
Abstract
Purpose
To investigate the efficacy and toxicity of cetuximab when added to radiochemotherapy for unresectable esophageal cancer.
Methods
This randomized phase 2 trial (clinicaltrials.gov, identifier NCT01787006) compared radiochemotherapy plus cetuximab (arm A) to radiochemotherapy (arm B) for unresectable esophageal cancer. Primary objective was 2‑year overall survival (OS). Arm A was considered insufficiently active if 2‑year OS was ≤40% (null hypothesis = H0), and promising if the lower limit of the 95% confidence interval was >45%. If that lower limit was >40%, H0 was rejected. Secondary objectives included progression-free survival (PFS), locoregional control (LC), metastases-free survival (MFS), response, and toxicity. The study was terminated early after 74 patients; 68 patients were evaluable.
Results
Two-year OS was 71% in arm A (95% CI: 55–87%) vs. 53% in arm B (95% CI: 36–71%); H0 was rejected. Median OS was 49.1 vs. 24.1 months (p = 0.147). Hazard ratio (HR) for death was 0.60 (95% CI: 0.30–1.21). At 2 years, PFS was 56% vs. 44%, LC 84% vs. 72%, and MFS 74% vs. 54%. HRs were 0.51 (0.25–1.04) for progression, 0.43 (0.13–1.40) for locoregional failure, and 0.43 (0.17–1.05) for distant metastasis. Overall response was 81% vs. 69% (p = 0.262). Twenty-six and 27 patients, respectively, experienced at least one toxicity grade ≥3 (p = 0.573). A significant difference was found for grade ≥3 allergic reactions (12.5% vs. 0%, p = 0.044).
Conclusion
Given the limitations of this trial, radiochemotherapy plus cetuximab was feasible. There was a trend towards improved PFS and MFS. Larger studies are required to better define the role of cetuximab for unresectable esophageal cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The prognosis of patients with locally advanced esophageal cancer is poor and requires improvement that may be achieved with the addition of new drugs [1]. For definitive and neoadjuvant treatment of locally advanced disease, radiochemotherapy with cisplatin and 5‑fluorouracil (5-FU) has been the standard regimen for more than 20 years [2]. The combination of radiotherapy and carboplatin/paclitaxel has been popular in the neoadjuvant setting since publication of a randomized trial in 2012 [3]. Combinations of radiotherapy or radiochemotherapy with newer systemic therapies such as antibodies to the epidermal growth factor receptor (EGFR) provide additional options. Overexpression of EGFR is frequent and associated with a poor prognosis in patients with squamous cell carcinoma (SCC) of the esophagus or adenocarcinoma of the gastroesophageal junction [4,5,6]. For cetuximab, a radiosensitizing effect was shown in preclinical studies and anticancer activity in small clinical studies [7,8,9,10,11]. These data led to the present trial that investigated the efficacy and feasibility of cetuximab added to radiochemotherapy for unresectable esophageal cancer.
Patients and methods
This multicenter open-label, randomized phase 2 trial evaluated radiochemotherapy plus cetuximab in patients treated for unresectable esophageal cancer between 09/2011 and 12/2016. It was approved by the local ethic committees (leading committee: University of Lübeck, reference: 11-104) and performed in accordance with the Helsinki Declaration. It was registered at clinicaltrials.gov (identifier: NCT01787006). Eligible patients had histologically confirmed unresectable esophageal cancer. Resectability was determined by a surgeon prior to randomization. Inclusion and exclusion criteria are listed in Table 1.
Patients were randomly assigned to radiochemotherapy plus cetuximab (experimental group, arm A) or radiochemotherapy alone (control group, arm B). Stratification was based on histology (SCC vs. adenocarcinoma), Karnofsky performance score (80–100% vs. 70%), and tumor stage (T1-3N0‑1 vs. T4 and/or N2 and/or M1a) [12]. Both histologies were allowed, since at the time of trial initiation, the standard treatment was the same.
