
Abstract
Gene-category analysis is one important knowledge integration approach in biomedical sciences that combines knowledge bases such as Gene Ontology with lists of genes or their products, which are often the result of high-throughput experiments, gained from either wet-lab or synthetic experiments. In this chapter, we will motivate this class of analyses and describe an often used variant that is based on Fisher’s exact test. We show that this approach has some problems in the context of Gene Ontology of which users should be aware. We then describe some more recent algorithms that try to address some of the shortcomings of the standard approach.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
The result of biological high-throughput methods is often a list consisting of several hundreds of biological entities, which are in case of gene expression profiling experiments identifiers of genes or their products. As a biological entity may have different context-specific functions, it is difficult for humans to interpret the outcome of an experiment on the basis of such a list. Computational approaches to access the biological knowledge about features of biological entities therefore play an important part in the successful realization of research based on high-throughput experiments. A practical way to address the question of what is going on? is to perform a gene-category analysis, i.e., to ask whether these responder genes share some biological features that distinguish them among the set of all genes tested in the experiment.
First of all, gene-category analysis involves a list of gene categories, in which genes with similar features are grouped together. The exact definition of the attribute similar depends on the provider of the categories. For instance, if Gene Ontology is the choice, then genes usually are grouped according to the terms, to which they are annotated. Another scheme is the KEGG database [1], in which genes are grouped according to the pathways in which they are involved. The second ingredient is a statistical method for identifying the really interesting categories.
In this chapter, we introduce some commonly used approaches for gene-category analysis. Throughout the remainder of this chapter, we refer to the set of items, which a study could possibly select, as the population set. We denote this set by the uppercase letter M while the size of the set, or its cardinality, is identified by its lowercase variant m. If, for example, a microarray experiment is conducted, the population set will comprise all genes whose expression can be measured with the microarray chip. The actual outcome of the study is referred to as the study set. It is denoted by N and has the cardinality n. In the microarray scenario the study set could consist of all genes that were detected to be differentially expressed.
2 Fisher’s Exact Test
One approach for gene-category analysis is to cast the problem as a statistical test. For this purpose, the study set is assumed to be a random sample that is obtained by drawing n items without replacement from the population. The population is dichotomic as the items can be characterized according to whether they are annotated to term t or not. In particular, the set M t with cardinality m t constitutes all items that are annotated to t. Denote the random variable that describes the number of items of the study set that are annotated to t in this random sample as X t . The hypergeometric distribution applies to X t , and the probably of observing exactly k items annotated to t, i.e., P(X t = k) is specified by

Furthermore, the set of items that are annotated to t and members of the study set are denoted by N t with cardinality n t . The objective is to assess whether the study set is enriched for term t, i.e., whether the observed n t is higher than one would expect. This forms the alternative hypothesis H 1 of the statistical test. The null hypothesis H 0 in this case is that there is no positive association between the observed occurrence of the items in the study set and the annotations of the items to the term t. Thus, the proportion of items annotated to term t is approximatively identical for the study set and the population set. In order to be able to reject H 0 in support of H 1 we conduct a one-tailed test, in which we ask for the probability of the event that we see n t or more annotated items given that H 0 is true:
If the probability obtained by this equationFootnote 1 is below a certain significance level α, e.g., α < 0. 05, we reject H 0 in favor of H 1. In that case, the tested term t is regarded as an interesting term that contributes to the characterization of the study set.
Example 2.1.
Suppose that we are given a population of m = 18 genes, of which m t = 4 genes are annotated to a term t. The outcome of an experiment yields a study set of 5 differentially expressed genes. A total of n t = 3 genes from the genes of the study set are annotated to t. Figure 1 illustrates the participating sets and how they are related to one another in that particular situation.

Sets and their relations in the standard approach. In this example the population consists of m = 18 genes and n = 5 of them are part of the study set. Exactly m t = 4 genes of the population are annotated to term t. This term has n t = 3 genes in common with the study set. The null hypothesis of the standard approach (term-for-term) is that there is no association between the number of genes that are in the study set and the number of genes that are annotated to the term t, i.e., the study set is a random sample of the population set. We therefore would expect that it contains the same proportion of annotated terms as the population set does. The probability under the null hypothesis of the event to see at least n t genes can be assessed via Eq. 1.
