
Abstract
This chapter reviews current research on how protein domain architectures evolve. We begin by summarizing work on the phylogenetic distribution of proteins, as this will directly impact which domain architectures can be formed in different species. Studies relating domain family size to occurrence have shown that they generally follow power law distributions, both within genomes and larger evolutionary groups. These findings were subsequently extended to multi-domain architectures. Genome evolution models that have been suggested to explain the shape of these distributions are reviewed, as well as evidence for selective pressure to expand certain domain families more than others. Each domain has an intrinsic combinatorial propensity, and the effects of this have been studied using measures of domain versatility or promiscuity. Next, we study the principles of protein domain architecture evolution and how these have been inferred from distributions of extant domain arrangements. Following this, we review inferences of ancestral domain architecture and the conclusions concerning domain architecture evolution mechanisms that can be drawn from these. Finally, we examine whether all known cases of a given domain architecture can be assumed to have a single common origin (monophyly) or have evolved convergently (polyphyly). We end by a discussion of some available tools for computational analysis or exploitation of protein domain architectures and their evolution.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others

Key words
- Protein domain
- Protein domain architecture
- Superfamily
- Monophyly
- Polyphyly
- Convergent evolution
- Domain evolution
- Kingdoms of life
- Domain co-occurrence network
- Node degree distribution
- Power law
- Parsimony
1 Introduction
1.1 Overview
By studying the domain architectures of proteins, we can understand their evolution as a modular phenomenon, with high-level events enabling significant changes to take place in a time span much shorter than required by point mutations only. This research field has become possible only now in the -omics era of science, as both identifying many domain families in the first place and acquiring enough data to chart their evolutionary distribution require access to many completely sequenced genomes. Likewise, the conclusions drawn generally consider properties averaged for entire species or organism groups or entire classes of proteins, rather than properties of single genes.
We will begin by introducing the basic concepts of domains and domain architectures, as well as the biological mechanisms by which these architectures can change. The remainder of the chapter is an attempt at answering, from the recent literature, the question of which forces shape domain architecture evolution and in what direction. The underlying issue concerns whether it is fundamentally a random process or whether it is primarily a consequence of selective constraints. We end by outlining some available software tools and resources for analysis of domain architectures and their evolution.
1.2 Protein Domains
Protein domains are high-level parts of proteins that either occur alone or together with partner domains on the same protein chain. Most domains correspond to tertiary structure elements and are able to fold independently. All domains exhibit evolutionary conservation, and many either perform specific functions or contribute in a specific way to the function of their proteins. The word domain strictly refers to a distinct region of a specific protein, an instance of a domain family. However, domain and domain family are often used interchangeably in the literature.
1.3 Domain Databases
By identifying recurring elements in experimentally determined protein 3D structures, the various domain families in structural domain databases such as SCOP [1] and CATH [2] were gathered. New 3D structures allow assignment to these classes from semiautomated inspection. The SUPERFAMILY [3] database assigns SCOP domains to all protein sequences by matching them to hidden Markov models (HMMs) that were derived from SCOP superfamilies, i.e., proteins whose evolutionary relationship is evidenced structurally. The Gene3D [4] database is similarly constructed but based on domain families from CATH.
This approach resembles the methodology used in pure sequence-based domain databases such as Pfam [5]. In these databases, conserved regions are identified from sequence analysis and background knowledge, to make multiple sequence alignments. From these, HMMs are built that are used to search new sequences for the presence of the domain represented by each HMM. All such instances are stored in the database. The HMM framework ensures stability across releases and high quality of alignments and domain family memberships. The stability allows annotation to be stored along with the HMMs and alignments. The InterPro database [6] is a meta-database of domains combining the assignments from several different source databases, including Pfam. The Conserved Domain Database (CDD) is a similar meta-database that also contains additional domains curated by the NCBI [7]. SMART [8] is a manually curated resource focusing primarily on signaling and extracellular domains. ProDom [9] is a comprehensive domain database automatically generated from sequences in UniProt [10]. Likewise, ADDA [11] is automatically generated by clustering subsequences of proteins from the major sequence databases, though it has not been updated for some time. Genome3D [12] is a recent consensus database which brings together several domain prediction tools as well as the SCOP and CATH databases for describing representative domain arrangements in a series of trusted, well-annotated genomes.
Since the domain definitions from different databases only partially overlap, results from analyses often cannot be directly compared. In practice, however, choice of database appears to have little effect on the main trends reported by the studies described here.
1.4 Domain Architectures
The terms “domain architecture” or “domain arrangement” generally refer to the domains in a protein and their order, reported in N- to C-terminal direction along the amino acid chain. Another recurring term is domain combinations. This refers to pairs of domains co-occurring in proteins, either anywhere in the protein (the “bag-of-domains” model) or specifically pairs of domains being adjacent on an amino acid chain, in a specific N- to C-terminal order [13]. The latter concept is expanded to triplets of domains, which are subsequences of three consecutive domains, with the N- and C-termini used as “dummy” domains. A domain X occurring on its own in a protein thus produces the triplet N-X-C [14].
1.5 Mechanisms for Domain Architecture Change
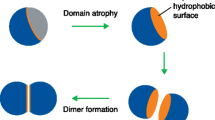
Most mutations are point mutations: substitutions, insertions, or deletions of single nucleotides. While conceivably enough of these might create a new domain from an old one or noncoding sequence or remove a domain from a protein, in practice we are interested in mechanisms whereby the domain architecture of a protein changes instantly or nearly so (but see below for an overview of recent work on the origin of new domains). Figure 1 shows some examples of ways in which domain architectures may mutate. In general, adding or removing domains requires genetic recombination events. These can occur either through errors made by systems for repairing DNA damage such as homologous [16, 17] or nonhomologous (illegitimate) [18, 19] recombination or through the action of mobile genetic elements such as DNA transposons [20] or retrotransposons [21, 22]. Recombination can cause loss or duplication of parts of genes, entire genes or much longer chromosomal regions.

Examples of mutations that can change domain architectures. Adapted from Buljan et al. [25]. (a) Gene fusion by a mobile element. LINE refers to a Long Interspersed Nuclear repeat Element, a retrotransposon. The reverse transcriptase encoded within the LINE causes its mRNA to be reverse-transcribed into DNA and integrated into the genome, making the domain-encoding blue exon from the donor gene integrate along with it in the acceptor gene. (b) Gene fusion by loss of a stop signal or deletion of much of the intergenic region. Genes 1 and 2 are joined together into a single, longer gene. (c) Domain insertion through recombination. The blue domain from the donor gene is inserted within the acceptor gene by either homologous or illegitimate recombination. (d) Right: Gene fission by introduction of transcription stop (the letter Ω) and start (the letter A). Left: Domain loss by introduction of a stop codon (exclamation mark) with subsequent degeneration of the now untranslated domain
In organisms that have introns, exon shuffling [23, 24] refers to the integration of an exon from one gene into another, for instance, through chromosomal crossover, gene conversion, or mobile genetic elements. Exons could also be moved around by being brought along by mobile genetic elements such as retrotransposons [24, 25].
Two adjacent genes can be fused into one if the first one loses its transcription stop signals. Point mutations can cause a gene to lose a terminal domain by introducing a new stop codon, after which the “lost” domain slowly degrades through point mutations as it is no longer under selective pressure [26]. Alternatively, a multi-domain gene might be split into two genes if both a start and a stop signal are introduced between the domains. Novel domains could arise, for instance, through exonization, whereby an intronic or intergenic region becomes an exon, after which subsequent mutations would fine-tune its folding and functional properties [25, 27].
Recent literature (see, e.g., [28]) has discussed the possibility of de novo domain creation through a variety of mutational mechanisms, with some support for this occurring more often than previously thought [29, 30]. The majority of such new domains arise as novel genes from noncoding sequence but may subsequently recombine to join with older domains. Furthermore, young domains in vertebrates tend more often to occur at the N-terminal of a protein and tend to experience higher relative rates of non-synonymous substitution than older domains, which may reflect the nature of the mechanisms through which novel domains arise. Moore, Bornberg-Bauer et al. explore the relative prevalence of domain loss, duplication, and de novo origination in arthropods [31] and plants [32], suggesting such novel domains most frequently are associated with environmental adaptations.
2 Distribution of the Sizes of Domain Families
Domain architectures are fundamentally the realizations of how domains combine to form multi-domain proteins with complex functions. Understanding how these combinations come to be requires first that we understand how common the constituent domains of those architectures are and whether there are selective pressures determining their abundances. Because of this, the body of work concerning the sizes and species distributions of domain families becomes important to us.
Comprehensive studies of the distributions and evolution of protein domains and domain architectures are possible as genome sequencing technologies have made many entire proteomes available for bioinformatic analysis. Initial work [33,34,35] focused on the number of copies that a protein family, either single domain or multi-domain, has in a species. Most conclusions from these early studies appear to hold true for domains, for supra-domains (see below) and for domain architectures [36,37,38]. In particular, these all exhibit a dominance of the population by a selected few [35], i.e., a small number of domain families are present in a majority of the proteins in a genome, whereas most domain families are found only in a small number of proteins.
