
Abstract
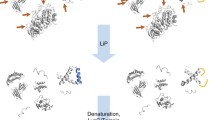
Limited proteolysis coupled to mass spectrometry (LiP-MS) is a recent proteomics technique that allows structure-based target engagement profiling on a proteome-wide level. To achieve this, native lysates are first incubated with a compound, followed by a short incubation with a nonspecific protease. Binding of a compound can change accessibility at the binding site or induce other structural changes in the target. This leads to treatment-specific proteolytic fingerprints upon limited proteolysis, which can be analyzed by standard bottom-up MS-based proteomics. Here, we describe a basic LiP-MS protocol using the natural product rapamycin as an example compound. Along with the provided LiP-MS reference data available via ProteomeXchange with identifier PXD035183, this enables the straightforward implementation of the method by scientists with a basic biochemistry and mass spectrometry background. We describe how the procedure can easily be adapted to other protein samples and small molecules.
Access this chapter
Tax calculation will be finalised at checkout
Purchases are for personal use only
Similar content being viewed by others


References
Molina DM, Jafari R, Ignatushchenko M et al (2013) Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 341(6141):84–87. https://doi.org/10.1126/science.1233606
Piazza I, Beaton N, Bruderer R et al (2020) A machine learning-based chemoproteomic approach to identify drug targets and binding sites in complex proteomes. Nat Commun 11(1):4200. https://doi.org/10.1038/s41467-020-18071-x
Meissner F, Geddes-McAlister J, Mann M, Bantscheff M (2022) The emerging role of mass spectrometry-based proteomics in drug discovery. Nat Rev Drug Discov 21(9):637–654. https://doi.org/10.1038/s41573-022-00409-3
Feng Y, De Franceschi G, Kahraman A et al (2014) Global analysis of protein structural changes in complex proteomes. Nat Biotechnol 32(10):1036–1044. https://doi.org/10.1038/nbt.2999
Hendricks JA, Beaton N, Chernobrovkin A et al (2022) Mechanistic insights into a CDK9 inhibitor via orthogonal proteomics methods. ACS Chem Biol 17(1):54–67. https://doi.org/10.1021/acschembio.1c00488
Gillet LC, Navarro P, Tate S et al (2012) Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics 11(6):O111.016717. https://doi.org/10.1074/mcp.O111.016717
Bruderer R, Bernhardt OM, Gandhi T et al (2015) Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues. Mol Cell Proteomics 14(5):1400–1410. https://doi.org/10.1074/mcp.M114.044305
Quast J-P, Schuster D, Picotti P (2022) Protti: an R package for comprehensive data analysis of peptide- and protein-centric bottom-up proteomics data. Bioinforma Adv 2(1):vbab041. https://doi.org/10.1093/BIOADV/VBAB041
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B 57(1):289–300. https://doi.org/10.1111/j.2517-6161.1995.tb02031.x
Perez-Riverol Y, Bai J, Bandla C et al (2022) The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res 50(D1):D543–D552. https://doi.org/10.1093/nar/gkab1038
Acknowledgements
We would like to acknowledge valuable inputs from Ludovic Gillet and the rest of the Picotti lab for optimising this protocol. This work was supported by the Innovative Medicines Initiative 2 Joint Undertaking (JU) under grant agreement No 875510. The JU receives support from the European Union’s Horizon 2020 research and innovation program and EFPIA and Ontario Institute for Cancer Research, Royal Institution for the Advancement of Learning McGill University, Kungliga Tekniska Hoegskolan, Diamond Light Source Limited.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Science+Business Media, LLC, part of Springer Nature
About this protocol

Cite this protocol
Reber, V., Gstaiger, M. (2023). Target Deconvolution by Limited Proteolysis Coupled to Mass Spectrometry. In: Merk, D., Chaikuad, A. (eds) Chemogenomics. Methods in Molecular Biology, vol 2706. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-3397-7_13
Download citation
DOI: https://doi.org/10.1007/978-1-0716-3397-7_13
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-3396-0
Online ISBN: 978-1-0716-3397-7
eBook Packages: Springer Protocols