In order to check whether term t can be used to characterize the experiment, we ask whether term t is overrepresented in the study set. The application of Eq. 1 yields a p-value for t
Thus, the null hypothesis is rejected and the term is said to be overrepresented among the differentially expressed genes and is thus likely to reflect an association between the term and the experiment.
3 Multiple Testing Problem
In hypothesis-generating studies it is a priori not clear, which terms should be tested. Therefore, the procedure is not only conducted using a single term but also applied to many, often all terms that Gene Ontology provides and to which at least one gene is annotated. The result of the entire analysis is then a list of terms that were found to be significant. This, however, implies that the number of false-positive terms is high.
To see this, suppose that there are T tests to be performed. We assume that the null hypothesis is true for all of those tests. Before its actual determination, any p-value can be considered as a random variable as well, for which P(p ≤ α | H 0) ≤ α holds [2]. This implies that it can be expected that α × T tests lead to the rejection of a null hypothesis although it is true.
Example 3.1.
If there are 10,000 null hypotheses that are true and all of them are tested, then we expect that we reject the null hypotheses for about 500 tests. Obviously, describing the result of experiment with 500 random terms is not useful.
Therefore, the result of a term enrichment analysis shall be further subjected to a multiple test correction. The most simple is the Bonferroni correction [3]. Here, each p-value is simply multiplied by the number of tests saturated at a value of 1.0. Bonferroni controls the so-called family-wise error rate, which is the probability of making one or more false discoveries. It is a very conservative approach because it handles all p-values as independent. But as we see later, this is not a typical case of gene-category analysis, so this approach often goes along with a reduced statistical power. In contrast, the Westfall–Young [4] procedure also takes dependencies into account. This correction, however, is computationally more costly as it is based on resampling schemes. In particular in the gene category setting, this scheme involves randomly sampling study sets of the same size as the original study set from the population. Each set is subjected to the test procedure yielding a set of p-values for each term, also referred to as the null distribution of that term. By relating the original p-value to the null distribution, an adjusted p-value is derived. There are other types of multiple test corrections that do not aim to control the family-wise error rate. For instance, the Benjamini–Hochberg [5] approach controls the expected false discovery rate (FDR), which is the proportion of false discoveries among all rejected null hypotheses. This has a positive effect on the statistical power at the expense of having less strict control over false discoveries. Controlling the FDR is considered by the American Physiological Society as “the best practical solution to the problem of multiple comparisons” [6].
Note that less conservative corrections usually yield a higher amount of significant terms, which may be not desirable after all. In the following section, we further explore the structural origin of the correlations of the p-values in the setting of enrichment tests for ontology terms.
4 Gene Propagation
While the application of multiple testing correction aims to reduce the number of false-positives in a rather universal manner, one can also try to tackle the problem at a more basic level. The root of the problem is that if a term shares genes with a second term, and one of the terms is overrepresented, then it is not too surprising that the other term is also detected as overrepresented. That the gene sharing of terms of an ontology is more a rule than an exception can be deduced from the principles of how ontologies are designed. Within an ontology, terms describe concepts of a domain that can be related to other terms by various types of relationships. The most prominent relationship thereby is the is a relationship, which effectively propagates the membership of the subject (source) of the relationship to the object (destination). That means, if a term T 1 is related to a term T 2 by the is a relationship, and a gene is annotated to T 1, then it is implicitly annotated also to term T 2 (see Chap. 1 [7]). In the context of GO overrepresentation analysis, we refer to this as the gene propagation problem.Footnote 2
Example 4.1 (Continuation of Example 2.1).
There is another term s, which is the only parent of t. For s we know that m s = 6 and n s = 4. Figure 2 shows this structure graphically. There, it is also indicated that the p-values of terms t and s are 0.044 and 0.022, respectively, which means that both terms are considered as significant for α < 0.05 if no multiple test correction is performed. Obviously, both terms share the majority of items that are also part of the study set. One can argue that the fact that term t is identified as overrepresented is a consequence of the fact that s is overrepresented.

Extended example with three terms. This depicts the situation of Example 2.1 with two more terms. Term t is a s and therefore s is a parent of t. Term r is the root of the ontology. It is the only parent of s. As indicated in the last row, the procedure based on Fisher’s exact test determines a p-value below 0.05 for both terms. Thus, both terms will be considered as a meaningful summary of the underlying experiment.