Looking at the frequency N of families of size X (defined as the number of members in the genome), in the earliest studies, this frequency was modeled as the power law
where a is an exponent parameter. The power law is a special case of the generalized Pareto distribution (GPD) [39]:
Power law distributions arise in a vast variety of contexts: from human income distributions, connectivity of internet routers, word usage in languages, and many other situations ([34, 35, 40, 41], see also [42], for a conflicting view). Luscombe et al. [35] described a number of other genomic properties that also follow power law distributions, such as the occurrence of DNA “words,” pseudogenes, and levels of gene expression. These distributions fit much better than the alternative they usually are contrasted against, an exponential decay distribution. The most important difference between exponential and power law distributions in this context concerns the fact that the latter has a “fat tail,” that is, while most domain families occur only a few times in each proteome, most domains in the proteome still belong to one of a small number of families.
Later work ([39, 43], see also [44]) demonstrated that proteome-wide domain occurrence data fit the general GPD better than the power law but that it also asymptotically fits a power law as X ≫ i. The deviation from strict power law behavior depends on proteome size in a kingdom-dependent manner [43]. Regardless, it is mostly appropriate to treat the domain family size distribution as approximately (and asymptotically) power law-like, and later studies typically assume this.
The power law, but not the GPD, is scale-free in the sense of fulfilling the condition
where f(x) and g(x) are some functions of a variable x and where a is a scaling parameter, that is, studying the data at a different scale will not change the shape of function. This property has been extensively studied in the literature and is connected to other attributes, notably when it occurs in network degree distributions (i.e., frequency distributions of edges per node). Here it has been associated with properties such as the presence of a few central and critical hubs (nodes with many edges to other nodes), the similarity between parts and the whole (as in a fractal), and the growth process called preferential attachment, under which nodes are more likely to gain new links the more links they already have. However, the same power law distribution may be generated from many different network topologies with different patterns of connectivity. In particular, they may differ in the extent that hubs are connected to each other [42]. It is possible to extend the analysis by taking into account the distribution of degree pairs along network edges, but this is normally not done.
What kind of evolutionary mechanisms give rise to this kind of distribution of gene or domain family sizes within genomes? In one model by Huynen and van Nimwegen [33], every gene within a gene family will be more or less likely to duplicate, depending on the utility of the function of that gene family within the particular lineage of organisms studied, and they showed that such a model matches the observed power laws. While they claimed that any model that explains the data must take into account family-specific probabilities of duplication fixation, Yanai and coworkers [45] proposed a simpler model using uniform duplication probability for all genes in the genome and also reported a good fit with data.
Later, more complex birth-death [43] and birth-death-and-innovation (BDIM) [29, 34, 39, 46] models were introduced to explain the observed distributions, and from investigating which model parameter ranges allow this fit, the authors were able to draw several far-ranging conclusions. First, the asymptotic power law behavior requires that the rates of domain gain and loss are asymptotically equal. Karev et al. [39] interpreted this as support for a punctuated equilibrium-type model of genome evolution, where domain family size distributions remain relatively stable for long periods of time but may go through stages of rapid evolution, representing a shift between different BDIM evolutionary models and significant changes in genome complexity. Like Huynen and van Nimwegen [33], they concluded that the likelihood of fixated domain duplications or losses in a genome directly depend on family size. The family will however only grow as long as new copies can find new functional niches and contribute to a net benefit for survival, i.e., as long as selection favors it.
Aside from Huynen and van Nimwegen’s, none of the models discussed depend very strongly on family-specific selection to explain the abundances of individual gene families, nor do they exclude such selection. Some domains may be highly useful to their host organism’s lifestyle, such as cell-cell connectivity domains to an organism beginning to develop multicellularity. Expansion of these domain families might therefore become more likely in some lineages than in others. To what extent these factors actually affect the size of domain families remains to be fully explored. Karev et al. [39] suggested that the rates of domain-level change events themselves—domain duplication and loss rates, as well as the rate of influx of novel domains from other species or de novo creation—must be evolutionarily adapted, as only some such parameters allow the observed distributions to be stable. Van Nimwegen [47] investigated how the number of genes increases in specific functional categories as total genome size increases. He found that the relationship matches a power law, with different coefficients for each functional class remaining valid over many bacterial lineages. Ranea et al. found similar results. Also, Ranea et al. [48] showed that, for domain superfamilies inferred to be present in the last universal common ancestor (LUCA), domains associated with metabolism have significantly higher abundance than those associated with translation, further supporting a connection between the function of a domain family and how likely it is to expand.
Extending the analysis to multi-domain architectures, Apic et al. [37] showed that the frequency distribution of multi-domain family sizes follows a power law curve similar to that reported for individual domain families. It therefore seems likely that the basic underlying mechanisms should be similar in both cases, i.e., that duplication of genes, and thus their domain architectures, is the most important type of event affecting the evolution of domain architectures.
Have the trends described above stood the test of time as more genomes have been sequenced and more domain families have been identified? We considered the 1943 UniProt proteomes covered by version 30.0 of Pfam, plotted the frequency Y of domain families that have precisely X members as a function of X, and fit a power law curve to this. Figure 2a shows the resulting plots for three representative species, one complex eukaryote (Homo sapiens), one simple eukaryote (Saccharomyces cerevisiae), and one prokaryote (Escherichia coli). Figure 2b shows the corresponding plots for all domains in all complete eukaryotic, bacterial, and archaeal proteomes. The power law curve fits decently well, with slopes becoming less steep for the more complex organisms, whose distributions have relatively more large families. The power law-like behavior suggests that complex organisms with large proteomes were formed by heavily duplicating domains from relatively few families. Figures 3a, b show equivalent plots, not for single domains but for entire multi-domain architectures. The curve shapes and the relationship between both species and organism groups are similar, indicating that the evolution of these distributions have been similar.

(a) Distribution of domain family sizes in three selected species. Power law distributions were fitted to these curves such that for frequency f of families of size X, f = cX a. For S. cerevisiae, a = −1.9, for E. coli, a = −1.7, and for H. sapiens, a = −1.5. (b) Distribution of domain family sizes across the three kingdoms. Power law distributions were fitted to these curves such that for frequency f of families of size X, f = cX a. For bacteria, a = −0.9, for archaea, a = −1.1, for eukaryotes, a = −0.8, and for viruses, a = −1.9

(a) Distribution of multi-domain (architecture) family sizes in three selected species. Power law distributions were fitted to these curves such that for frequency f of families of size X, f = cX a. For S. cerevisiae, a = −2.0, for E. coli, a = −1.8, and for H. sapiens, a = −1.5. (b) Distribution of multi-domain (architecture) family sizes across the three kingdoms. Power law distributions were fitted to these curves such that for frequency f of families of size X, f = cX a. For bacteria, a = −1.0, for archaea, a = −1.1, for eukaryotes, a = −1.1, and for viruses, a = −2.0
3 Kingdom and Age Distribution of Domain Families and Architectures
How old are specific domain families or domain architectures? With knowledge of which organism groups they are found in, it is possible to draw conclusions about their age and whether lineage-specific selective pressures have determined their kingdom-specific abundances. Domain families and their combinations have arisen throughout evolutionary history, presumably by new combinations of pre-existing elements that may have diverged beyond recognition or by processes such as exonization. We can estimate the age of a domain family by finding the largest clade of organisms within which it is found, excluding organisms with only xenologs, i.e., horizontally transferred genes [14]. The age of this lineage’s root is the likely age of the family. The same holds true for domain combinations and entire domain architectures. This methodology allows us to determine how changing conditions at different points in evolutionary history, or in different lineages, have affected the evolution of domain architectures.
Apic et al. [36] analyzed the distribution of SCOP domains across 40 genomes from archaea, bacteria, and eukaryotes. They found that a majority of domain families are common to all three kingdoms of life and thus likely to be ancient. Kuznetsov et al. [43] performed a similar analysis using InterPro domains and found that only about one fourth of all such domains were present in all three kingdoms, but a majority was present in more than one of them. Lateral gene transfer or annotation errors can cause a domain family to be found in one or a few species in a kingdom without actually belonging to that kingdom. To counteract this, one can require that a family must be present in at least a reasonable fraction of the species within a kingdom for it to be considered anciently present there. For instance, using Gene3D assignments of CATH domains to 114 complete genomes, mainly bacterial, Ranea et al. [48] isolated protein superfamily domains that were present in at least 90% of all the genomes and at least 70% of the archaeal and eukaryotic genomes, respectively. Under these stringent cutoffs for considering a domain to be present in a kingdom, 140 domains, 15% of the CATH families found in at least one prokaryote genome, were inferred to be ancient. Chothia and Gough [49] performed a similar study on 663 SCOP superfamily domains evaluated at many different thresholds and found that while 516 (78%) superfamilies were common to all three kingdoms at a threshold of 10% of species in each kingdom, only 156 (24%) superfamilies were common to all three kingdoms at a threshold of 90%. They also showed that for prokaryotes, a majority of domain instances (i.e., not domain families but actual domain copies) belong to common superfamilies at all thresholds below 90%.