Treatments
Radiotherapy was performed with photons from a linear accelerator using 3D treatment planning. Initially, 50.4 Gy in 28 fractions was planned for primary tumor plus locoregional lymph nodes. Reevaluation assessing resectability was performed after 4–4.5 weeks. If resectability was achieved and the patient agreed to surgery, radiotherapy was stopped after 45 Gy and the patient underwent surgery. This applied to 8 patients (25%) of the experimental group and 17 patients (47%) of the control group (p = 0.079, Fisher’s exact test). If resectability was not achieved or the patient refused surgery, radiotherapy was continued until 50.4 Gy and followed by a boost of 9.0 Gy to primary tumor and involved lymph nodes (Fig. 1). Treatment planning and quality assurance were performed according to the standard operating procedures of the contributing centers. Generally, the initial clinical target volume (CTV) included the gross tumor volume (GTV) plus margins of 3–5 cm in the superior–inferior direction and 1 cm in the lateral and anterior–posterior directions. The CTV for the boost included the GTV plus margins of 2 cm in the superior–inferior direction and 1 cm in lateral and anterior–posterior directions. For the involved lymph nodes, a GTV-to-CTV margin of 0.5–1.0 cm was suggested. The margin from the CTV to the planning target volume (PTV) was 0.5–1.0 cm. In accordance with the QUANTEC (Quantitative Analyses of Normal Tissue Effects in the Clinic) data, the mean doses for heart, lung, liver, and kidney (bilateral) should be <26 Gy, ≤7 Gy, <30–32 Gy, and <15 Gy, respectively [13]. Moreover, the dose to the spinal cord should not exceed 45 Gy. A brachytherapy boost was not implemented in the protocol, since this is not a standard therapy for the primary treatment of esophageal cancer. However, it can be a reasonable option for a local recurrence or symptom relief in a palliative situation [14].

Flowchart of the treatments administered in this trial. 5‑FU 5-fluorouracil
Two courses of 5‑FU (1000 mg/m2/d) were administered as a continuous infusion over 96 h during the first and fifth weeks of radiotherapy (Fig. 1; [2, 15]). Two cycles of 750 mg/m2/d of 5‑FU (over 96 h) were administered after radiotherapy, 5 and 9 weeks after the second course. Cisplatin (20 mg/m2/d) was administered after saline hydration as an intravenous bolus over 60 min on the same days as 5‑FU. Patients received antiemetic therapy prior to cisplatin, including 5HT3 antagonists and dexamethasone.
Cetuximab was administered as an intravenous infusion with a loading dose of 400 mg/m2 over 120 min (day 1), followed by weekly doses of 250 mg/m2 over 60 min for a total of 14 weeks (Fig. 1). Patients were pretreated with antihistamines and glucocorticoids.
Statistical considerations
The primary objective was to determine the 2‑year OS rate of the two cohorts. OS was referenced from the day of randomization and analyzed using the Kaplan–Meier method and the univariate Cox proportional hazards method. For the primary hypothesis of 2‑year OS, OS rate and 95% confidence interval (CI) were calculated for the 2‑year timepoint. The 2‑year OS of 40% for the reference group was expected based on prior studies [2, 16, 17]. If the observed 2‑year OS rate was ≤40% (null hypothesis H0) for the experimental therapy (arm A), this therapy would be deemed insufficiently active to pursue in further research. The alternative hypothesis (H1) was that adding cetuximab would result in 2‑year OS >40%. The decision for rejection of H0 was based on the 95% CI of the 2‑year OS. H0 could be rejected if the lower limit of the 95% CI was >40%. If the lower limit was >45%, radiochemotherapy plus cetuximab would be considered a promising enough therapy to justify further investigation. The probability of accepting the experimental therapy as promising (2-year OS >45%) when the true OS rate was ≤40%, was 5% (type I error). The probability of rejecting the experimental therapy as insufficiently active (≤40%) when the true OS rate was promising (>45%), was 20% (type II error, power of 80%).
Secondary objectives included determination of 1‑year OS, progression-free survival (PFS), locoregional control (LC), metastases-free survival (MFS), overall response (OR; RECIST v11 [18]), and toxicities (CTCAE v4.03 [19]). In addition, OS, PFS, and MFS were assessed irrespectively of specific timepoints. For PFS, the event was defined as first occurrence of radiologically proven or clinical progression or death due to progressive disease. Locoregional failure was defined as progressive primary tumor and/or regional lymph nodes on endoscopy, endoscopic ultrasound, or computed tomography. For MFS, the event was defined as first occurrence of distant metastasis. Both groups were compared for these endpoints using the Kaplan–Meier method and the log-rank test. Differences between OR rates were compared with the chi-square test. If frequencies in one group were ≤5, Fisher’s exact test was used. The comparisons of toxicities were performed with the Fisher’s exact test.
When using a standard single-stage phase 2 design according to Fleming [20], 124 evaluable patients were required to determine efficacy. The standard treatment (control group) served to reduce some of the result variability typically encountered in single-arm phase 2 trials. To cover potential dropouts, 134 patients should be recruited.
