
Abstract
The study of protein production and degradation in a quantitative and time-dependent manner is a major challenge to better understand cellular physiological response. Among available technologies bioorthogonal noncanonical amino acid tagging (BONCAT) is an efficient approach allowing for time-dependent labeling of proteins through the incorporation of chemically reactive noncanonical amino acids like l-azidohomoalanine (L-AHA). The azide-containing amino-acid derivative enables a highly efficient and specific reaction termed click chemistry, whereby the azide group of the L-AHA reacts with a reactive alkyne derivate, like dibenzocyclooctyne (DBCO) derivatives, using strain-promoted alkyne–azide cycloaddition (SPAAC). Moreover, available DBCO containing reagents are versatile and can be coupled to fluorophore (e.g., Cy7) or affinity tag (e.g., biotin) derivatives, for easy visualization and affinity purification, respectively.
Here, we describe a step-by-step BONCAT protocol optimized for the model archaeon Haloferax volcanii , but which is also suitable to harness other biological systems. Finally, we also describe examples of downstream visualization, affinity purification of L-AHA-labeled proteins and differential expression analysis.
In conclusion, the following BONCAT protocol expands the available toolkit to explore proteostasis using time-resolved semiquantitative proteomic analysis in archaea .
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others

Key words
- BONCAT
- l-Azidohomoalanine
- L-AHA
- Archaea
- Time-dependent
- Translation
- Translatome
- Proteostasis
- Haloferax volcanii
- Sulfolobus acidocaldarius
- Escherichia coli
- Click chemistry
1 Introduction
Cellular adaptation depends on efficient and timely controlled variation of the cellular protein synthesis and degradation capacity [1,2,3]. Accordingly, the resulting qualitative and quantitative ensemble of proteins present at a given time, in a given cell or tissue, or in a given environmental condition is instrumental for proper cell fate determination. Hence, proteome plasticity is a key feature in enabling suitable gene expression modulation. Depending on the nature of intra- and extracellular cues, changes in the protein repertoire and individual protein levels may occur at different time-scales, and need to be experimentally captured with accuracy to understand the underlying cellular and physiological mechanisms [1,2,3].
Over the last decade(s) various approaches have been used to explore protein synthesis and degradation dynamics [1,2,3]. Among these methods, ribosome profiling , which takes advantage of high-throughput sequencing of ribosome associated mRNA , has emerged as the method of choice to assess the translational capacity and properties of various cellular systems across different conditions [1,2,3]. Even though extremely powerful, this method remains an indirect measurement of protein synthesis, as it fails to directly measure the product of translation itself . Moreover, protein degradation is not monitored using this experimental setup. An alternative, but still emerging method, is the use of time-dependent semiquantitative proteomic approaches, which allow for analysis of both protein synthesis and degradation [1]. While less popular, and still technically challenging, various mass spectrometry–based approaches have been developed and significantly improved over the years and are now able to provide semiquantitative and direct analysis of cellular protein composition in a time-dependent manner [1, 4,5,6,7,8]. To achieve such in-depth analysis various experimental setups can be used depending on the time resolution and desired extensiveness of the analyses [1, 3]. Early on, time-dependent proteomics have relied on isotope labeling (e.g., SILAC ), whereby differential incorporation of heavy and light isotope metabolites like amino acids are used over a specified time to sort protein populations [1, 3, 9,10,11]. However, isotope labeling strategies have different drawbacks: (1) some labeled metabolites can be rather expensive, (2) efficiency of labeling is highly dependent on cellular uptake and metabolism capacity, and usually requires defined synthetic growth medium, (3) multiplexing is limited due to the lack of different reagents, (4) short kinetic pulse, low-abundant or proteins with a high turnover rate are difficult to be properly analyzed due to the lack of a specific enrichment step of the isotopically labeled peptides [1, 3, 9,10,11].