Extending to domain combinations, Apic et al. [36] reported that a majority of SCOP domain pairs are unique to each kingdom but also that more kingdom-specific domain combinations than expected were composed only of domain families shared between all three kingdoms. This would imply a scenario where the independent evolution of the three kingdoms mainly involved creating novel combinations of domains that existed already in their common ancestor.
Several studies have reported interesting findings on domain architecture evolution in lineages closer to ourselves: in metazoa and vertebrates. Ekman et al. [50] claimed that new metazoa-specific domains and multi-domain architectures have arisen roughly once every 0.1–1 million years in this lineage. According to their results, most metazoa-specific multi-domain architectures are a combination of ancient and metazoa-specific domains. The latter category are however mostly found as novel single-domain proteins. Much of the novel metazoan multi-domain architectures involve domains that are versatile (see below) and exon-bordering (allowing for their insertion through exon shuffling). The novel domain combinations in metazoa are enriched for proteins associated with functions required for multicellularity—regulation, signaling, and functions involved in newer biological systems such as immune response or development of the nervous system, as previously noted by Patthy [23]. They also showed support for exon shuffling as an important mechanism in the evolution of metazoan domain architectures. Itoh et al. [51] added that animal evolution differs significantly from other eukaryotic groups in that lineage-specific domains played a greater part in creating new domain combinations. Nasir et al. [52] analyzed the age and taxonomic distribution of domains drawing on species phylogenies reconstructed from domain repertoires, concluding among other things that most widespread domains are relatively old and suggesting high numbers of both domain gain and loss in the evolution of the three organismal superkingdoms. Bacterial and archaeal genes have tended to gain or lose domains encoding aspects of metabolic capacity, whereas those of eukaryotes—including multicellular ones—have gained domains enabling more elaborate extracellular processes such as immunity and regulatory capacities.
In the most recent datasets, what is the distribution of domains and domain combinations across the three kingdoms of life? Looking at the set of UniProt proteomes represented in version 30.0 of Pfam, the distribution of domains across the three kingdoms are as displayed in the Venn diagram of Fig. 4a. Figure 4b, c show the equivalent distributions of immediate neighbors and triplets of domains, respectively, and Fig. 4d the distribution of multi-domain architectures across kingdoms. The numbers are somewhat biased toward bacteria as 56% of the UniProt proteomes are from this kingdom. However, with this high coverage of all kingdoms (506 eukaryotic, 94 archaeal, and 1090 bacterial proteomes, as well as 253 viral entities), the results should be robust in this respect. Compared to most previous reports, we see a striking difference in that a much smaller portion of domains are shared between all kingdoms. There are some potential artifacts which could affect this analysis. If lateral gene transfer is very widespread, we may overestimate the number of families present in all three kingdoms. Moreover, there are cases where separate Pfam families are actually distant homologs of each other, which could lead to underestimation of the number of ancient families. To counteract this, we make use of Pfam clans, considering domains in the same clan to be equivalent. While not all distant homologies have yet been registered in the clan system, performing the analysis on the clan level reduces the risk of such underestimation.

(a) Kingdom distribution of unique domains. Values are given as percentages of the total, 10,330 domains. (b) Kingdom distribution of unique domain pairs. Values are given as percentages of the total, 31,287 domain pairs. (c) Kingdom distribution of unique domain triplets. Values are given as percentages of the total, 33,662 domain triplets. (d) Kingdom distribution of unique multi-domain architectures. Values are given as percentages of the total, 23,238 multi-domain architectures
Our finding that 10% of all Pfam-A domains are present in all three main kingdoms is strikingly lower than in the earlier works and is even lower than reported by Ranea et al. [48], who used very stringent cutoffs. However, a direct comparison of statistics for Pfam domains/clans and CATH superfamilies is difficult. The decrease in ancient families that we observe may be a consequence of the massive increase in sequenced genomes and/or that the recent growth of Pfam has added relatively more kingdom-specific domains. We further found that only 1.5% of all domains or domain combinations are unique to archaea, suggesting that known representatives of this lineage have undergone very little independent evolution and/or that most archaeal gene families have been horizontally transferred to other kingdoms. The trend when going from domain via domain combinations to whole architectures is clear—the more complex patterns are less shared between the kingdoms. In other words, each kingdom has used a common core of domains to construct its own unique combinations of multi-domain architectures.
4 Domain Co-occurrence Networks
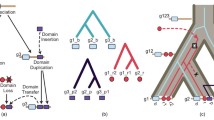
A multi-domain architecture connects individual domains with each other. There are several ways to derive these connections and quantify the level of co-occurrence. The simplest method is to consider all domains on the same amino acid chain to be connected, but we can also limit the set of co-occurrences we consider to, e.g., immediate neighbor pairs or triplets. Regardless of which method is used, the result is a domain co-occurrence network, where nodes represent domains and where edges represent the existence of proteins in which members of these families co-occur. Figure 5 shows an example of such a network and the set of domain architectures which defines it. This type of explicit network representation is explored in several studies, notably by Itoh et al. [51], Przytycka et al. [53], and Kummerfeld and Teichmann [13]. It is advantageous as it allows the introduction of powerful analysis tools developed within the engineering sciences for use with artificial network structures such as the World Wide Web. The patterns of co-occurrences that we observe should be a direct consequence of the constraints and conditions under which domain architectures evolve, and because of this, the study of these patterns becomes relevant for understanding such factors.

Example of protein domain co-occurrence network, adapted from Kummerfeld and Teichmann [13]. (a) Sample set of domain architectures. The lines represent proteins and the boxes their domains in N- to C-terminal order. (b) Resulting domain co-occurrence (neighbor) network. Nodes correspond to domains and are linked by an edge if at least one domain exists where the two domains are found adjacent to each other along the amino acid chain
The frequency distribution of node degrees in the domain co-occurrence network has been fitted to a power law [36] and a more general GPD as well [40]. The closer this approximation holds, the more the network will have the scale-free property. This property can be thought of as a hierarchy in the network, where the more centrally connected nodes link to more peripheral nodes with the same relative frequency at each level. In the context of domains, this means that a small number of domains co-occur with a high number of other domains, whereas most domains only have a few neighbors—usually some of the highly connected hubs. The most highly connected domains are referred to as promiscuous [54], mobile, or versatile [14, 55, 56]. Many such hub domains are involved in intracellular or extracellular signaling, protein-protein interactions and catalysis, and transcription regulation. In general, these are domains that encode a generic function, e.g., phosphorylation, which is reused in many contexts by additional domains that confer substrate specificity or localization. Table 1 shows the domains (or clans) with the highest numbers of immediate neighbors in Pfam 30.0.
One way of evolving a domain co-occurrence network that follows a power law is by “preferential attachment” [53, 57]. This means that new edges (corresponding to proteins where two domains co-occur) are added with a probability that is higher the more edges these nodes (domains) already have, resulting in a power law distribution.
Apic et al. [37] considered a null model for random domain combination, in which a proteome contains domain combinations with a probability based on the relative abundances of the domains only. They showed that this model does not hold and that far fewer domain combinations than expected under it are actually seen. If most domain duplication events are gene duplication events that do not change domain architecture—or at the very least do not disrupt domain pairs—then this finding is not unexpected, nor does it require or exclude any particular selective pressure to keep these domains together in proteins. There is growing support for the idea that separate instances of a given domain architecture in general descend from a single ancestor with that architecture [58], with polyphyletic evolution of domain architectures occurring only in a small fraction of cases [53, 59, 60].
Itoh et al. [51] performed reconstruction of ancestral domain architectures using maximum parsimony, as described in the next section. This allowed them to study the properties of the ancestral domain co-occurrence network and thus explore how network connectivity has altered over evolutionary time. Among other things, they found increased connectivity in animals, particularly of animal-specific domains, and suggest that this phenomenon explains the high connectivity for eukaryotes reported by Wuchty [40]. For non-animal eukaryotes, they reported a correlation between connectivity and age, such that older domains had relatively higher connectivity, with domains preceding the divergence of eukaryotes and prokaryotes being the most highly connected, followed by early eukaryotic domains. In other words, early eukaryotic evolution saw the emergence of some key hub proteins, while the most prominent eukaryotic hubs emerged in the animal lineage. Parikesit et al. [61] studied the functional annotation of co-occurring domains in eukaryotes, concluding that while these may have different associated functional descriptors, these descriptors usually tend to fall within the same overall category within the gene ontology. Co-occurring domains thus tend to contribute to the same overall process type rather than have very widely divergent functional annotations. Hsu et al. [62] constructed a network linking domain architectures (i.e., each node is a multi-domain architecture, as opposed to in a regular domain co-occurrence network) where parsimonious reconstruction suggests evolution of one from the other, identifying “highly evolvable” architectures as hubs in this network. Proteins with such architectures were reported to be more widespread, less often essential, more often duplicated, and more often associated with gene functions involved in specific adaptation of organisms.
