotometer, USA) at 260 nm. The absorption of the 0.9 % saline solution containing the same concentration of CA was corrected.
Measurement of intracellular and extracellular ATP concentrations
The method described by Turgis et al. [18] was followed with some modifications. The working culture of ATCC 25923 was centrifuged for 5 min at 4,500 rpm/min, and the supernatant was removed. The cell pellets were washed three times with 0.9 % saline solution and then cells were collected by centrifugation. A cell suspension (OD600 = 0.5) was prepared with 50 ml of 0.9 % saline solution and 4 ml of cell solution was taken into the eppendorf tube for CA treatment. Then, the CA was added to each tube and the final concentrations were 0 (control), MIC, 2MIC, and 4MIC, respectively. Samples were maintained at 37 °C for 30 min and centrifuged for 5 min at 4,500 rpm/min. Then the cells (lower layer) and supernatant (upper layer) were let in ice to prevent ATP loss until measurement. The extracellular and the intracellular ATP concentrations were measured using an ATP assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, Jiangsu, China). The ATP concentration of the supernatants, which represents the extracellular concentration, was determined by the kit following instructions by manufacture with a spectrophotometer (SmartSpec™ Plus Spectrophotometer, USA). To determine the intracellular ATP concentrations, cell pellets were washed with 0.9 % saline solution and centrifuged for 5 min at 4,500 rpm/min, and the supernatant was removed. The resulting cell pellets were maintained in 60 μl of 0.9 % saline solution at 100 °C for 10 min to disrupt the microorganisms. The intracellular ATP concentrations were then measured as described for the extracellular ATP concentration.
pHin measurements
pHin was determined according to a modified method of Breeuwer et al. [19]. For loading of cells with fluorescent probe, LB broth (25 ml) was inoculated with 250 μl of working culture (OD600 = 0.5) and incubated at 37 °C for 8 h. Cells were then harvested by centrifugation (4,500 rpm/min, 10 min) and washed twice with 50 mM HEPES buffer (containing 5 mM EDTA, pH 8). The cell pellet was resuspended in 10 ml of HEPES buffer with 1.0 μM fluorescent probe, carboxyfluorescein diacetate succinimidyl ester (cFDA-SE; Molecular Probes, Sigma, USA). Cells were then incubated for 10 min at 37 °C, washed once in 50 mM potassium phosphate buffer with 10 mM MgCl2, pH 7.0, and resuspended in 10 ml buffer. To eliminate nonconjugated cFDA-SE, glucose (10 mM, final concentration) was added and cells were incubated for an additional 30 min at 37 °C. Cells were then washed twice, resuspended in 50 mM phosphate buffer (pH 7), and placed on ice until use.
An aliquot (4 ml) of fluorescently labeled cell suspensions was dispensed in a 15-ml tube with CA at three different concentrations (0, 2MIC and 4MIC). After 30 min, fluorescence intensities were measured with two different excitation wavelengths (440 and 490 nm) by rapidly alternating the monochromator between both wavelengths using the spectrofluorometer (RF-5301, SHIMADZU, Kyoto, Japan). The emission was collected at 525 nm, and the excitation and emission widths were 10 nm for both slits. The pHin of bacteria was determined from the ratio of the fluorescence signal at the pH-sensitive wavelength (490 nm) and the fluorescence signal at the pH-insensitive wavelength (440 nm). During the assay, the system was maintained at 25 °C.
Calibration curves were determined for cFDA-SE-loaded cells with different pH buffers. Controls included the following buffers: glycine (50 mM), citric acid (50 mM), Na2HPO4·2H2O (50 mM), and KCl (50 mM) adjusted to various pH values (4, 5, 6, 7, 8, 9, and 10). The fluorescence intensity was measured at 25 °C after equilibrating the pHin and pHout by addition of valinomycin (1 μM) and nigericin (1 μM). In this assay, a drop in relative fluorescence occurred when the cytoplasmatic pH decreased.