A simple synthetic experiment, in which a term will be artificially overrepresented, demonstrates the extent of the problem. Let’s select the term localization for this purpose. We create a study set that consists of all genes that are annotated to that term with probability 0.8. This corresponds to false-negative rate β = 0. 2. Furthermore, to introduce some background noise, each gene that is not annotated to the term is added to that study set with a false-positive rate of α = 0. 1. In this example, the procedure yields a set of 1542 genes. For each considered term, this set is subjected to Fisher’s exact test resulting in a list of 4549 p-valuesFootnote 3. Finally, the p-values are adjusted using the Bonferroni correction.
The analysis correctly identifies the term localization as significantly enriched. In addition to that, it identifies 275 other terms as significantly enriched. In particular, 6 of the 6 children, to which at least one gene is annotated, are significant. Among the 681 possible descendants of localization, we find 172 significant ones. These figures suggest that descendants come up only because their annotations converge in the term localization. Although, in the statistical sense, this is a correct result, it is not desirable to use that huge amount of terms to characterize the study set, especially as it is sufficient to use the term localization for this purpose, and what is more, the result suggests a specificity that we did not put in there. It makes sense to consider each of the additional 275 significant terms as a false-positive and in the next sections we will briefly describe methods that attempt to reduce that number.
5 Parent–Child Approach
The parent–child approach [8] is still based on Fisher’s exact test, but the probability of t being overrepresented is conditioned on properties of the parental terms. In the following, let pa(t) be the set of parents of term t, which are, for instance, those terms, to which t is connected by a is a relation. In order to introduce the principal ideas of the parent–child approaches, we initially assume that there is only a single parent of t, i.e., pa(t) = {s}.
Instead of drawing the items from the population M, items will be drawn just from the set of items that are annotated to the parent of t, which is written as M pa(t) and whose size is m pa(t). This consideration yields the following equation:
The right part of Fig. 3 shows the setting of the parent–child approaches. Effectively, in the parent–child approaches, we change the population that underlies Fisher’s exact test to the items annotated to the parents. Obviously, this also alters the involved sets for the study set. As previously, we ask for the probability of seeing the observed number of items or a more extreme event:

Sets and their relations in the parent–child approaches. Part (a) depicts the model of the term-for-term approach as it was shown in Fig. 1. This is contrasted in part (b) with the model of the parent–child approaches. In this approach, we shift the focus to a smaller set of genes, for instance to the genes that are annotated to at least one of the parents of term t. In this particular situation it is the set whose size is m pa(t) = 6 with pa(t) = {s} following Example 4.1. Genes that are not part of this set do not contribute to the calculation. This has an effect on the involved proportions, and thus on the outcome of the test. Effectively, for each term, we alter the population of the association test. Eq. 2 quantifies the probability.
Example 5.1 (Continuation of Example 4.1).
As shown in Fig. 2, the parent of term s is the root r of the ontology, which is always annotated to all genes of the population. Therefore, the p-value for s is the same for previous approach and for parent–child approach, i.e., \( {p}_{s}^{\mathrm{pc}}={p}_{s}=0.22 \) However, for term t, Eq. 3 yields
Thus, the null hypothesis for term t is not rejected, which is in contrast to the result of the previous approach. Given the initial observations that the study set is already skewed to the parent s of t makes the enrichment of term t less surprising, which the parent–child approaches reflect by returning a higher p-value.
If term t has more than one parent term, then it is not immediately apparent how to calculate m pa(t) and the observation n pa(t) in Eqs. 2 and 3. In Grossmann et al. [8] we examined two variants in detail, the union and the intersection of genes that are annotated to each of the parents.
6 Topology-Based Algorithms
Alexa et al. devised another method to address the gene propagation problem. The authors propose calculating a score for the term that depends on the relevance of the children of the term [9]. They argue that capturing the meaning in that way is biologically more interesting as the definitions of children are more specific. Following this argumentation, the authors formulated two concrete algorithms that try to provide a more suitable, i.e., less correlated, distribution of terms that get flagged as important. While the first approach which they called the elim-algorithm strictly favors significance of the most specific levels of the GO graph, their second algorithm called weight relaxes this restriction such that terms that are most significant are favored.