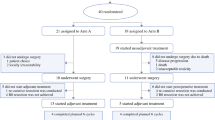
The trial was terminated after randomization of 74 patients (35 arm A, 39 arm B) due to slow accrual. Sixty-eight patients (32 arm A, 36 arm B) were evaluable for efficacy and toxicity (Fig. 2 and Table 2). This number of patients was sufficient to detect a difference of approximately 33% with a statistical power of 80% at a two-sided significance level of 0.05. Median follow-up was 18 (0–61) months in the entire cohort, and 26 (7–61) months in patients alive at last contact.

Consolidated Standards of Reporting Trials (CONSORT) diagram
Results
Two-year OS rates were 71% in arm A (95% CI: 55–87%) vs. 53% in arm B (95% CI: 36–71%). Since the lower limit of the 95% CI of the 2‑year OS rate in arm A was 55% (>40%), H0 was rejected. Since this value was also >45%, radiochemotherapy plus cetuximab was deemed promising.
Median OS was 49.1 months in arm A and 24.1 months in arm B (p = 0.147); 1‑year OS rates were 74% and 70%, respectively (Fig. 3). Hazard ratio (HR) for death was 0.60 (95% CI: 0.30–1.21). Since less than half of the patients in arm A experienced progression, the median time of PFS could not be estimated; in arm B, median time of PFS was 17.6 months (p = 0.060). PFS was 64% vs. 58% at 1 year and 56% vs. 44% at 2 years (Fig. 3). HR for progression was 0.51 (95% CI: 0.25–1.04). LC was 89% vs. 81% at 1 year and 84% vs. 72% at 2 years (p = 0.151; Fig. 3). HR for locoregional failure was 0.43 (95% CI: 0.13–1.40). Since less than 50% of patients in arm A experienced distant metastasis, the median time of MFS could not be estimated; in arm B, median time of MFS was 31.3 months (p = 0.057). MFS was 79% vs. 70% at 1 year and 74% vs. 54% at 2 years (Fig. 3). HR for distant metastasis was 0.43 (95%-CI: 0.17–1.05). OR rates were 81.3% vs. 69.4% (p = 0.262).

Comparison of the two treatment groups (Kaplan–Meier analysis and log-rank test) with respect to overall survival (a), progression-free survival (b), locoregional control (c), and metastases-free survival (d)
All 68 patients who received at least one dose of study medication experienced at least one adverse event (AE). Of all AEs when counted separately, 74.5% were mild or moderate. Significantly more AEs (any grade) were observed in arm A for leukopenia (50.0% vs. 22.2%, p = 0.023), hypocalcemia (28.1% vs. 5.6%, p = 0.019), hypomagnesemia (40.6% vs. 8.3%, p = 0.003), acneiform rash (34.4% vs. 0%, p < 0.001), radiation dermatitis (28.1% vs. 2.8%, p = 0.005), maculopapular rash (21.9% vs. 2.8%, p = 0.022), and allergic reactions (12.5% vs. 0%, p = 0.044). Non-significantly higher rates were found in arm A for thrombopenia (34.4% vs. 19.4%, p = 0.182), hypopotassemia (50.0% vs. 33.3%, p = 0.219), oral mucositis (25.0% vs. 11.1%, p = 0.203), weight loss (28.1% vs. 8.3%, p = 0.054), and pneumonitis (6.3% vs. 2.8%, p = 0.598). Non-significantly higher rates (any grade) were observed in arm B for fatigue (50.0% vs. 28.1%, p = 0.085), constipation (30.6% vs. 18.8%, p = 0.401), and lung infection (25.0% vs. 9.4%, p = 0.118).
Twenty-six patients (81.3%) in arm A and 27 patients (75.0%) in arm B (p = 0.573) experienced at least one AE grade ≥3 (Table 3). A significant difference was found only for allergic reactions (12.5% vs. 0%, p = 0.044). All allergic reactions were managed and resolved without sequelae. Two patients in arm B died during the treatment phase due to underlying disease and cardiovascular disorders, respectively.
In arm A, cetuximab was discontinued after the loading dose due to allergic reactions in 4 patients. In the remaining 28 patients, cetuximab was stopped after 6 doses, since patients went to surgery. In another 6 patients, cetuximab was discontinued (reason not specified) after 7 (n = 4), 8 (n = 1), and 11 (n = 1) doses, respectively.