An emerging complementary strategy allowing to circumvent some of these limitations and facilitating time-dependent proteomic analysis is bioorthogonal noncanonical amino acids tagging (BONCAT ) [12, 13]. In brief, BONCAT exploits the power of in vivo incorporation of bioorthogonal molecules into polypeptides and of click chemistry which enables enrichment and/or visualization of the labeled proteins synthesized over a defined time window [4, 5, 8, 13,14,15,16,17,18]. Most importantly, BONCAT can be easily combined with isotope labeling and/or multiplexing (e.g., [4, 5, 8, 17]), thereby allowing to fully harness the potential of time-dependent proteomic analysis.
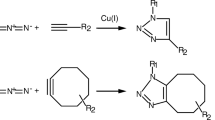
Most commonly BONCAT experiments utilize, l-azidohomoalanine (L-AHA) , a methionine analogue containing a functional azide group (Fig. 1a). L-AHA is recognized, albeit with less efficiency, by the methionyl-tRNAMet acyl transferase, and is charged onto tRNAMet, thereby allowing for incorporation of L-AHA into polypeptides instead of methionine during protein synthesis [19]. The resulting L-AHA-modified polypeptides can be further altered using strain-promoted alkyne–azide cycloaddition (SPAAC ) as outlined in Fig. 1b [20]. Alkyne derivative of biotin or various fluorophores can be efficiently and quantitatively coupled to the azide-containing proteins, thereby facilitating their further visualization and/or subsequent specific enrichment for downstream analysis (Fig. 1b) and exemplary results shown in Figs. 2, 3 and 4).

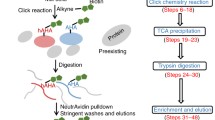
Optimized BONCAT workflow in H. volcanii . (a) Chemical structures of L-methionine and L-azidohomoalanine (b) Click chemistry: Strain Promoted Alkyne–Azide Cycloaddition (SPAAC ) reaction. Azide-containing polypeptide, DBCO derivate, and the results of click chemistry reaction are schematically depicted (c). Workflow summary of BONCAT in H. volcanii . Typical workflow as described in this protocol. The numbering below the different steps refers to the corresponding protocol steps described in the Methods section (see Subheading 3)

Exemplary BONCAT of prokaryotic cells. (a) L-AHA-based labeling of proteins in E. coli and H. volcanii . The corresponding cells (E. coli K12 and H. volcanii H26) were grown in synthetic medium lacking methionine (M9 and Hv-min, respectively) and pulse labeled with increasing amounts of L-AHA (0.1 mM and 1 mM) or 1 mM Methionine for 3 h. Cells were lysed, proteins were extracted and subjected to SPAAC click chemistry with DBCO-Cy7 as described in Subheading 3. Around 150 μg of proteins were subsequently fractionated on a 4–12% polyacrylamide gradient gel and the fluorescence signal was acquired with an Odyssey Infrared Imager (right panel). Coomassie staining (loading control) is depicted in the left panel (b) Inhibition of L-AHA-based labeling in presence of translation inhibitor in H. volcanii . Same as in (a) except that cells (H. volcanii H26) were grown in synthetic medium lacking methionine and supplemented with 0.1 mM methionine or 0.1 mM of L-AHA with or without 2.5 μM of the translation inhibitor Thiostrepton (Th) [31] for 1 h. (c) Short-time L-AHA labeling in H. volcanii . Same as in (a) except that cells (H. volcanii H26) were grown in synthetic medium lacking methionine and supplemented with 1 mM L-AHA for the indicated time. The depicted fluorescence signal intensities across single lanes were quantified using Fiji [32] and visualized in Microsoft Excel. Adapted from [33] under CC-BY License