As before, we understand the top of the graph as the root of the ontology, while the bottom of the graph consists of the most specific terms. The idea of the elim algorithm is to traverse the graph representation of the ontology in bottom-up fashion, which, for instance, can be accomplished by utilizing the backtrack phase of a depth-first search (DFS) [10].
The elim procedure awaits a term t as a variable parameter and returns a set of flagged genes. On its initial invocation, it begins with the root of the ontology. For the current term t, we apply Fisher’s exact test in order to relate the genes of the study set to the genes of the population with respect to the genes that are annotated to term t. As in the parent–child approaches, not all genes of the study set contribute to the calculation. For elim, a set of previously determined genes is subtracted from the set of the study set before the calculation for p t is carried out. This set is constructed by recursively applying the elim procedure for all children of t and taking the union of the result. If p t is significant, we add all genes of t to the set of flagged genes. Finally, we return the set of flagged genes to the caller. Note that when the DFS reaches a leaf node of the ontology, Fisher’s exact test is performed exactly as in the standard approach.
Obviously, the complexity of the algorithm is the same as the complexity of a depth-search algorithm if we assume that the number of genes that are annotated to a term is constant. Note in the original publication of the elim, the algorithm was based on an iteration over the levels of the GO DAG, which partitions the nodes according to their longest distance to the root. The algorithm as outlined here yields an equivalent result without the need to explicitly keep track of the DAG levels.
Example 6.1 (Continuation of Example 5.1).
The p-value of term t matches the p-value of term t of the standard approach, i.e., \( {p}_{t}^{\mathrm{elim}}={p}_{t}^{\mathrm{tft}}=0.044 \) . As this is a significant result, at least, if correction for multiple testing is omitted, all four genes that are annotated to t are removed in the consideration of upper terms, i.e., we assume that those four genes are not annotated to them. This leaves two genes for the computation of term s, of which only one is member of the study set (Fig. 3 b). With m s = 2, n s = 1, and the rest as before, Eq. 1 yields
Hence, theelim method doesn’t report term s as important.
An equivalent characterization of the elim method is the following: If a term t is identified as significant, all genes that are annotated to t are no longer considered in the computation of the relevance of the ancestors of t. As it was discussed in Example 2.1 at page 2.1 and as can also be seen in Fig. 2, the term-for-term approach assigns term s a lower p-value than it does for term t. One may conclude that it is more appropriate to take term s than to take term t in order to provide a compact description of the study set. However, in Example 13.6.1 we saw that the application of the elim method results in usage of term t to describe the outcome, which is contrary to that conclusion.
This concern is addressed by weight method. It compares significance scores of a family terms (a parent and its child) to identify the locally most significant terms and down-weight genes in less significant neighbors. This effectively decorrelates the p-values of the related terms such that their differences are enforced while the existence of the most significant terms is still maintained.
7 Model-Based Approaches
The previously described procedures that address gene propagation problem have in common that they successively test overrepresentation for each of the terms. They all use some form of the Fisher’s exact test. In contrast to this, model-based gene set analysis (MGSA) models the gene response in a genome-wide experiment as the result of an activation of a number of terms [11].Footnote 4
The approach is based on a model that can nicely be expressed using a Bayesian network with three layers of Boolean random variables. The term layer consists of m Boolean nodes corresponding to m terms of the ontology. A term can be active or inactive. A parameter p, usually much less than 0.5, represents the prior probability of a term being active. The hidden layer contains n Boolean nodes representing the n hidden state of the genes. The hidden state of a gene is a consequence of the states the terms to which the gene is annotated: The gene is on if and only if at least one term to which the gene is annotated is active, otherwise it is off. The third layer, the observed layer, contains Boolean nodes reflecting the experimentally observed state of all genes. For instance, in the setting of a microarray experiment, the on state would correspond to differential expression, and the off state would correspond to a lack of differential expression of a gene. The observed gene state depends on the corresponding hidden gene state in a one-to-one fashion with a false-positive (α) and false-negative rates (β) that is identical and independent for all genes. A simple instance of the model is depicted in Fig. 4.