What is the degree distribution of current domain co-occurrence networks? We again used the domain architectures from all complete proteomes in version 30.0 of Pfam and considered the network of immediate neighbor relationships, i.e., nodes (domains) have an edge between them if there is a protein where they are adjacent. Each domain was assigned a degree as its number of links to other domains. We then counted the frequency with which each degree occurs in the co-occurrence network. Figure 6a shows this relationship for the set of domain architectures found in the same species as for Figs. 2a, and 6b shows the equivalent plots for the three kingdoms as found among the complete proteomes in Pfam. Regressions to a power law have been added to the plots. The presence of a power law-like behavior of this type implies that few domains have very many immediate neighbors, while most domains have few immediate neighbors. Note that the observed degrees in our dataset were strongly reduced by removing all sequences with a stretch longer than 50 amino acids lacking domain annotation.

(a) Distribution of domain co-occurrence network node degrees in three selected species. Power law distributions were fitted to these curves such that for frequency f of families of size X, f = cX a. For S. cerevisiae, a = −2.2, for E. coli, a = −2.0, and for H. sapiens, a = −1.9. (b) Distribution of domain co-occurrence network node degrees across the three kingdoms. This corresponds to a network where two domains are connected if any species within the kingdom has a protein where these domains are immediately adjacent. Power law distributions were fitted to these curves such that for frequency f of families of size X, f = cX a. For bacteria, a = −1.6, for archaea, a = −1.7, for eukaryotes, a = −1.5, and for viruses a = −2.0
5 Supra-domains and Conserved Domain Order
As we have seen, whole multi-domain architectures or shorter stretches of adjacent domains are often repeated in many proteins. These only cover a small fraction of all possible domain combinations. Are the observed combinations somehow special? We would expect selective pressure to retain some domain combinations but not others, since only some domains have functions that would synergize together in one protein. Often, co-occurring domains require each other structurally or functionally, for instance, in transcription factors where the DNA-binding domain provides substrate specificity, whereas the trans-activating domain recruits other components of the transcriptional machinery [63]. Vogel et al. [38] identified series of domains co-occurring as a fixed unit with conserved N- to C-terminal order but flanked by different domain architectures and termed them supra-domains. By investigating their statistical overrepresentation relative to the frequency of the individual domains in the set of nonredundant domain architectures (where “nonredundant” is crucial, as otherwise, e.g., whole-gene duplication would bias the results), they identified a number of such supra-domains. Many ancient domain combinations (shared by all three kingdoms) appear to be such selectively preserved supra-domains.
How conserved is the order of domains in multi-domain architectures? In a recent study, Kummerfeld and Teichmann [13] built a domain co-occurrence network with directed edges, allowing it to represent the order in which two domains are found in proteins. As in other studies, the distribution of node degrees fits a power law well. Most domain pairs were only found in one orientation. This does not seem required for functional reasons, as flexible linker regions should allow the necessary interface to form also in the reversed case [58], but may rather be an indication that most domain combinations are monophyletic. Weiner and Bornberg-Bauer [64] analyzed the evolutionary mechanisms underlying a number of reversed domain order cases and concluded that independent fusion/fission is the most frequent scenario. Although domain reversals occur in only a few proteins, it actually happens more often than was expected from randomizing a co-occurrence network [13]. That study also observed that the domain co-occurrence network is more clustered than expected by a random model and that these clusters are also functionally more coherent than would be expected by chance.
6 Domain Mobility, Promiscuity, or Versatility
While some protein domains co-occur with a variety of other domains, some are always seen alone or in a single architecture in all proteomes where they are found. A natural explanation is that some domains are more likely to end up in a variety of architectural contexts than others due to some intrinsic property they possess. Is such domain versatility or promiscuity a persistent feature of a given domain, and does it correlate with certain functional or biological properties of the domain?
Several ways of measuring domain versatility have been suggested. One measure, NCO [40], counts the number of other domains found in any architectures where the domain of interest is found. Another measure, NN [37], instead counts the number of distinct other domains that a domain is found adjacent to. Yet another measure, NTRP [65], counts the number of distinct triplets of consecutive domains where the domain of interest is found in the middle. All of these measures can be expected to be higher for common domains than for rare domains, i.e., variations in domain abundance (the number of proteins a domain is found in) can hide the intrinsic versatility of domains. Therefore, three different studies [14, 55, 66] formulated relative domain versatility indices that aim to measure versatility independently of abundance. It is worth noting that most studies have considered only immediately adjacent domain neighbors in these analyses, a restriction based on the assumption that those are more likely to interact functionally than domains far apart on a common amino acid chain. More recent work [67] introduced a network versatility metric which can classify domains as being central or peripheral with regard to the large-scale structure of their bigram network (i.e., the network-linking domains found adjacent in proteins), observing how peripheral such domains exhibit relatively higher primary sequence conservation suggestive of adaptation to more specific functions, whereas the core domains may be more multifunctional.
The first relative versatility study was presented by Vogel et al. [66], who used as their domain dataset the SUPERFAMILY database applied to 14 eukaryotic, 14 bacterial, and 14 archaeal proteomes. They modeled the number of unique immediate neighbor domains as a power law function of domain abundance, performed a regression on this data, and used the resulting power law exponent as a relative versatility measure. Basu et al. [55] used Pfam and SMART [8] domains and measured relative domain versatility for 28 eukaryotes as the immediate neighbor pair frequency normalized by domain frequency. They then defined promiscuous domains as a class according to a bimodality in the distribution of the raw numbers of unique domain immediate neighbor pairs. Weiner et al. [14] used Pfam domains for 10,746 species in all kingdoms and took as their relative versatility measure the logarithmic regression coefficient for each domain family across genomes, meaning that it is not defined within single proteomes.
To what extent is high versatility an intrinsic property of a certain domain? Vogel et al. [66] only examined large groups of domains together and therefore did not address this question for single domains. Basu et al. [55] and Weiner et al. [14] instead analyzed each domain separately and concluded that there are strong variations in relative versatility at this level. Their results are very different in detail, however, reflected by the fact that only one domain family (PF00004, AAA ATPase family) is shared between the ten most versatile domains reported in the two studies. As they used fairly similar domain datasets, it would appear that the results strongly depend on the definition of relative versatility. Another potential reason for the different results is that Basu’s list was based on eukaryotes only, while Weiner’s analysis was heavily biased toward prokaryotes. Furthermore, the top ten list in Basu et al. [55] and their follow-up paper [56] only overlap by four domains, yet the main difference is that in the latter study all 28 eukaryotes were considered, while the former study was limited to the subset of 20 animal, plant, and fungal species. The choice of species thus seems pivotal for the results when using this method. They also used different methods for calculating the average value of relative versatility across many species, which may influence the results.
Does domain versatility vary between different functional classes of domains? Vogel et al. [66] found no difference in relative versatility between broad functional or process categories or between SCOP structural classes. In contrast to this, Basu et al. [55] reported that high versatility was associated with certain functional categories in eukaryotes. However, no test for the statistical significance of these results was performed. Weiner et al. [14] also noted some general trends but found no significant enrichment of gene ontology terms in versatile domains. This does not necessarily mean that no such correlation exists, but more research is required to convincingly demonstrate its strength and its nature. More recently, Cromar et al. [68] analyzed domain architectures in eukaryotic extracellular matrix proteomes, noting that these structures are organized around a set of versatile domains under the weighted bigram metric of Basu et al. [55].
Another important question is to what extent domain versatility varies across evolutionary lineages. Vogel et al. [66] reported no large differences in average versatility for domains in different kingdoms. The versatility measure of Basu et al. [55] can be applied within individual genomes, which means that according to this measure domains may be versatile in one organism group but not in another, as well as gain or lose versatility across evolutionary time. They found that more domains were highly versatile in animals than in other eukaryotes. Modeling versatility as a binary property defined for domains in extant species, they further used a maximum parsimony approach to study the persistence of versatility for each domain across evolutionary time and concluded that both gain and loss of versatility are common during evolution. Inferring ancestral domain architectures, Cohen-Gihon et al. [69] report an increase in versatility in many domains during eukaryotic evolution, in particular around the divergence of Bilateria. Weiner at al. [14] divided domains into age categories based on distribution across the tree of life and reported that the versatility index is not dependent on age, i.e., domains have equal chances of becoming versatile at different times in evolution. This is consistent with the observation by Basu et al. [55] that versatility is a fast-evolving and varying property. When measuring versatility as a regression within different organism groups, Weiner et al. [14] found slightly lower versatility in eukaryotes, which is in conflict with the findings of Basu et al. [55]. Again, this underscores the strong dependence of the method and dataset on the results.
Further properties reported to correlate with domain versatility include sequence length, where Weiner et al. [14] found that longer domains are significantly more versatile within the framework of their study, while at the same time, shorter domains are more abundant and hence may have more domain neighbors in absolute numbers. Basu et al. [55] further reported that more versatile domains have more structural interactions than other domains. To determine which of these reported correlations that genuinely reflect universal biological trends, further comprehensive studies are needed using more data and uniform procedures. This would hopefully allow the results from the studies described here to be validated and any conflicts between them to be resolved.