Enrichment of L-AHA-containing protein by affinity purification . H26 cells were grown in synthetic medium lacking methionine and supplemented with 0.1 mM of L-AHA or methionine (Met) for 5 h. Cells were lysed and proteins were extracted and subjected to click chemistry with 1 μM DBCO-PEG4-Biotin as described in Subheading 3.4. Affinity purification was performed as described in Subheading 3.6. Sample aliquots for input, unbound fraction (Flow), wash fraction and elution were taken at each step of the AP. Elution was performed as described in Subheading 3.6.4. 0.5% of the input, unbound (Flow) and 0.25% of the wash fraction and 20% of the eluate were loaded on two SDS-PAGE gels respectively. The fractionated proteins were visualized by silver staining (a) and transferred onto a nylon membrane (b). Membrane was incubated with IRDye-coupled streptavidin to visualize the biotinylated proteins. Images were acquired either with the Epson Scanner (silver staining) (a) or the Li-COR Odyssey Infrared Imager (IRDye signal) (b). Asterisk indicates streptavidin monomers that are coeluted

Differential 2D gel analysis of L-AHA-labeled proteins. (a) Differential 2D gel experimental workflow. Exponentially growing wildtype (WT) and △yfg (your favorite gene mutant/or perturbation) cells grown in Hv-min lacking methionine were pulse-labeled with 1 mM L-AHA for 45 min (representing approx. 20% of the H. volcanii doubling time in these growth conditions). Cells were lysed, protein were extracted and subjected to click chemistry with DBCO-Cy7 and DBCO-Cy5.5 as described in Subheading 3.4. Equal protein amounts of WT cells labeled with DBCO-Cy7 and △yfg cells labeled with DBCO-Cy5.5 and vice versa (dye swap to control fluorescence bias) were mixed cells with DBCO-Cy5.5 and vice versa to control fluorescence bias. Sample mixtures were used to rehydrate immobilized pH stripe and isoelectric focusing as describe in Subheading 3.7.2. Second dimension was resolved using 4–12% polyacrylamide gradient gel and the fluorescence signals were acquired with the Li-COR Odyssey Infrared Imager. (b) Exemplary results of differentially expressed proteins. Full gel scan (upper part) for the 2D gel analysis obtained for the wild-type (Cy7-red channel) and mutant H. volcanii cells (Cy5.5-green channel) mixture are provided. Zoom in of the 2D gel (boxing) showing representative differential expression between wildtype and exemplary mutant cell used in this study is provided in the lower part. Note that the dye-swap experiment (WT-Cy5.5/Δyfg-Cy7) provided very similar results (data not shown). (Adapted from [33] under CC-BY License)
Interestingly, BONCAT has been successfully applied in a wide variety of organisms and/or tissues across the tree of life , thereby highlighting its potential [4, 5, 8, 13,14,15,16,17,18,19]. However, despite its broad application potential, proteomics analysis in general and time-dependent analysis of protein synthesis and degradation in particular, remains in its early days in archaea .
Haloferax volcanii , an extreme halophile Euryarchaeota , has emerged as a very powerful model organism to perform genetic and functional analysis in archaea [21, 22]. Its handling simplicity has attracted several scientists who aim to push the experimentational limits of this organism and crack open its molecular biology. Efforts in proteomics analysis do not escape this trend. In fact, proteomics analyses in H. volcanii have been making significant progress over the last years [23,24,25,26,27], and are currently being improved by a concerted action of several groups working on H. volcanii (see Archaeal Proteome Project—ArcPP—https://archaealproteomeproject.org and https://github.com/arcpp/ArcPP—[28]). Finally, recent methodological breakthroughs using isotope labeling, SILAC reagents or 15N-labeled nitrogen source, in H. volcanii [24, 27] offer a unique opportunity to perform cutting-edge time-dependent proteomics analysis in this model archaeon.
Here we expand the H. volcanii proteomics toolkit and describe a step-by-step BONCAT protocol and downstream visualization/enrichment procedures of labeled proteins optimized for H. volcanii (summarized in Fig. 1c). In addition, this protocol has been validated in another model archaeon (i.e., Sulfolobus acidocaldarius —Ferreira-Cerca lab unpublished), another model bacterium (i.e., Escherichia coli —see below) and is likely to be suitable for various additional biological systems [4, 5, 8, 13,14,15,16,17,18,19].