The graphical representation of an MGSA network. An example structure for four terms and three genes with a possible realizations is displayed. Terms (T i ) that constitute the first layer can be either active (light) or inactive (dark). Terms that are active enable the hidden state (H j ) of all genes annotated to them, the other genes remaining off. The observed states (O j ) of the genes are noisy observations of their true hidden state. In this example, the observed states for gene 1 and 3 match the hidden state while for some unknown reasons the measurement of gene 2 doesn’t correspond to the hidden state. It’s a false-negative.
The model describes how the activity of terms leads to the observed stats of genes. This, however, is not the direction we are interested in. We are interested in the set of terms that explain the experimentally obtained data best, and the mathematical tool that can be applied to and such sets is probabilistic inference. The optimization problem that finds the term state configuration that explains the observed gene pattern best is NP-hard [12]. However, it is easily possible to find nearby solutions by sampling from the state space. This procedure additionally allows to determine the so-called marginal probability for each term, which is a measure how good the particular term will explain the observed genes with respect to all the other terms. The value ranges between 0 and 1 with 0 being the lowest possible support and 1 being the best possible support for a term. As all terms compete with one another, the inference takes dependencies both due to gene propagation and due to similarity of annotations into account. For example, if two unrelated terms are annotated to the same set of genes that matches the observation, the marginal probability for both terms will be 0.5. Consequently, it is advisable to run MGSA for each of the subontologies separately as they are designed to express orthogonal features.
8 Gene Set Enrichment Analysis
In addition to approaches that take a fixed subset of the population as input, procedures that take the measurements of the genes into account are also widely in use. This is attractive as it frees the investigator from the need to define a sometimes arbitrary cutoff that is used to construct the study set.
A first version of the so-called Gene Set Enrichment Analysis (GSEA) that received much attention of the scientific community was published by Mootha et al. [13]. In this approach, genes are ranked according to an interesting feature (e.g., the difference of the mean of their expression values for two experimental conditions). The null hypothesis is that the genes of the interesting set (e.g., genes annotated to a term) have no association with that list, in which case they would be randomly ordered. The alternative hypothesis is that the genes of the interesting set have an association. For instance, if the genes of the set are grouped together on the top of the list, we would tend to believe that there is such an association.
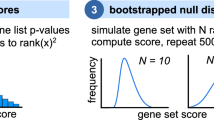
To capture the association via statistical means, the authors proposed a normalized Kolmogorov–Smirnov (KS) test statistic. Let r i ∈ M be the gene of the population M that has rank i in the gene list that is sorted according to the interesting gene feature. Using the previously established notation, i.e., that m is the total number of genes and N t is the set of cardinality n t that contains only genes that are annotated to t, the score is defined as:
Thus, the score is the maximum of a running sum that is increased if the gene is annotated to t and decreased if the gene is not annotated to t. In order to check if the obtained score is significant, the calculation is repeated for k randomly chosen sets N t 1, …, N t k, which all are subsets of M with size n t . The p-value for a term t is calculated as
The GSEA method went a slight revision Subramanian et al. [14], where ad-hoc modifications are implemented that are supposed to countervail the well-known lack of sensitivity of the KS test [15, 16].
9 Software
Gene-category analysis is a very prominent use case of Gene Ontology. It shouldn’t come as a surprise that users can choose among a variety of software implementations that will perform this sort of analysis. For instance, current version of the web site of Gene Ontology Consortium (geneontology.org) provides access to the method of the basic Fisher’s exact test directly on the front page. There are also graphical tools that integrate into existing frameworks such as BiNGO [17], standalone graphical clients such as Ontologizer Footnote 5 [18] or packages for Bioconductor such as topGo [19], mgsa [20], or gCMAP [21], just to name a few of them.
10 Exercises
-
1.
Repeat the random experiment outlined in the text that was used to show the influence of the gene propagation. When doing this in R/Bioconductor, it is advisable to use the GO.db and org.Sc.sgd.db packages that provide the structure and the annotations. The calculation involving the hypergeometric distribution can be expressed directly in R using dhyper and phyper. Now repeat this experiment with other approaches based on study sets that were outlined in this chapter and compare the results. For the topology-based algorithms the topGo package can be used and for the model-based approach the mgsa package is well suited.
-
2.
Apply the approach now to an arbitrary example or on real world data. Compare the results.
Funding Open Access charges were funded by the University College London Library, the Swiss Institute of Bioinformatics, the Agassiz Foundation, and the Foundation for the University of Lausanne.