The proportions of patients requiring a reduction of cisplatin during cycles 1–4 were 3.4%, 10.7%, 18.9%, and 8.3%, respectively, in arm A vs. 5.6%, 14.3%, 27.3%, and 10.0%, respectively, in arm B. Toxicity-associated dose reductions of 5‑FU in cycles 1–4 were required by 3.4%, 11.5%, 13.3%, and 8.3% of patients in arm A vs. 2.8%, 14.8%, 10.0%, and 0% of patients in arm B, respectively. Using the Fisher’s exact test, the differences between the two groups regarding the reduction of cisplatin and 5‑FU were not significant. In arm A, 3 patients (9.4%) who experienced allergic reactions to cetuximab did not receive radiotherapy. In the irradiated patients, interruption of radiotherapy for >7 days was required in 0% (arm A) and 11.1% (arm B) of patients (p = 0.122, Fisher’s exact test), and a reduction of the planned radiotherapy dose in 10.3% and 19.4%, respectively (p = 0.492).
Discussion
During recent years, a considerable amount of research has been performed to improve the prognoses of patients with esophageal cancer [21,22,23,24]. The present trial investigated the impact of adding cetuximab to radiochemotherapy for unresectable esophageal cancer. In the experimental arm (arm A), 2‑year OS was 71% (95% CI: 55–87%) compared to 53% in arm B (95% CI: 36–71%). The lower limit of the 95% CI met the trial criteria. Therefore, H0 was rejected and radiochemotherapy plus cetuximab was deemed promising. Two-year OS was also higher in arm B than in the reference studies [2, 16, 17]. However, the 95% CI in arm B included the threshold of 40%. Median OS was longer in arm A (49.1 vs. 24.1 months), although significance was not achieved.
Our trial suggested a trend towards improved PFS and MFS when cetuximab was added. Conflicting results were reported from other studies. In the SCOPE‑1 trial, patients were randomized to radiochemotherapy with 50 Gy in 25 fractions plus four cycles of cisplatin 60 mg/m2/d1 and capecitabine 625 mg/m2 twice daily/d1-21, or to the same regimen plus cetuximab (400 mg/m2/d1 followed by 250 mg/m2 weekly) [25]. In 2017, long-term results of this trial were published [26]. Median OS was 34.5 months without and 24.7 months with cetuximab (p = 0.137). Median PFS was 24.1 vs. 15.9 months (p = 0.114).
The RTOG 0436 trial compared definitive radiochemotherapy with 50.4 Gy in 28 fractions plus weekly cisplatin (50 mg/m2) and paclitaxel (25 mg/m2) with or without cetuximab for unresectable esophageal cancer [27]. OS rates at 2 and 3 years were 45% and 34% with vs. 44% and 28% without cetuximab (p = 0.47). Local failure rates at 2 and 3 years were 47% and 49% vs. 49% and 49% (p = 0.65).
A third randomized trial adding cetuximab to radiochemotherapy in patients with resectable esophageal cancer was favorable [28]. It compared chemotherapy (two cycles of 75 mg/m2 docetaxel and 75 mg/m2 cisplatin) followed by radiochemotherapy (45 Gy in 25 fractions plus weekly 20 mg/m2 docetaxel and 25 mg/m2 cisplatin) and surgery to the same regimen plus cetuximab (neoadjuvant: 250 mg/m2 weekly; adjuvant: 500 mg/m2 every second week). Median PFS was 2.9 years with and 2.0 years without cetuximab (p = 0.13). Median OS was 5.1 vs. 3.0 years (p = 0.055). Time to locoregional failure after R0 resection was significantly longer in the cetuximab group (p = 0.017).
The results of available prospective studies regarding toxicity were also conflicting. In the present trial, significantly more AEs (any grade) were found in the experimental arm for leucopenia, hypocalcemia, hypomagnesemia, acneiform rash, radiation dermatitis, maculopapular rash, and allergic reactions. When counted separately, the majority of all AEs were mild or moderate and managed without problems. A significant difference was observed for allergic reactions that resolved without sequelae. Rates of patients experiencing at least one AE grade ≥3 were not significantly different. This is consistent with the majority of prospective studies that rated radiochemotherapy plus cetuximab for esophageal cancer as feasible or well tolerated [15, 29,30,31,32,33,34].
In contrast, one randomized and four non-randomized studies concluded that radiochemotherapy plus cetuximab was associated with considerable toxicity [28, 30, 31, 35, 36]. The non-randomized studies used radiochemotherapy regimens different from the two most common regimens [2, 3]. In two studies, radiochemotherapy included cisplatin and irinotecan, in one study preoperative oxaliplatin/5-FU followed by postoperative docetaxel, and in one study induction chemotherapy with epirubicin, cisplatin, and capecitabine [37,38,39,40]. Therefore, the results of these studies are difficult to interpret. The SCOPE‑1 trial included two cycles of induction chemotherapy with cisplatin and capecitabine followed by radiochemotherapy with two concurrent cycles, which is also different from the most common regimens in use today [2, 3]. Moreover, the addition of cetuximab to radiochemotherapy resulted in increased toxicity in trials performed in patients with other cancer types such as head and neck cancer and anal carcinoma [41, 42].