Basu et al. [55] further analyzed the phylogenetic spread of all immediate domain neighbor pairs (“bigrams”) containing domains classified as promiscuous. The main observation this yielded was that although most such combinations occurred in only a few species, most promiscuous domains are part of at least one combination that is found in a majority of species. They interpreted this as implying the existence of a reservoir of evolutionarily stable domain combinations from which lineage-specific recombination may draw promiscuous domains to form unique architectures. Later work by Hsu et al. [70] analyzed the domain co-occurrence networks centered on each domain family, classifying such subnetworks as being either mostly starlike, taillike, or tetragon-like, with promiscuous domains forming cores of starlike architecture networks in this representation.
7 Principles of Domain Architecture Evolution
What mutation events can generate new domain architectures, and what is their relative predominance? The question can be approached by comparing protein domain architectures of extant proteins. This is based on the likely realistic assumption that most current domain architectures evolved from ancestral domain architectures that can still be found unchanged in other proteins. Because of this, in pairs of most similar extant domain architectures, one can assume that one of them is ancestral. This agrees well with results indicating that most groups of proteins with identical domain architectures are monophyletic. By comparing the most similar proteins, several studies have attempted to chart the relative frequencies of different architecture-changing mutations.
Björklund et al. [71] used this particular approach and came to several conclusions. First, changes to domain architecture are much more common by the N- and C-termini than internally in the architecture. This is consistent with several mechanisms for architecture changes such as introduction of new start or stop codons or mergers with adjacent genes, and similar results have been found in several other studies [15, 25, 26]. Furthermore, insertions or deletions of domains (“indels”) are more common than substitutions of domains, and the events in question mostly concern just single domains, except in cases with repeats expanding with many domains in a row [72]. In a later study, the same group made use of phylogenetic information as well, allowing them to infer directionality of domain indels [50]. They then found that domain insertions are significantly more common than domain deletions.
Weiner et al. [26] performed a similar analysis on domain loss and found compatible results—most changes occur at the termini (see also discussion in [28]). Moreover, they demonstrated that terminal domain loss seldom involves losing only part of a domain, or rather, that such partial losses quickly progress into loss of the entire domain. However, it is important to ensure such observations are not confounded by cases where errors in gene boundary recognition make domain detection less accurate [73].
There is some support [23, 74, 75] for exon shuffling to have played an important part in domain evolution, and there are a number of domains that match intron borders well, for example, structural domains in extracellular matrix proteins. While it may not be a universal mechanism, exon shuffling is suggested to have been particularly important for vertebrate evolution [23].
Recognizing the potential role of gene duplications in domain architecture evolution, Grassi et al. [76] analyzed domain architecture shifts following either whole-genome duplication (WGD) or smaller-scale gene duplication events in yeast. Surviving WGD duplicates had retained ancestral architecture in ca 95% of cases, with approximately the same chance of architecture change in WGD as under local duplication. Genes retained over time from either type of duplication were enriched for a core of commonly occurring domains but with a subset of rarer domains additionally enriched in retained WGD duplicates compared to locally duplicated genes. The former category more often was associated with housekeeping-type gene functions, whereas the latter more often involved adaptive functions. Functional change was generally larger than architectural change following duplication. Zhang et al. [77] similarly studied domain architecture evolution in plants, noting that lineage-specific architecture expansions largely can be explained from differential retention of genes following successive whole-genome duplications. Another form of domain duplication particularly relevant in plants is amplification of the numbers of domain repeats in proteins, discussed, e.g., by Sharma and Pandey [78].
8 Inferring Ancestral Domain Architectures
The above analyses, based on pairwise comparison of extant protein domain architectures, cannot tally ancestral evolutionarily events nearer the root of the tree of life. With ancestral architectures, one can directly determine which domain architecture changes have taken place during evolution and precisely chart how mechanisms of domain architecture evolution operate, as well as gauge their relative frequency. A drawback is that since we can only infer ancestral domain architectures from extant proteins, the result will depend somewhat on our assumptions about evolutionary mechanisms. On the upside, it should be possible to test how well different assumptions fit the observed modern-day protein domain architecture patterns.
Attempts at such reconstructions have been made using parsimony. Given a gene tree and the domain architectures at the leaves, dynamic programming can be used in order to find the assignment of architectures to internal nodes that require the smallest number of domain-level mutation events. This simple model can be elaborated by weighting loss and gain differently or by requiring that a domain or an architecture can only be gained at most once in a tree (Dollo parsimony) [79].
An early study of Snel et al. [80] considered 252 gene trees across 17 fully sequenced species and used parsimony to minimize the number of gene fission and fusion events occurring along the species tree. Their main conclusion, that gene fusions are more common than gene fissions, was subsequently supported by a larger study by Kummerfeld and Teichmann [81], where fusions were found to be about four times as common as fissions in a most parsimonious reconstruction. Fong et al. [82] followed a similar procedure on yet more data and concluded that fusion was 5.6 times as likely as fission.
Buljan and Bateman [15] performed a similar maximum parsimony reconstruction of ancestral domain architectures. They too observed that domain architecture changes primarily take place at the protein termini, and the authors suggested that this might largely occur because terminal changes to the architecture are less likely to disturb overall protein structure. Moreover, they concluded from reconciliation of gene and species trees that domain architecture changes were more common following gene duplications than following speciation but that these cases did not differ with respect to the relative likelihood of domain losses or gains.
Recently, Buljan et al. [25] presented a new ancestral domain architecture reconstruction study which assumed that gain of a domain should take place only once in each gene tree, i.e., Dollo parsimony [79]. Their results also support gene fusion as a major mechanism for domain architecture change. The fusion is generally preceded by a duplication of either of the fused genes. Intronic recombination and insertion of exons are observed but relatively rarely. They also found support for de novo creation of disordered segments by exonization of previously noncoding regions. More recently still a method for domain architecture history reconstruction using a network construct called a plexus was described [83]. Yang and Bourne [84] further described another parsimony-based reconstruction approach, as did Wu et al. [85], reporting that histories of signaling and development proteins are enriched for gene fusion/fission events. Stolzer et al. [86] present another method for domain architecture history inference, made available through the Notung software.
9 Polyphyletic Domain Architecture Evolution
There appears to be a “grammar” for how protein domains are allowed to be combined. If nature continuously explores all possible domain combinations, one would expect that the allowed combinations would be created multiple times throughout evolution. Such independent creation of the same domain architecture can be called convergent or polyphyletic evolution, whereas a single original creation event for all extant examples on an architecture would be called divergent or monophyletic evolution. This is relevant for several reasons, not least because it determines whether or not we can expect two proteins with identical domain architectures to have the same history along their entire length.
A graph theoretical approach to answer this question was taken by Przytycka et al. [53], who analyzed the set of all proteins containing a given superfamily domain. The domain architectures of these proteins define a domain co-occurrence network, where edges connect two domains both found in a protein, regardless of sequential arrangement. The proteins of such a set can also be placed in an evolutionary tree, and the evolution of all multi-domain architectures containing the reference domain can be expressed in terms of insertions and deletions of other domains along this tree to form the extant domain architectures. The question, then, is whether or not all leaf nodes sharing some domain arrangement (up to and including an entire architecture) stem from a single ancestral node possessing this combination of domains. For monophyly to be true for all architectures containing the reference domain, the same companion domain cannot have been inserted in more than one place along the tree describing the evolution of the reference domain. By application of graph theory and Dollo parsimony [79], they showed that monophyly is only possible if the domain co-occurrence network defined by all proteins containing the reference domain is chordal, i.e., it contains no cycles longer than three edges.
Przytycka et al. [53] then evaluated this criterion for all superfamily domains in a large-scale dataset. For domains where the co-occurrence network contained fewer than 20 nodes (domains), the chordal property and hence the possibility of complete monophyly of all domain combinations and domain architectures containing that domain held. By comparing actual domain co-occurrence networks with a preferential attachment null model, they showed that far more architectures are potentially monophyletic than would be expected under a pure preferential attachment process. This finding is analogous to the observation by Apic et al. [37] that most domain combinations are duplicated more frequently (or reshuffled less) than expected by chance. In other words, gene duplication is much more frequent than domain recombination [66]. However, for many domains that co-occurred with more than 20 other different domains, particularly for domains previously reported as promiscuous, the chordal property was violated, meaning that multiple independent insertions of the same domain, relative to the reference domain phylogeny, must be assumed.
A more direct approach is to do complete ancestral domain architecture reconstruction of protein lineages and to search for concrete cases that agree with polyphyletic architecture evolution. There are two conceptually different methodologies for this type of analysis. Either one only considers architecture changes between nodes of a species tree, or one considers any node in a reconstructed gene tree. The advantage of using a species tree is that one avoids the inherent uncertainty of gene trees, but on the other hand, only events that take place between examined species can be observed.