Notes
- 1.
The superscript tft in p t tft stands for term-for-term. It allows to distinguish this p-value with other measures that are described later.
- 2.
Note that in addition to this gene sharing that is due to the graph structure of the ontology, also unrelated terms can be annotated to similar sets of genes, for instance, if the same gene plays a role in distinct biological processes.
- 3.
This corresponds to the number of terms from the biological process subontology that are annotated by at least one gene.
- 4.
We use the word term here because we primarily work with GO, but the method can be applied to any other structured or unstructured vocabulary.
- 5.
http://ontologizer.de
References
Kanehisa M, Goto S (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28(1):27–30
Ewens WJ, Grant GR (2005) Statistical methods in bioinformatics: an introduction, 2nd edn. Springer, Berlin. ISBN 978-0387400822
Abdi H (2007) Bonferroni and Sidak corrections for multiple comparisons. Sage, Thousand Oaks, CA
Westfall PH, Young SS (1993) Resampling-based multiple testing: examples and methods for P-value adjustment. Wiley, London. ISBN 978-0471557616
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B 57:289–300
Curran-Everett D, Benos DJ (2004) Guidelines for reporting statistics in journals published by the American Physiological Society. Adv Physiol Educ 28:85–87
Hastings J (2016) Primer on ontologies. In: Dessimoz C, Škunca N (eds) The gene ontology handbook. Methods in molecular biology, vol 1446. Humana Press. Chapter 1
Grossmann S, Bauer S, Robinson PN, Vingron M (2007) Improved detection of overrepresentation of Gene-Ontology annotations with parent child analysis. Bioinformatics 23:3024–3031
Alexa A, Rahnenführer J, Lengauer T (2006) Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22(13):1600–1607. doi:10.1093/bioinformatics/btl140
Cormen TH, Leiserson CE, Rivest RL, Stein C (2001) Introduction to algorithms, 2nd edn. MIT Press, Cambridge, MA. ISBN 978-0262531962
Bauer S, Gagneur J, Robinson PN (2010) GOing Bayesian: model-based gene set analysis of genome-scale data. Nucleic Acids Res 38(11):3523–3532
Bauer S (2012) Algorithms for knowledge integration in biomedical sciences. PhD thesis
Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC (2003) PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34(3):267–273. doi:10.1038/ng1180
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102(43):15545–15550. doi:10.1073/pnas.0506580102
Mason DM, Schuenemeyer JH (1983) A modified Kolmogorov-Smirnov test sensitive to tail alternatives. Ann Stat 11(3):933–946
Irizarry RA, Wang C, Zhou Y, Speed TP (2009) Gene set enrichment analysis made simple. Stat Methods Med Res 18(6):565–575. ISSN 1477-0334
Maere S, Heymans K, Kuiper M (2005) Bingo: a cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21:3448–3449
Bauer S, Grossmann S, Vingron M, Robinson PN (2008) Ontologizer 2.0–a multifunctional tool for go term enrichment analysis and data exploration. Bioinformatics 24(14):1650–1651. doi:10.1093/bioinformatics/btn250
Alexa A, Rahnenführer J (2010) topGO: enrichment analysis for Gene Ontology. R package version 2.22.0
Bauer S, Robinson NP, Gagneur J (2011) Model-based Gene Set Analysis for Bioconductor. Bioinformatics 27
Sandmann T, Kummerfeld SK, Gentleman R, Bourgon R (2014) gcmap: user-friendly connectivity mapping with r. Bioinformatics 30(1):127–128
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
This chapter is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, a link is provided to the Creative Commons license and any changes made are indicated.
The images or other third party material in this chapter are included in the work’s Creative Commons license, unless indicated otherwise in the credit line; if such material is not included in the work’s Creative Commons license and the respective action is not permitted by statutory regulation, users will need to obtain permission from the license holder to duplicate, adapt or reproduce the material.
Copyright information
© 2017 The Author(s)
About this protocol
Cite this protocol
Bauer, S. (2017). Gene-Category Analysis. In: Dessimoz, C., Škunca, N. (eds) The Gene Ontology Handbook. Methods in Molecular Biology, vol 1446. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3743-1_13
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3743-1_13
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3741-7
Online ISBN: 978-1-4939-3743-1
eBook Packages: Springer Protocols