Membrane potential determination
The method described by Sánchez et al. [20] was followed with minor exceptions. Cells were grown in 20 ml of LB broth at 37 °C to an optical density of 0.5 (approximately 107 CFU/ml) at 600 nm. Then, 1 μM of membrane potential sensitive fluorescent probe bis-(1,3-dibutylbarbituric acid) trimethine oxonol [DiBAC4(3); Molecular Probes, Sigma, USA] was added for 5 min, followed by the addition of CA (0, 2MIC, and 4MIC). After 5 min, fluorescence was measured at the excitation and emission wavelengths of 492 nm and 515 nm, respectively, using the spectrofluorometer (RF-5301, Japan). The excitation and emission slit widths were 3 and 5 nm, respectively. Background fluorescence resulting from the medium was corrected.
Field emission scanning electron microscope (FESEM) analysis
FESEM assays were carried out as previously reported [21]. Cells (OD600 = 0.5) were treated with CA at 0, 2MIC, and 4MIC, respectively. The samples were incubated at 37 °C. After incubation for 2 or 4 h, cells were harvested by centrifugation for 10 min at 4,500 rpm/min and washed twice with 0.9 % saline solution, and then were resuspended in water containing 2.5 % glutaraldehyde and kept at −4 °C for 10 h to fix the cells. After centrifugation, the cells were further dehydrated in water–alcohol solutions at various alcohol concentrations (30, 50, 70, 80, 90, and 100 %) for 10 min. Finally, the samples were fixed on CLSM support, and then sputter coated with gold under vacuum, followed by microscopic examinations using a field emission scanning electron microscope (S-4800, Hitachi, Tokyo, Japan).
Statistical analysis
All experiments were performed in triplicate. Statistical analyses were performed using the SPSS software (version 19.0; SPSS, Inc., Chicago, IL). The data were presented as the mean values ± SD (n = 6). Results were analyzed with an analysis of variance test, and an independent Student’s t test was used for the analysis. Differences between means were considered significant at P ≤ 0.05.
Results and discussion
Growth inhibitory effect of CA on S. aureus
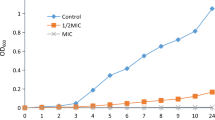
The MICs of CA against eight S. aureus strains were determined and ranged from 2.5 to 5.0 mg/ml. The MIC of CA against S. aureus ATCC 25923 was found to be 2.5 mg/ml (Table 1). At the concentrations of 1/16, 1/8, 1/4, and 1/2MIC, CA had no obvious effects on the growth of S. aureus. However, the growth of S. aureus was attenuated remarkably in the presence of CA treatment at concentrations of MIC or 2MIC. The OD600 values of treated culture with CA at these two concentrations were 40–60 % of the untreated culture after 9 h (Fig. 1).

Growth curves for S. aureus ATCC 25923 cultured in LB with various concentrations of chlorogenic acid. Each value represents the average of three reproducible experiments
Cell constituents’ release
The cell constituents’ release was determined by the measurement of the absorbance at 260 nm of the filtrates of S. aureus cultures. After CA was added to S. aureus, a significant (P ≤ 0.05) increase in the cell constituents’ release was observed when S. aureus were treated with CA at 2MIC. The absorbance was 0.18 ± 0.05 for CA (2MIC)-treated cells, while it was 0.04 ± 0.01 for the control. When the concentration of CA increased from 2MIC to 4MIC, the release of cell constituents increased gradually with the increase in the CA’s concentration (Fig. 2).

Effect of chlorogenic acid on cell constituents’ release of S. aureus ATCC 25923. Values represent the average of three reproducible experiments. Bars represent the standard deviation (n = 6). *P < 0.05, **P < 0.01
A loss of 260-nm absorbing materials has been observed after CA treatments. Compounds, such as oil, oregano, carvacrol, nobiletin, and tangeretin, induced a loss of cytoplasmic constituents inside of bacteria including K+, nucleic acids, protein, and ATP [7, 18, 22, 23]. Ultee et al. [24] demonstrated that 0.25 and 1 mM carvacrol induced release of potassium ions in Bacillus cereus (B. cereus). Lv et al. [21] found that essential oil, derived from ten types of plants including Pogostemon cablin, Salvia sclarea, and Rosmarinus officinalis, damaged the membrane structure of Pseudomonas aeruginosa (P. aeruginosa) with the significant increase in the cell constituents’ release. Yao et al. [22] observed that nobiletin and tangeretin destroyed the permeability of the membrane of P. aeruginosa and S. aureus, with release of alanine transaminase and aspartate transaminase. The release of those constituents indicated that the membrane structure of microorganism has been damaged by natural compounds.