Median OS of the cetuximab group in the LEOPARD-2 trial was better than in the SCOPE‑1 trial (49.1 vs. 24.7 months) [26]. This may be explained by the fact that in the LEOPARD-2 trial, no patient in the cetuximab arm required interruption of radiotherapy for >7 days, and only 9.4% of the patients did not receive radiotherapy. In the SCOPE‑1 trial, radiotherapy was not given to 19% of patients in the cetuximab arm, and interruptions of radiotherapy appeared more frequent in the cetuximab group. In head and neck cancer patients, interruptions of radiotherapy decrease disease control and survival [35, 43]. Moreover, the proportion of patients in the cetuximab group with SCC was higher in the LEOPARD-2 than in the SCOPE‑1 trial (84% vs. 71%). In a phase 1b/2 trial, SCC was associated with significantly better outcomes than adenocarcinoma regarding LC (96% vs. 74%, p < 0.001) and OS (58% vs. 25%, p = 0.001) [31]. In another prospective study, where all patients had SCC, CR was 69% and 2‑year PFS 75% [29]. Another important aspect is the low rate of acneiform skin reactions and rash (only 18%) in the SCOPE‑1 trial. This is important, since rash was associated with better outcomes in head and neck cancer patients receiving cetuximab [44]. In the LEOPARD-2 trial, 56% of patients experienced acneiform or maculopapular rash.
The results of the LEOPARD-2 trial regarding the addition of cetuximab also appeared more promising than in the RTOG 0436 trial (2-year OS: 71% vs. 45%) [26]. This may be explained by a considerably higher proportion of SCC in the cetuximab arm (84% vs. 37%) in the LEOPARD-2 trial. Interruptions of radiotherapy were not reported for the RTOG 0436 trial. Moreover, the chemotherapy regimen in the LEOPARD-2 trial was more intensive than in the RTOG 0436 trial. Although this regimen was associated with more grade ≥3 toxicities, many patients received the planned treatment due to intensive patient care including administration of chemotherapy and cetuximab under inpatient conditions.
Since different radiochemotherapy regimens were used in the available trials, the treatments were performed in different settings, and patient characteristics varied, it is difficult to compare the results of the LEOPARD-2 and other trials. When interpreting the results of the LEOPARD-2 trial, one should keep in mind its limitations, particularly the comparatively small sample size after premature termination. Additional limitations included lack of a central review, the fact that less than 20% of the patients had adenocarcinomas, no assessment of the expression of EGFR and mutations in the vast majority of the patients, and lack of standardized procedures of treatment planning and quality assurance. Moreover, one has to consider that the proportions of patients who underwent surgery after 45 Gy were different in both groups. This difference might have introduced a bias. Due to these limitations, the results of the present study appear less meaningful than the results of previous larger trials [25,26,27,28].
Conclusion
Given the limitations of the LEOPARD-2 trial, radiochemotherapy with cisplatin/5-FU plus cetuximab is feasible for unresectable esophageal cancer. Although not statistically significant, there was a trend towards improved PFS and MFS. Larger studies are required to better define the role of the addition of cetuximab to radiochemotherapy for unresectable esophageal cancer.