Together with the isotope-labeling strategy mentioned above and additional multiplexing [4, 5, 8], the following BONCAT protocol allows for global inspection of protein synthesis capacity in Haloferax volcanii and paves the way to time-dependent proteomics analysis in this ever more versatile model archaeon [21, 22]. Moreover, these additional experimental possibilities position H. volcanii as a unique experimental system to explore the molecular principles of proteostasis in archaea .
2 Materials
There are no specific preferences of sources of chemical reagents or materials, unless stated otherwise. Use ultrapure water with 18 MΩ·cm resistivity at 25 °C.
2.1 Microbiological Cultures
2.1.1 Strains
2.2 Haloferax volcanii Minimal Medium (Hv-min)
-
1.
1 M Tris–HCl pH 7.0.
-
2.
1 M Tris–HCl pH 7.5.
-
3.
30% (w/v) Salt Water: 4.1 M NaCl; 147.5 mM MgCl2; 142 mM MgSO4; 94 mM KCl; 20 mM Tris–HCl pH 7.5.
-
4.
Haloferax volcanii minimal carbon source: 10% sodium dl-lactate; 9% succinic acid; 1% glycerol; pH 7.5. Sterilize by filtration.
-
5.
Haloferax volcanii minimal salts: 417 mM NH4Cl; 250 mM CaCl2; 152 μM MnCl2; 127 μM ZnSO4; 689 μM FeSO4; 16.7 μM CuSO4. Sterilize by filtration.
-
6.
Hv-Trace elements solution: 1.8 mM MnCl2; 1.53 mM ZnSO4; 8.27 mM FeSO4; 0.2 mM CuSO4 (see step 4 Subheading 3.1).
-
7.
0.5 M KPO4 buffer, pH 7.0.
-
8.
1 mg/L thiamine solution. Sterilize by filtration.
-
9.
1 mg/L biotin solution. Sterilize by filtration.
-
10.
1,000× uracil stock solution (50 mg/mL in DMSO).
-
11.
200× methionine stock solution (10 mg/mL in H2O).
-
12.
Sterile filters (0.22 μm) and syringes.
2.3 Noncanonical Amino Acid Pulse Reagents
-
1.
l-azidohomoalanine (L-AHA) stock solution (100 mM).
-
2.
200× methionine stock solution (10 mg/mL in H2O).
-
3.
Haloferax volcanii methionine free minimal medium (see Note 1 ).
2.4 Cell Lysis
-
1.
Extraction Buffer (EB) + 1% SDS : 150 mM NaCl, 100 mM EDTA, 50 mM Tris–HCl pH 8.5, 1 mM MgCl2, 1% SDS .
-
2.
Extraction Buffer (EB) without detergent: 150 mM NaCl, 100 mM EDTA, 50 mM Tris–HCl pH 8.5, 1 mM MgCl2.
-
3.
Thermoblock.
-
4.
Benchtop centrifuge.
2.5 Protein Reduction
-
1.
β-mercaptoethanol.
-
2.
Dark box.
2.6 Acetone Precipitation of Total Proteins
-
1.
Precooled (−20 °C) Acetone p.a.
-
2.
2 mL reagent tubes.
-
3.
Precooled benchtop centrifuge.
2.7 Protein Alkylation
-
1.
Extraction Buffer (EB) + 1% SDS : 150 mM NaCl, 100 mM EDTA, 50 mM Tris–HCl pH 8.5, 1 mM MgCl2, 1% SDS .
-
2.
Extraction Buffer (EB) without detergent: 150 mM NaCl, 100 mM EDTA, 50 mM Tris–HCl pH 8.5, 1 mM MgCl2.
-
3.
10× Alkylation buffer: 10× PBS, 2 M 2-chloroacetamide.
-
4.
Thermoblock.
-
5.
Benchtop centrifuge.
-
6.
Black 1.5 mL reagent tubes.
-
7.
Rotating wheel.
2.8 Click-Chemistry Reagents—Strain-Promoted Alkyne–Azide Cycloaddition (SPAAC )
-
1.