Intracellular and extracellular ATP concentrations
The intracellular and extracellular ATP concentrations in the untreated S. aureus cells were 75.25 ± 6.00 and 9.50 ± 2.10 pM, respectively (Fig. 3). Compared with control, S. aureus treated with CA from MIC to 4MIC showed a significant (P ≤ 0.05) reduction in intracellular ATP concentration, while no significant (P > 0.05) difference was observed in the extracellular ATP concentration. When the concentration of CA increased from MIC, 2MIC to 4MIC, intracellular ATP concentrations were 4.50 ± 4.40, 7.35 ± 3.7, and 2.25 ± 1.8 pM, respectively, and extracellular ATP concentrations were 6.50 ± 2.10, 9.50 ± 0.7, and 8.50 ± 0.7 pM, respectively. However, no significant (P > 0.05) difference was observed in the intracellular and extracellular ATP concentration by increasing the concentration of CA from MIC to 4MIC.

Effect of chlorogenic acid on intracellular and extracellular ATP concentrations of S. aureus ATCC 25923. Values represent the means of three reproducible experiments. Bars represent the standard deviation (n = 6). *P < 0.05, **P < 0.01
We found that exposure to CA led to a decrease in the intracellular ATP concentration. No proportional increase in the extracellular ATP was observed. It is possible that CA does not enhance the permeability of the membrane for ATP, but it could interact with the membranes of S. aureus and change its permeability for cations like H+ and K+. Depletion of the internal ATP pool may result from a reduced rate of ATP synthesis or increased ATP hydrolysis. Our results were consistent with Ultee et al. [24] who reported that the level of intracellular ATP decreased, while there was no proportional increase outside the cell after B. cereus cell was exposed to carvacrol. In contrast, Lee et al. [25] observed that linolenic acid with or without monoglyceride (glycerol laurate or glycerol myristate) changed both the intracellular and extracellular ATP concentrations of B. cereus and S. aureus. Turgis et al. [18] reported that mustard essential oil induced a decrease in the intracellular ATP concentration and increased the extracellular ATP concentration. The differences in the permeability of the cell membrane caused by natural compounds may be due to the various degrees of damages of cell membranes which are dependent on the membrane structure of pathogens and antimicrobial effects of different phytochemicals.
pHin
A clear change in cytoplasmic pH was observed after addition of CA (Fig. 4). The pHin of untreated S. aureus was 9.22 ± 0.77. The addition of CA at 2MIC caused a significant (P ≤ 0.05) decrease in pHin of S. aureus from 9.22 ± 0.77 to 5.71 ± 1.07. And the addition of CA at 4MIC significantly (P ≤ 0.05) lowered the pHin from 9.22 ± 0.77 to 4.50 ± 0.72. However, there was no significant (P > 0.05) difference in pHin by increasing the concentration of CA from 2MIC to 4MIC.

Effect of chlorogenic acid on the intracellular pH (pHin) of S. aureus ATCC 25923. Values represent the means of three reproducible experiments. Bars represent the standard deviation (n = 6). *P < 0.05, **P < 0.01
The pHin is critical for the control of many cellular processes, such as DNA transcription, protein synthesis, and enzyme activity and also controlled by the membrane [26]. Changes in pHin are an indicator of membrane integrity. Turgis et al. [18] demonstrated that mustard essential oil at concentration of the MIC lowered the pHin from 6.23 to 5.20 in E. coli O157:H7 and from 6.59 to 5.44 in S. typhi. Sanchez et al. [20] showed that all the plant extracts induced changes in the pHin of Vibrio cholerae (V. cholerae). The results indicated that plants compounds generally damaged the membrane integrity and caused a decrease in pHin.
Membrane potential
Staphylococcus aureus cells treated with CA displayed cell membrane hyperpolarization, as evidenced by a decrease in fluorescence (negative values) (Fig. 5). After the addition of CA at 2MIC or 4MIC, the membrane potential was decreased, and a higher rate of reduction was observed by increasing concentrations from 2MIC to 4MIC.