Abbreviations
- CI:
-
Confidence interval
- CONSORT:
-
Consolidated Standards of Reporting Trials
- CTCAE:
-
Common Terminology Criteria for Adverse Events
- EGFR:
-
Epidermal growth factor receptor
- 5‑FU:
-
5‑Fluorouracil
- HR:
-
Hazard ratio
- LC:
-
Locoregional control
- MFS:
-
Metastases-free survival
- OR:
-
Overall response
- OS:
-
Overall survival
- PFS:
-
Progression-free survival
- RECIST:
-
Response Evaluation Criteria in Solid Tumors
- RTOG:
-
Radiation Therapy Oncology Group
- SCC:
-
Squamous cell carcinoma
References
Siegel RL, Miller KD, Jemal A (2020) Cancer statistics, 2020. CA Cancer J Clin 70:7–30. https://doi.org/10.3322/caac.21590
Herskovic A, Martz K, al-Sarraf M et al (1992) Combined chemotherapy and radiotherapy compared with radiotherapy alone in patients with cancer of the esophagus. N Engl J Med 326:1593–1598. https://doi.org/10.1056/NEJM199206113262403
van Hagen P, Hulshof MC, van Lanschot JJ et al (2012) Preoperative chemoradiotherapy for esophageal or junctional cancer. N Engl J Med 366:2074–2084. https://doi.org/10.1056/NEJMoa1112088
Hirai T, Kuwahara M, Yoshida K et al (1998) Clinical results of transhiatal esophagectomy for carcinoma of the lower thoracic esophagus according to biological markers. Dis Esophagus 11:221–225. https://doi.org/10.1093/dote/11.4.221
Itakura Y, Sasano H, Shiga C et al (1994) Epidermal growth factor receptor over-expression in esophageal carcinoma. An immunohistochemical study correlated with clinicopathologic findings and DNA amplification. Cancer 74:795–804
Kitagawa Y, Ueda M, Ando N et al (1996) Further evidence for prognostic significance of epidermal growth factor receptor gene amplification in patients with esophageal squamous cell carcinoma. Clin Cancer Res 2:909–914
Vanhoefer U, Tewes M, Rojo F et al (2004) Phase I study of the humanized anti-epidermal growth factor receptor monoclonal antibody EMD72000 in patients with advanced solid tumors that express the epidermal growth factor receptor. J Clin Oncol 22:175–184. https://doi.org/10.1200/JCO.2004.05.114
Foon KA, Yang XD, Weiner LM et al (2004) Preclinical and clinical evaluations of ABX-EGF, a fully human anti-epidermal growth factor receptor antibody. Int J Radiat Oncol Biol Phys 58:984–990. https://doi.org/10.1016/j.ijrobp.2003.09.098
Lorenzen S, Schuster T, Porschen R et al (2009) Cetuximab plus cisplatin/5-fluorouracil versus cisplatin/5-fluorouracil alone in first-line metastatic squamous cell carcinoma of the esophagus: a randomized phase II study of the Arbeitsgemeinschaft Internistische Onkologie (AIO). Ann Oncol 20:1667–1673. https://doi.org/10.1093/annonc/mdp069
Saleh MN, Raisch KP, Stackhouse MA et al (1999) Combined modality therapy of A431 human epidermoid cancer using anti-EGFR antibody C225 and radiation. Cancer Biother Radiopharm 14:451–463. https://doi.org/10.1089/cbr.1999.14.451
Milas L, Mason K, Hunter N et al (2000) In vivo enhancement of tumor radioresponse by C225 antiepidermal growth factor receptor antibody. Clin Cancer Res 6:701–708
Sobin LH, Gospodarowicz MK, Wittekind C (eds) (2009) TNM classification of malignant tumors, 7th edn. Wiley-Blackwell, Hoboken, New Jersey
Marks LB, Yorke ED, Jackson A et al (2010) Use of normal tissue complication probability models in the clinic. Int J Radiat Oncol Biol Phys 76(3):S10–19. https://doi.org/10.1016/j.ijrobp.2009.07.1754
Lancellotta V, Cellini F, Fionda B et al (2020) The role of palliative interventional radiotherapy (brachytherapy) in esophageal cancer: an AIRO (Italian association of radiotherapy and clinical oncology) systematic review focused on dysphagia-free survival. Brachytherapy 19:104–110. https://doi.org/10.1016/j.brachy.2019.09.005
Rades D, Maderer A, Panzner A et al (2017) Phase I study of definitive radio-chemotherapy with cisplatin, 5-fluorouracil and cetuximab for unresectable locally advanced esophageal cancer. Anticancer Res 37:2703–2708, https://doi.org/10.21873/anticanres.11620
Bedenne L, Michel P, Bouché O et al (2007) Chemoradiation followed by surgery compared with chemoradiation alone in squamous cancer of the esophagus: FFCD 9102. J Clin Oncol 25:1160–1168. https://doi.org/10.1200/JCO.2005.04.7118
Stahl M, Stuschke M, Lehmann N et al (2005) Chemoradiation with and without surgery in patients with locally advanced squamous cell carcinoma of the esophagus. J Clin Oncol 23:2310–2317. https://doi.org/10.1200/JCO.2005.00.034
Eisenhauer EA, Therasse P, Bogaerts J et al (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45:228–247. https://doi.org/10.1016/j.ejca.2008.10.026