Gough [59] applied the former species-tree-based methodology to SUPERFAMILY domain architectures and concluded that polyphyletic evolution is rare, occurring in 0.4–4% of architectures. The value depends on methodological details, with the lower bound considered more reliable.
The latter gene-tree-based methodology was applied by Forslund et al. [60] to the Pfam database. Ancestral domain architectures were reconstructed through maximum parsimony of single-domain phylogenies which were overlaid for multi-domain proteins. This strategy yielded a higher figure, ranging between 6% and 12% of architectures depending on dataset and whether or not incompletely annotated proteins were removed. The two different approaches thus give very different results. The detection of polyphyletic evolution is in both frameworks dependent on the data that is used—its quality, coverage, filtering procedures, etc. The studies used different datasets which makes it hard to compare. However, given that their domain annotations are more or less comparable, the major difference ought to be the ability of the gene-tree method to detect polyphyly at any point during evolution, even within a single species. It should be noted that domain annotation is by no means complete—only a little less than half of all residues are assigned to a domain [5]—and this is clearly a limiting factor for detecting architecture polyphyly. The numbers may thus be adjusted considerably upwards when domain annotation reaches higher coverage. A later study by Zmasek and Godzik [87] reports much higher rates (25–75%) still of polyphyletic evolution of eukaryotic multi-domain architectures, arguing that previous datasets were too small to have the power to reveal this.
Future work will be required to provide more reliable estimates of how common polyphyletic evolution of domain architectures is. Any estimate will depend on the studied protein lineage, the versatility of the domains, and methodological factors. A comprehensive and systematic study using more complex phylogenetic methods than the fairly ad hoc parsimony approach, as well as effective ways to avoid overestimating the frequency of polyphyletic evolution due to incorrect domain assignments or hidden homology between different domain families, may be the way to go. At this point all that can be said is that polyphyletic evolution of domain architectures definitely does happen, but relatively rarely, and that it is more frequent for complex architectures and versatile domains. A detailed case study was made recently of netrin domain-containing proteins, where polyphyletic evolution in metazoa seems well-supported [88]; these authors further suggest the term merology for such polyphyletic evolution. A series of papers by Nagy and Patthy et al. [73, 89, 90] further elaborates on challenges faced within this line of research; they report strong confounding influence of gene prediction errors. They further propose the term epaktology for gene similarity resulting from the independent acquisition of two proteins by the same additional domain. The authors suggest such cases inflate both estimates of terminal domain changes and estimates of gene fusion-driven changes in domain architecture. Beyond such changes, whether correctly inferred or not, the authors describe internal domain shuffling as an important mechanism for how domain architecture evolution has occurred.
10 Conclusions
As access to genomic data and to increasing amounts of compute power has grown during the last decade-and-a-half, so has our knowledge of the overall patterns of domain architecture evolution. Still, no study is better than its underlying assumptions, and differences in the representation of data and hypotheses mean that results often cannot be directly compared. Overall, however, the current state of the field appears to support some broad conclusions.
Domain and multi-domain family sizes, as well as numbers of co-occurring domains, all approximately follow power laws, which implies a scale-free hierarchy. This property is associated with many biological systems in a variety of ways. In this context, it appears to reflect how a relatively small number of highly versatile components have been reused again and again in novel combinations to create a large part of the domain and domain architecture repertoire of organisms. Gene duplication is the most important factor to generate multi-domain architectures, and as it outweighs domain recombination, only a small fraction of all possible domain combinations is actually observed. This is probably further modulated by family-specific selective pressure, though more work is required to demonstrate to what extent. Most of the time, all proteins with the same architecture or domain combination stem from a single ancestor where it first arose, but there remains a fraction of cases, particularly with domains that have very many combination partners, where this does not hold.
Most changes to domain architectures occur following a gene duplication and involve the addition of a single domain to either protein terminus. The main exceptions to this occur in repeat regions. Exon shuffling played an important part in animals by introducing a great variety of novel multi-domain architectures, reusing ancient domains as well as domains introduced in the animal lineage.
In this chapter, we have reexamined with the most up-to-date datasets many of the analyses done previously on less data and found that the earlier conclusions still hold true. Even though we are at the brink of amassing enormously much more genome and proteome data thanks to the new generation of sequencing technology, there is no reason to believe that this will alter the fundamental observations we can make today on domain architecture evolution. However, it will permit a more fine-grained analysis, and also there will be a greater chance to find rare events, such as independent creation of domain architectures. Furthermore, careful application of more complex models of evolution with and without selection pressure may allow us to determine more closely to what extent the process of domain architecture evolution was shaped by selective constraints.
11 Materials and Methods
Updated statistics were generated from the data in Pfam 30.0. All UniProt proteins in the SwissPfam set for Pfam 30.0 were included. These span 1090 bacteria, 506 eukaryotes, and 94 archaea. All Pfam-A domains regardless of type were included. However, as stretches of repeat domains are highly variable, consecutive subsequences of the same domain were collapsed into a single pseudo-domain, if it was classified as type Motif or Repeat, as in several previous works [50, 60, 66, 82].
Domains were ordered within each protein based on their sequence start position. In the few cases of domains being inserted within other domains, this was represented as the outer domain followed by the nested domain, resulting in a linear sequence of domain identifiers. As long regions without domain assignments are likely to represent the presence of as-yet uncharacterized domains, we excluded any protein with unassigned regions longer than 50 amino acids (more than 95% of Pfam-A domains are longer than this). This approach is similar to that taken in previous works [59, 60, 71]. Other studies [50, 72] have instead performed additional, more sensitive domain assignment steps, such as clustering the unassigned regions to identify unknown domains within them.
Pfam domains are sometimes organized in clans, where clanmates are considered homologous. A transition from a domain to another of the same clan is thus less likely to be a result of domain swapping of any kind and more likely to be a result of sequence divergence from the same ancestor. Because of this, we replaced all Pfam domains that are clan members with the corresponding clan.
The statistics and plots were generated using a set of Perl and R scripts, which are available upon request. Power law regressions were done using the R nls function. For reasons of scale, the regression for a power law relation such as
was performed on the equivalent relationship
for the parameters a and c, with the exception of the data for Fig. 6, where instead the relationship
was used. Moreover, because species or organism group datasets were of very different size, raw counts of domains were converted to frequencies before the regression was performed.
12 Online Domain Database Resources
For further studies or research into this field, the first and most important stop will be the domain databases. Table 2 presents a selection of domain databases in current use.
13 Domain Architecture Analysis Software
Several software tools have been described and made available that allow for analysis and visualization of domain architectures and their evolution. A selection of such tools is shown in Table 3.
A few of these tools allow domain architecture evolution analysis by visualizing each protein’s domain architecture along a protein sequence tree. An example is the web tool TreeDom [96] which, given a protein domain family and an anchor sequence, fetches the family from Pfam and builds a tree with the nearest neighbors of the anchor sequence. An example output from TreeDom is shown in Fig. 7, in which a nonredundant set of representative proteomes were queried. Here one can see that while the NUDIX domain of the anchor sequence tends to co-occur with two other domains (zf-NADH-PPase and NUDIX-like), it also has recombined with many other domains over the course of evolution.

TreeDom output using as query the NUDIX domain (PF00293), the human NUDT12 (Q9BQG2) protein, 30 closest sequences, and RP15 (representative proteomes at 15% co-membership). The domains are green, NUDIX; blue, NUDIX-like (PF09296); yellow, zf-NADH-PPase (PF09297); red, Ocnus (PF05005); cyan, Ank_2 (PF12796); black, Ank_5 (PF13857); orange, Prefoldin (PF02996); and pink, Fibrinogen_C (PF00147)
Other tools allow different types of analyses, for instance, searching for similar domain architectures or showing taxonomic distributions. Some of the protein domain databases listed in Table 2 include variants of such analyses, while external tools typically offer more specialized functionality. For example, the Pfam website allows searching for domain content, while the java tool PfamAlyzer allows searching Pfam for particular domain architecture patterns specified with a given domain order and spacing [94].
The RAMPAGE/RADS tools [95] make use of domain assignments for rapid homology searching. DoMosaics [92] is a software tool that can act as a wrapper for domain annotation tools, allowing detailed visualization and analysis of domain architectures, as does DomArch [97]. The DAAC algorithm [98] explicitly transfers functional annotation to query sequences based on domain architectural similarity to annotated homologs, as does FACT [93]. In the same vein, similarity measures between architectures are available using the WDAC [99] tool and in ADASS [100]. Domain architecture similarity is used for orthology detection in the porthoDom software [68]. The DOGMA tool makes use of domain content data to assess completeness of a proteome or transcriptome [101].
14 Exercises/Questions
-
Which aspects of domain architecture evolution follow from properties of nature’s repertoire of mutational mechanisms, and which follow from selective constraints?
-
What trends have characterized the evolution of domain architectures in animals?
-
Discuss approaches to handle limited sampling of species with completely sequenced genomes. How can one draw general conclusions or test the robustness of the results? Apply, e.g., to the observed frequency of domain architectures that have emerged multiple times independently in a given dataset.