Effect of chlorogenic acid on the membrane potentials of S. aureus ATCC 25923. Values represent the means of three reproducible experiments. Bars represent the standard deviation (n = 6)
A reduction in fluorescence and a hyperpolarization of cellular membranes have been observed after CA treatment. The fluorescence emitted by DiBAC4(3), a fluorescent membrane potential dye, is enhanced when the dye crosses the cell membrane as a result of membrane depolarization (less negative charge inside the cell). Lou et al. [13] found that CA caused depolarization of cell membranes of S. dysenteria and S. pneumoniae. Although a depolarization effect was expected, hyperpolarization has also been reported as an important type of membrane damage [27]. Consistent with our results, Sanchez et al. [20] reported that extracts from edible and medicinal plants were able to disrupt the cell membranes of V. cholerae cells, causing hyperpolarization of cell membrane. There are two reasons to explain the phenomenons: (1) a pH change (from acidic to neutral); and (2) by increasing movement of ions, specifically K+, which diffuses out of the cell membrane through K+ channels and affects cellular homeostasis [28].
FESEM observation
Cells of S. aureus had a typical structure of gram-positive microorganism, showing an irregular and striated membrane (Fig. 6). Cells treated with CA showed disrupted membranes and deformed cell walls. Cells treated with higher concentrations of CA or longer time exhibited more damage to cell membrane than those untreated cells.

Scanning electron micrographs of S. aureus ATCC 25923 untreated for 2 h (a) and 4 h (b), treated with chlorogenic acid at 2MIC for 2 h (c) and 4 h (d), and treated with chlorogenic acid at 4MIC for 2 h (e) and 4 h (f)
Microstructural observations showed that the cell membrane structures of cells were severely affected and damaged by CA. The findings are in good agreement with other studies. Observed by scanning electron microscope, Rhayour et al. [17] demonstrated that cell walls and membranes of E. coli and B. subtilis treated by oregano and clove essential oils were significantly damaged. Turgis et al. [18] reported that a variety of morphological changes in E. coli O157:H7 and S. typhi caused by the mustard EO had been observed by SEM.
Conclusion
In conclusion, CA effectively inhibited the growth of S. aureus partially by affecting the cell membrane, which was evidenced by the damaged membrane structure, the release of cytoplasmic constituents, and changes in the pHin and membrane potential. The findings reveal the potential of CA as an alternative therapeutic agent against S. aureus or as a food preservative. Prior to the application of CA in either area, issues of safety and toxicity of CA need to be evaluated in future studies.
References
Qiu J, Wang D, Xiang H, Feng H, Jiang Y, Xia L, Dong J, Lu J, Yu L, Deng X (2010) Subinhibitory concentrations of thymol reduce enterotoxins A and B and α-hemolysin production in Staphylococcus aureus isolates. PLoS ONE 5(3):e9736
Redelman CV, Maduakolam C, Anderson GG (2012) Alcohol treatment enhances Staphylococcus aureus biofilm development. FEMS Immunol Med Microbiol 66(3):411–418
Kurlenda J, Grinholc M (2012) Alternative therapies in Staphylococcus aureus diseases. Acta Biochim Pol 59(2):171–184
Hennekinne J-A, De Buyser M-L, Dragacci S (2012) Staphylococcus aureus and its food poisoning toxins: characterization and outbreak investigation. FEMS Microbiol Rev 36(4):815–836
Smith J, Buchanan R, Palumbo S (1983) Effect of food environment on Staphylococcal enterotoxin synthesis: a review. J Food Prot 46:545–555
Al-Zoreky N (2009) Antimicrobial activity of pomegranate (Punica granatum L.) fruit peels. Int J Food Microbiol 134(3):244–248
Carson C, Hammer K, Riley T (2006) Melaleuca alternifolia (tea tree) oil: a review of antimicrobial and other medicinal properties. Clin Microbiol Rev 19(1):50–62
Ankri S, Mirelman D (1999) Antimicrobial properties of allicin from garlic. Microbes Infect 1(2):125–129
Gyawali R, Ibrahim SA (2012) Impact of plant derivatives on the growth of foodborne pathogens and the functionality of probiotics. Appl Microbiol Biotechnol 95(1):29–45