National Institutes of Health/National Cancer Institute (2010) Common terminology criteria for adverse events (CTCAE) version 4.03
Fleming TR (1982) One-sample multiple testing procedure for phase II clinical trials. Biometrics 38:143–151
Hegemann NS, Koepple R, Walter F et al (2018) Neoadjuvant chemoradiation for esophageal cancer: surgery improves locoregional control while response based on FDG-PET/CT predicts survival. Strahlenther Onkol 194:435–443. https://doi.org/10.1007/s00066-018-1261-y
Nachbar M, Mönnich D, Kalwa P et al (2019) Comparison of treatment plans for a high-field MRI-linac and a conventional linac for esophageal cancer. Strahlenther Onkol 195:327–334. https://doi.org/10.1007/s00066-018-1386-z
Haefner MF, Lang K, Verma V et al (2018) Neoadjuvant versus definitive chemoradiotherapy for locally advanced esophageal cancer: outcomes and patterns of failure. Strahlenther Onkol 194:116–124. https://doi.org/10.1007/s00066-017-1211-0
Vitz S, Göbel H, Leibl B et al (2018) Adenocarcinoma of the oesophagus: neoadjuvant chemoradiation and radical surgery: long-term results. Strahlenther Onkol 194:1007–1016. https://doi.org/10.1007/s00066-018-1320-4
Crosby T, Hurt CN, Falk S et al (2013) Chemoradiotherapy with or without cetuximab in patients with oesophageal cancer (SCOPE1): a multicenter, phase 2/3 randomised trial. Lancet Oncol 14:627–637. https://doi.org/10.1016/S1470-2045(13)70136-0
Crosby T, Hurt CN, Falk S et al (2017) Long-term results and recurrence patterns from SCOPE-1: a phase II/III randomized trial of definitive chemoradiotherapy +/− cetuximab in oesophageal cancer. Br J Cancer 116:709–716. https://doi.org/10.1038/bjc.2017.21
Suntharalingam M, Winter K, Ilson D et al (2017) Effect of the addition of cetuximab to paclitaxel, cisplatin, and tadiation therapy for patients with esophageal cancer—the NRG oncology RTOG 0436 phase 3 randomized clinical trial. JAMA Oncol 3:1520–1528. https://doi.org/10.1001/jamaoncol.2017.1598
Ruhstaller T, Thuss-Patience P, Hayoz S et al (2018) Neoadjuvant chemotherapy followed by chemoradiation and surgery with and without cetuximab in patients with resectable esophageal cancer: a randomized, open-label, phase III trial (SAKK 75/08). Ann Oncol 29:1386–1393. https://doi.org/10.1093/annonc/mdy105
Chen Y, Wu X, Bu S et al (2012) Promising outcomes of definitive chemoradiation and cetuximab for patients with esophageal squamous cell carcinoma. Cancer Sci 103:1979–1984. https://doi.org/10.1111/j.1349-7006.2012.02393.x
Lledo G, Huguet F, Chibaudel B et al (2016) Chemoradiotherapy with FOLFOX plus cetuximab in locally advanced oesophageal cancer: the GERCOR phase II trial ERaFOX. Eur J Cancer 56:115–121. https://doi.org/10.1016/j.ejca.2015.12.020
Brenner B, Purim O, Gordon N et al (2019) The addition of cetuximab to preoperative chemoradiotherapy for locally advanced esophageal squamous cell carcinoma is associated with high rate of long-term survival: mature results from a prospective phase Ib/II trial. Radiother Oncol 134:74–80. https://doi.org/10.1016/j.radonc.2019.01.013
Ruhstaller T, Pless M, Dietrich D et al (2011) Cetuximab in combination with chemoradiotherapy before surgery in patients with resectable, locally advanced esophageal carcinoma: a prospective, multicenter phase IB/II trial (SAKK 75/06). J Clin Oncol 29:626–631. https://doi.org/10.1200/JCO.2010.31.9715
Safran H, Suntharalingam M, Dipetrillo T et al (2008) Cetuximab with concurrent chemoradiation for esophagogastric cancer: assessment of toxicity. Int J Radiat Oncol Biol Phys 70:391–395. https://doi.org/10.1016/j.ijrobp.2007.07.2325
De Vita F, Orditura M, Martinelli E et al (2011) A multicenter phase II study of induction chemotherapy with FOLFOX‑4 and cetuximab followed by radiation and cetuximab in locally advanced oesophageal cancer. Br J Cancer 104:427–432. https://doi.org/10.1038/sj.bjc.6606093
Fesinmeyer MD, Mehta V, Blough D et al (2010) Effect of radiotherapy interruptions on survival in medicare enrollees with local and regional head-and-neck cancer. Int J Radiat Oncol Biol Phys 78:675–681. https://doi.org/10.1016/j.ijrobp.2009.08.004
Luo HS, Huang HC, Lin LX (2019) Effect of modern high-dose versus standard-dose radiation in definitive concurrent chemo-radiotherapy on outcome of esophageal squamous cell cancer: a meta-analysis. Radiat Oncol 14:178. https://doi.org/10.1186/s13014-019-1386-x