-
Describe the principle of “preferential attachment” for evolving networks. In what protein domain-related contexts does this seem to model the evolutionary process, and what distribution of node degrees does it produce?
-
What protein properties correlate with domain versatility? Can the versatility of a domain be different in different species (groups) and change over evolutionary time?
-
What protein domain-related properties differ between prokaryotes and eukaryotes?
References
Chandonia J-M, Fox NK, Brenner SE (2017) SCOPe: manual curation and artifact removal in the structural classification of proteins – extended database. Comput Res Mol Biol 429(3):348–355
Dawson NL, Lewis TE, Das S, Lees JG, Lee D, Ashford P et al (2017) CATH: an expanded resource to predict protein function through structure and sequence. Nucleic Acids Res 45(D1):D289–D295
Wilson D, Pethica R, Zhou Y, Talbot C, Vogel C, Madera M et al (2009) SUPERFAMILY—sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic Acids Res 37(suppl_1):D380–D386
Lam SD, Dawson NL, Das S, Sillitoe I, Ashford P, Lee D et al (2016) Gene3D: expanding the utility of domain assignments. Nucleic Acids Res 44(D1):D404–D409
Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE et al (2010) The Pfam protein families database. Nucleic Acids Res 38(suppl_1):D211–D222
Finn RD, Attwood TK, Babbitt PC, Bateman A, Bork P, Bridge AJ et al (2017) InterPro in 2017—beyond protein family and domain annotations. Nucleic Acids Res 45(D1):D190–D199
Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY et al (2015) CDD: NCBI’s conserved domain database. Nucleic Acids Res 43(D1):D222–D226
Letunic I, Doerks T, Bork P (2015) SMART: recent updates, new developments and status in 2015. Nucleic Acids Res 43(D1):D257–D260
Bru C, Courcelle E, Carrère S, Beausse Y, Dalmar S, Kahn D (2005) The ProDom database of protein domain families: more emphasis on 3D. Nucleic Acids Res 33(suppl_1):D212–D215
UniProt Consortium (2017) UniProt: the universal protein knowledgebase. Nucleic Acids Res 45(D1):D158–D169
Heger A, Wilton CA, Sivakumar A, Holm L (2005) ADDA: a domain database with global coverage of the protein universe. Nucleic Acids Res 33(suppl_1):D188–D191
Lewis TE, Sillitoe I, Andreeva A, Blundell TL, Buchan DWA, Chothia C et al (2015) Genome3D: exploiting structure to help users understand their sequences. Nucleic Acids Res 43(D1):D382–D386
Kummerfeld SK, Teichmann SA (2009) Protein domain organisation: adding order. BMC Bioinformatics 10(1):39
Weiner J, Moore AD, Bornberg-Bauer E (2008) Just how versatile are domains? BMC Evol Biol 8(1):285
Buljan M, Bateman A (2009) The evolution of protein domain families. Biochem Soc Trans 37(4):751
Orozco-Mosqueda M d C, Altamirano-Hernandez J, Farias-Rodriguez R, Valencia-Cantero E, Santoyo G (2009) Homologous recombination and dynamics of rhizobial genomes. Res Microbiol 160(10):733–741
Heyer W-D, Ehmsen KT, Liu J (2010) Regulation of homologous recombination in eukaryotes. Annu Rev Genet 44:113–139
Brissett NC, Doherty AJ (2009) Repairing DNA double-strand breaks by the prokaryotic non-homologous end-joining pathway. Biochem Soc Trans 37(3):539
van Rijk A, Bloemendal H (2003) Molecular mechanisms of exon shuffling: illegitimate recombination. Genetica 118(2):245–249
Feschotte C, Pritham EJ (2007) DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet 41:331–368
Cordaux R, Batzer MA (2009) The impact of retrotransposons on human genome evolution. Nat Rev Genet 10(10):691–703
Gogvadze E, Buzdin A (2009) Retroelements and their impact on genome evolution and functioning. Cell Mol Life Sci 66(23):3727
Patthy L (2003) Modular assembly of genes and the evolution of new functions. In: Long M (ed) Origin and evolution of new gene functions. Springer, Dordrecht, pp 217–231
Liu M, Grigoriev A (2004) Protein domains correlate strongly with exons in multiple eukaryotic genomes – evidence of exon shuffling? Trends Genet 20(9):399–403
Buljan M, Frankish A, Bateman A (2010) Quantifying the mechanisms of domain gain in animal proteins. Genome Biol 11(7):R74
Weiner J, Beaussart F, Bornberg-Bauer E (2006) Domain deletions and substitutions in the modular protein evolution. FEBS J 273(9):2037–2047
Schmidt EE, Davies CJ (2007) The origins of polypeptide domains. Bioessays 29(3):262–270
Bornberg-Bauer E, Huylmans A-K, Sikosek T (2010) How do new proteins arise? Nucl Acids Seq Topol 20(3):390–396
Demuth JP, Hahn MW (2009) The life and death of gene families. Bioessays 31(1):29–39
Toll-Riera M, Albà MM (2013) Emergence of novel domains in proteins. BMC Evol Biol 13(1):47
Moore AD, Bornberg-Bauer E (2012) The dynamics and evolutionary potential of domain loss and emergence. Mol Biol Evol 29(2):787–796
Kersting AR, Bornberg-Bauer E, Moore AD, Grath S (2012) Dynamics and adaptive benefits of protein domain emergence and arrangements during plant genome evolution. Genome Biol Evol 4(3):316–329
Huynen MA, van Nimwegen E (1998) The frequency distribution of gene family sizes in complete genomes. Mol Biol Evol 15(5):583–589
Qian J, Luscombe NM, Gerstein M (2001) Protein family and fold occurrence in genomes: power-law behaviour and evolutionary model11Edited by J. Thornton. J Mol Biol 313(4):673–681
Luscombe NM, Qian J, Zhang Z, Johnson T, Gerstein M (2002) The dominance of the population by a selected few: power-law behaviour applies to a wide variety of genomic properties. Genome Biol 3(8):research0040.1
Apic G, Gough J, Teichmann SA (2001) Domain combinations in archaeal, eubacterial and eukaryotic proteomes11Edited by G. von Heijne. J Mol Biol 310(2):311–325
Apic G, Huber W, Teichmann SA (2003) Multi-domain protein families and domain pairs: comparison with known structures and a random model of domain recombination. J Struct Funct Genomics 4(2–3):67–78
Vogel C, Berzuini C, Bashton M, Gough J, Teichmann SA (2004) Supra-domains: evolutionary units larger than single protein domains. J Mol Biol 336(3):809–823
Karev GP, Wolf YI, Rzhetsky AY, Berezovskaya FS, Koonin EV (2002) Birth and death of protein domains: a simple model of evolution explains power law behavior. BMC Evol Biol 2:18–18
Wuchty S (2001) Scale-free behavior in protein domain networks. Mol Biol Evol 18(9):1694–1702
Rzhetsky A, Gomez SM (2001) Birth of scale-free molecular networks and the number of distinct DNA and protein domains per genome. Bioinformatics (Oxford, England) 17(10):988–996
Li L, Alderson D, Doyle JC, Willinger W (2005) Towards a theory of scale-free graphs: definition, properties, and implications. Internet Math 2(4):431–523
Kuznetsov VA, Pickalov VV, Senko OV, Lnott GD (2002) Analysis of the evolving proteomes: predictions of the number of protein domains in nature and the number of genes in eukaryotic organisms. J Biol Syst 10(04):381–407
Koonin EV, Wolf YI, Karev GP (2002) The structure of the protein universe and genome evolution. Nature 420(6912):218–223
Yanai I, Camacho CJ, DeLisi C (2000) Predictions of gene family distributions in microbial genomes: evolution by gene duplication and modification. Phys Rev Lett 85(12):2641–2644
Eirin-Lopez JM, Rebordinos L, Rooney AP, Rozas J (2012) The birth-and-death evolution of multigene families revisited. Genome Dyn 7:170–196
van Nimwegen E (2003) Scaling laws in the functional content of genomes. Trends Genet 19(9):479–484
Ranea JAG, Sillero A, Thornton JM, Orengo CA (2006) Protein superfamily evolution and the last universal common ancestor (LUCA). J Mol Evol 63(4):513–525
Chothia C, Gough J (2009) Genomic and structural aspects of protein evolution. Biochem J 419(1):15
Ekman D, Björklund ÅK, Elofsson A (2007) Quantification of the elevated rate of domain rearrangements in metazoa. J Mol Biol 372(5):1337–1348
Itoh M, Nacher JC, Kuma K, Goto S, Kanehisa M (2007) Evolutionary history and functional implications of protein domains and their combinations in eukaryotes. Genome Biol 8(6):R121
Nasir A, Kim KM, Caetano-Anollés G (2014) Global patterns of protein domain gain and loss in superkingdoms. PLoS Comput Biol 10(1):e1003452