Farah A, de Paulis T, Trugo LC, Martin PR (2005) Effect of roasting on the formation of chlorogenic acid lactones in coffee. J Agric Food Chem 53(5):1505–1513
Li S-Y, Chang C-Q, Ma F-Y, Yu C-L (2009) Modulating effects of chlorogenic acid on lipids and glucose metabolism and expression of hepatic peroxisome proliferator-activated receptor-α in golden hamsters fed on high fat diet. Biomed Environ Sci 22(2):122–129
Wang G-F, Shi L-P, Ren Y-D, Liu Q-F, Liu H-F, Zhang R-J, Li Z, Zhu F-H, He P-L, Tang W (2009) Anti-hepatitis B virus activity of chlorogenic acid, quinic acid and caffeic acid in vivo and in vitro. Antivir Res 83(2):186–190
Lou Z, Wang H, Zhu S, Ma C, Wang Z (2011) Antibacterial activity and mechanism of action of chlorogenic acid. J Food Sci 76(6):M398–M403
Sung WS, Lee DG (2010) Antifungal action of chlorogenic acid against pathogenic fungi, mediated by membrane disruption. Pure Appl Chem 82(1):219–226
EUCAST (2010) Determination of minimum inhibitory concentration (MICs) of antibacterial agents by agar dilution. Clin Microbiol Infect 6:509–515
Qiu J, Feng H, Xiang H, Wang D, Xia L, Jiang Y, Song K, Lu J, Yu L, Deng X (2010) Influence of subinhibitory concentrations of licochalcone A on the secretion of enterotoxins A and B by Staphylococcus aureus. FEMS Microbiol Lett 307(2):135–141
Rhayour K, Bouchikhi T, Tantaoui-Elaraki A, Sendide K, Remmal A (2003) The mechanism of bactericidal action of oregano and clove essential oils and of their phenolic major components on Escherichia coli and Bacillus subtilis. J Essent Oil Res 15(4):286–292
Turgis M, Han J, Caillet S, Lacroix M (2009) Antimicrobial activity of mustard essential oil against Escherichia coli O157: H7 and Salmonella typhi. Food Control 20(12):1073–1079
Breeuwer P, Drocourt J, Rombouts FM, Abee T (1996) A novel method for continuous determination of the intracellular pH in bacteria with the internally conjugated fluorescent probe 5 (and 6-)-carboxyfluorescein succinimidyl ester. Appl Environ Microbiol 62(1):178–183
Sánchez E, García S, Heredia N (2010) Extracts of edible and medicinal plants damage membranes of Vibrio cholerae. Appl Environ Microbiol 76(20):6888–6894
Lv F, Liang H, Yuan Q, Li C (2011) In vitro antimicrobial effects and mechanism of action of selected plant essential oil combinations against four food-related microorganisms. Food Res Int 44(9):3057–3064
Yao X, Zhu X, Pan S, Fang Y, Jiang F, Phillips GO, Xu X (2012) Antimicrobial activity of nobiletin and tangeretin against Pseudomonas. Food Chem 132(4):1883–1890
Lambert R, Skandamis PN, Coote PJ, Nychas GJ (2001) A study of the minimum inhibitory concentration and mode of action of oregano essential oil, thymol and carvacrol. J Appl Microbiol 91(3):453–462
Ultee A, Kets E, Smid E (1999) Mechanisms of action of carvacrol on the food-borne pathogen Bacillus cereus. Appl Environ Microbiol 65(10):4606–4610
Lee J-Y, Kim Y-S, Shin D-H (2002) Antimicrobial synergistic effect of linolenic acid and monoglyceride against Bacillus cereus and Staphylococcus aureus. J Agric Food Chem 50(7):2193–2199
Biteau B, Labarre J, Toledano MB (2003) ATP-dependent reduction of cysteine–sulphinic acid by S. cerevisiae sulphiredoxin. Nature 425(6961):980–984
Yuroff A, Sabat G, Hickey W (2003) Transporter-mediated uptake of 2-chloro-and 2-hydroxybenzoate by Pseudomonas huttiensis strain D1. Appl Environ Microbiol 69(12):7401–7408
Bot C, Prodan C (2009) Probing the membrane potential of living cells by dielectric spectroscopy. Eur Biophys J 38(8):1049–1059
Acknowledgments
This work was supported by the International Science and Technology Collaboration Research Fund (No. A213021202) in Northwest A&F University.
Conflict of interest
None.
Compliance with Ethics Requirements
This article does not contain any studies with human or animal subjects.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, G., Wang, X., Xu, Y. et al. Antimicrobial effect and mode of action of chlorogenic acid on Staphylococcus aureus . Eur Food Res Technol 238, 589–596 (2014). https://doi.org/10.1007/s00217-013-2140-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-013-2140-5