Gibson MK, Catalano P, Kleinberg LR et al (2020) Phase II study of preoperative chemoradiotherapy with oxaliplatin, infusional 5‑fluorouracil, and cetuximab followed by postoperative docetaxel and cetuximab in patients with adenocarcinoma of the esophagus: a trial of the ECOG-ACRIN cancer research group (E2205). Oncologist 25:e53–59. https://doi.org/10.1634/theoncologist.2018-0750
Tomblyn MB, Goldman BH, Thomas CR Jr et al (2012) Cetuximab plus cisplatin, irinotecan, and thoracic radiotherapy as definitive treatment for locally advanced, unresectable esophageal cancer: a phase-II study of the SWOG (S0414). J Thorac Oncol 7:906–912. https://doi.org/10.1097/JTO.0b013e31824c7bed
Lee MS, Mamon HJ, Hong TS et al (2013) Preoperative cetuximab, irinotecan, cisplatin, and radiation therapy for patients with locally advanced esophageal cancer. Oncologist 18:281–287. https://doi.org/10.1634/theoncologist.2012-0208
Ubink I, van der Sluis P, Schipper M et al (2014) Adding preoperative radiotherapy plus cetuximab to perioperative chemotherapy for resectable esophageal adenocarcinoma: a single-center prospective phase II trial. Oncologist 19:32–33. https://doi.org/10.1634/theoncologist.2013-0254
Garg MK, Zhao F, Sparano JA et al (2017) Cetuximab plus chemoradiotherapy in immunocompetent patients with anal carcinoma: a phase II eastern cooperative oncology group - American college of radiology imaging network cancer research group trial (E3205). J Clin Oncol 35:718–726. https://doi.org/10.1200/JCO.2016.69.1667
Ang KK, Zhang Q, Rosenthal DI et al (2014) Randomized phase III trial of concurrent accelerated radiation plus cisplatin with or without cetuximab for stage III to IV head and neck carcinoma: RTOG 0522. J Clin Oncol 32:2940–2950. https://doi.org/10.1200/JCO.2013.53.5633
Rades D, Stoehr M, Kazic N et al (2008) Locally advanced stage IV squamous cell carcinoma of the head and neck: impact of pre-radiotherapy hemoglobin level and interruptions during radiotherapy. Int J Radiat Oncol Biol Phys 70:1108–1114. https://doi.org/10.1016/j.ijrobp.2007.07.2380
Bonner JA, Harari PM, Giralt J et al (2010) Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5‑year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol 11:21–28. https://doi.org/10.1016/S1470-2045(09)70311-0
Authors’ contributions
All authors contributed to study conception and design. All authors were involved in material preparation, data collection, and/or analyses. The first draft of the manuscript was written by D. Rades and S.E. Schild; all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Funding
The LEOPARD-2 trial was funded by Merck Serono GmbH, an affiliate of Merck KGaA, Darmstadt, Germany. MERCK Reviews prior to Submission, Merck KGaA, Darmstadt, Germany reviewed the manuscript for medical accuracy only prior to submission. The authors are fully responsible for the content of this manuscript, and the views and opinions described in the publication reflect solely those of the authors.
Funding
Open Access funding provided by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
D. Rades received speakers’ honoraria and travel grants from Merck Serono, Roche Pharma, Bristol Myers Squibb, Astra Zeneca, and Amgen until 2015. S.E. Schild edits and writes for UpToDate and has done consulting work for Noxopharm in the past. T. Bartscht, P. Hunold, H. Schmidberger, L. König, J. Debus, C. Belka, N. Homann, P. Spillner, C. Petersen, T. Kuhnt, R. Fietkau, K. Ridwelski, K. Karcher-Kilian, A. Kranich, S. Männikkö, A. Maderer, and M. Moehler declare that they have no competing interests.
Ethical standards
The study has been approved by the ethics committee of the University of Lübeck (reference: 11-104). The study was conducted in accordance with the principles laid out in the Declaration of Helsinki and in accordance with the principles of Good Clinical Practice. Informed consent was obtained from all individual participants included in the study.
Additional information
Data availability
The study has been registered at clinicaltrials.gov (identifier: NCT01787006), where data and results of the study are available as well.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rades, D., Bartscht, T., Hunold, P. et al. Radiochemotherapy with or without cetuximab for unresectable esophageal cancer: final results of a randomized phase 2 trial (LEOPARD-2). Strahlenther Onkol 196, 795–804 (2020). https://doi.org/10.1007/s00066-020-01646-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00066-020-01646-4