Przytycka T, Davis G, Song N, Durand D (2005) Graph theoretical insights into evolution of multidomain proteins. In: Miyano S, Mesirov J, Kasif S, Istrail S, Pevzner PA, Waterman M (eds) Res. Comput. Mol. Biol. 9th Annu. Int. Conf. RECOMB 2005 Camb. MA USA May 14-18 2005 Proc. Springer, Berlin, pp 311–325
Marcotte EM, Pellegrini M, Ng H-L, Rice DW, Yeates TO, Eisenberg D (1999) Detecting protein function and protein-protein interactions from genome sequences. Science 285(5428):751
Basu MK, Carmel L, Rogozin IB, Koonin EV (2008) Evolution of protein domain promiscuity in eukaryotes. Genome Res 18(3):449–461
Basu MK, Poliakov E, Rogozin IB (2009) Domain mobility in proteins: functional and evolutionary implications. Brief Bioinform 10(3):205–216
Barabási A-L, Albert R (1999) Emergence of scaling in random networks. Science 286(5439):509
Bashton M, Chothia C (2002) The geometry of domain combination in proteins11Edited by J. Thornton. J Mol Biol 315(4):927–939
Gough J (2005) Convergent evolution of domain architectures (is rare). Bioinformatics 21(8):1464–1471
Forslund K, Henricson A, Hollich V, Sonnhammer ELL (2008) Domain tree-based analysis of protein architecture evolution. Mol Biol Evol 25(2):254–264
Parikesit AA, Stadler PF, Prohaska SJ (2017) Large-scale evolutionary patterns of protein domain distributions in eukaryotes. BioRxiv
Hsu C-H, Chiang AWT, Hwang M-J, Liao B-Y (2016) Proteins with highly evolvable domain architectures are nonessential but highly retained. Mol Biol Evol 33(5):1219–1230
Brivanlou AH, Darnell JE (2002) Signal transduction and the control of gene expression. Science 295(5556):813
Weiner J III, Bornberg-Bauer E (2006) Evolution of circular permutations in multidomain proteins. Mol Biol Evol 23(4):734–743
Tordai H, Nagy A, Farkas K, Bányai L, Patthy L (2005) Modules, multidomain proteins and organismic complexity. FEBS J 272(19):5064–5078
Vogel C, Teichmann SA, Pereira-Leal J (2005) The relationship between domain duplication and recombination. J Mol Biol 346(1):355–365
Xie X, Jin J, Mao Y (2011) Evolutionary versatility of eukaryotic protein domains revealed by their bigram networks. BMC Evol Biol 11(1):242
Bitard-Feildel T, Kemena C, Greenwood JM, Bornberg-Bauer E (2015) Domain similarity based orthology detection. BMC Bioinformatics 16(1):154
Cohen-Gihon I, Fong JH, Sharan R, Nussinov R, Przytycka TM, Panchenko AR (2011) Evolution of domain promiscuity in eukaryotic genomes-a perspective from the inferred ancestral domain architectures. Mol Biosyst 7(3):784–792
Hsu C-H, Chen C-K, Hwang M-J (2013) The architectural design of networks of protein domain architectures. Biol Lett 9(4):20130268
Björklund ÅK, Ekman D, Light S, Frey-Skött J, Elofsson A (2005) Domain rearrangements in protein evolution. J Mol Biol 353(4):911–923
Björklund ÅK, Ekman D, Elofsson A (2006) Expansion of protein domain repeats. PLoS Comput Biol 2(8):e114
Nagy A, Szlama G, Szarka E, Trexler M, Banyai L, Patthy L (2011) Reassessing domain architecture evolution of metazoan proteins: major impact of gene prediction errors. Genes. 2(3):449–501
Doolittle RF, Bork P (1993) Evolutionarily mobile modules in proteins. Sci Am 269(4):50–56
Moore AD, Björklund ÅK, Ekman D, Bornberg-Bauer E, Elofsson A (2008) Arrangements in the modular evolution of proteins. Trends Biochem Sci 33(9):444–451
Grassi L, Fusco D, Sellerio A, Cora D, Bassetti B, Caselle M et al (2010) Identity and divergence of protein domain architectures after the yeast whole-genome duplication event. Mol Biosyst 6(11):2305–2315
Zhang X-C, Wang Z, Zhang X, Le MH, Sun J, Xu D et al (2012) Evolutionary dynamics of protein domain architecture in plants. BMC Evol Biol 12(1):6
Sharma M, Pandey GK (2016) Expansion and function of repeat domain proteins during stress and development in plants. Front Plant Sci 6:1218
Farris JS (1977) Phylogenetic analysis under Dollo’s law. Syst Zool 26(1):77–88
Snel B, Bork P, Huynen M (2000) Genome evolution. Trends Genet 16(1):9–11
Kummerfeld SK, Teichmann SA (2005) Relative rates of gene fusion and fission in multi-domain proteins. Trends Genet 21(1):25–30
Fong JH, Geer LY, Panchenko AR, Bryant SH (2007) Modeling the evolution of protein domain architectures using maximum parsimony. J Mol Biol 366(1):307–315
Wiedenhoeft J, Krause R, Eulenstein O (2010) Inferring evolutionary scenarios for protein domain compositions. In: Borodovsky M, Gogarten JP, Przytycka TM, Rajasekaran S (eds) Bioinforma. Res. Appl. 6th Int. Symp. ISBRA 2010 Storrs CT USA May 23-26 2010 Proc. Springer, Berlin, pp 179–190
Yang S, Bourne PE (2009) The evolutionary history of protein domains viewed by species phylogeny. PLoS One 4(12):e8378
Wu Y-C, Rasmussen MD, Kellis M (2012) Evolution at the subgene level: domain rearrangements in the drosophila phylogeny. Mol Biol Evol 29(2):689–705
Stolzer M, Siewert K, Lai H, Xu M, Durand D (2015) Event inference in multidomain families with phylogenetic reconciliation. BMC Bioinformatics 16(14):S8
Zmasek CM, Godzik A (2012) This Déjà Vu Feeling—analysis of multidomain protein evolution in eukaryotic genomes. PLoS Comput Biol 8(11):e1002701
Leclère L, Rentzsch F (2012) Repeated evolution of identical domain architecture in metazoan netrin domain-containing proteins. Genome Biol Evol 4(9):883–899
Nagy A, Bányai L, Patthy L (2011) Reassessing domain architecture evolution of metazoan proteins: major impact of errors caused by confusing paralogs and epaktologs. Genes. 2(3):516–561
Nagy A, Patthy L (2011) Reassessing domain architecture evolution of metazoan proteins: the contribution of different evolutionary mechanisms. Genes 2(3):578–598
Geer LY, Domrachev M, Lipman DJ, Bryant SH (2002) CDART: protein homology by domain architecture. Genome Res 12(10):1619–1623
Moore AD, Held A, Terrapon N, Weiner J III, Bornberg-Bauer E (2014) DoMosaics: software for domain arrangement visualization and domain-centric analysis of proteins. Bioinformatics 30(2):282–283
Koestler T, von Haeseler A, Ebersberger I (2010) FACT: functional annotation transfer between proteins with similar feature architectures. BMC Bioinformatics 11(1):417
Hollich V, Sonnhammer ELL (2007) PfamAlyzer: domain-centric homology search. Bioinformatics 23(24):3382–3383
Terrapon N, Weiner J, Grath S, Moore AD, Bornberg-Bauer E (2014) Rapid similarity search of proteins using alignments of domain arrangements. Bioinformatics 30(2):274–281
Haider C, Kavic M, Sonnhammer ELL (2016) TreeDom: a graphical web tool for analysing domain architecture evolution. Bioinformatics 32(15):2384–2385
Vera-Parra N, Gutiérrez-Ramirez M, Lopez-Sarmiento D (2016) Automatic construction and graph-making of functional domain architectures. Adv Nat Appl Sci 10(12):99–106
Doğan T, MacDougall A, Saidi R, Poggioli D, Bateman A, O’Donovan C et al (2016) UniProt-DAAC: domain architecture alignment and classification, a new method for automatic functional annotation in UniProtKB. Bioinformatics 32(15):2264–2271
Lee B, Lee D (2009) Protein comparison at the domain architecture level. BMC Bioinformatics 10(15):S5
Syamaladevi DP, Joshi A, Sowdhamini R (2013) An alignment-free domain architecture similarity search (ADASS) algorithm for inferring homology between multi-domain proteins. Bioinformation 9(10):491–499
Dohmen E, Kremer LPM, Bornberg-Bauer E, Kemena C (2016) DOGMA: domain-based transcriptome and proteome quality assessment. Bioinformatics 32(17):2577–2581
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2019 The Author(s)
About this protocol

Cite this protocol
Forslund, S.K., Kaduk, M., Sonnhammer, E.L.L. (2019). Evolution of Protein Domain Architectures. In: Anisimova, M. (eds) Evolutionary Genomics. Methods in Molecular Biology, vol 1910. Humana, New York, NY. https://doi.org/10.1007/978-1-4939-9074-0_15
Download citation
DOI: https://doi.org/10.1007/978-1-4939-9074-0_15
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-4939-9073-3
Online ISBN: 978-1-4939-9074-0
eBook Packages: Springer Protocols