Extraction Buffer (EB) + 1% SDS : 150 mM NaCl, 100 mM EDTA, 50 mM Tris–HCl pH 8.5, 1 mM MgCl2, 1% SDS .
-
2.
Extraction Buffer (EB) without detergent: 150 mM NaCl, 100 mM EDTA, 50 mM Tris–HCl pH 8.5, 1 mM MgCl2.
-
3.
Stock solution (100 μM) DBCO-reagents for strain-promoted alkyne–azide cycloaddition (e.g., DBCO-Cy5, DBCO-Cy7, DBCO-PEG4-Biotin) (see Note 2 ).
-
4.
l-Azidohomoalanine (L-AHA) stock solution (100 mM).
2.9 Methanol–Chloroform Extraction of Proteins
-
1.
Methanol p.a.
-
2.
Chloroform p.a.
-
3.
Benchtop cooled centrifuge.
2.10 Affinity Purification
-
1.
AP buffer: 300 mM NaCl, 10 mM MgCl2, 50 mM Tris–HCl pH 8.4.
-
2.
AP buffer + 2% SDS : 300 mM NaCl, 10 mM MgCl2, 50 mM Tris–HCl pH 8.4, 2% SDS .
-
3.
AP buffer + 1% SDS : 300 mM NaCl, 10 mM MgCl2, 50 mM Tris–HCl pH 8.4, 1% SDS .
-
4.
HiAP buffer + 1% SDS : 1 M NaCl, 10 mM MgCl2, 50 mM Tris–HCl pH 8.4, 1% SDS .
-
5.
HiAP buffer + 0.1% SDS : 1 M NaCl, 10 mM MgCl2, 50 mM Tris–HCl pH 8.4, 0.1% SDS .
-
6.
BSA Fraction V.
-
7.
Streptavidin-bound agarose beads.
-
8.
Gravity flow chromatography column.
-
9.
DBCO-PEG4-Biotin.
-
10.
2× HU-buffer: 100 mM Tris–HCl pH 6.8, 0.5 mM EDTA pH 8, 4 M urea, 0.75% β-mercaptoethanol, 2.5% SDS , bromophenol blue.
-
11.
1 M DTT.
-
12.
2× HU-buffer + DTT: 100 mM Tris–HCl pH 6.8, 0.5 mM EDTA pH 8, 4 M Urea, 200 mM DTT, 2.5% SDS .
-
13.
0.1 mM NH4OAc, 0.1 mM MgCl2.
-
14.
Competitive elution buffer: 1 mM biotin, 1% NH4OH.
2.11 1D/2D Gel Electrophoresis and Detection
-
1.
2× HU buffer (see item 10 Subheading 2.10).
-
2.
Immobilized pH strip and 2D gel reagents (see Note 3 ).
-
3.
Low-melting agarose.
-
4.
Gel fixation solution: 40% methanol, 10% acetic acid.
-
5.
Blocking buffer + 10% SDS : 1× PBS, 1 mM EDTA pH 8, 10% SDS .
-
6.
Blocking buffer + 1% SDS : 1× PBS, 1 mM EDTA pH 8, 1% SDS .
-
7.
Blocking buffer + 0.1% SDS : 1× PBS, 1 mM EDTA pH 8, 0.1% SDS .
-
8.
IRDye-coupled streptavidin (1 mg/mL).
3 Methods (Workflow Is Summarized in Fig. 1c)
Use ultra-pure water with 18 MΩ·cm resistivity at 25 °C.
3.1 Microbiological Methods
All solutions/media should be autoclaved, unless stated otherwise.
3.1.1 Haloferax volcanii Media and Cultivation
-
1.
Prepare 30% (w/v) salt water by dissolving in 4 L of Millipore water 1,200 g NaCl; 150 g MgCl2·6H2O; 175 g MgSO4·7H2O; 35 g KCl; 100 mL 1 M Tris–HCl pH 7.5. Fill up with H2O to 5 L. Autoclave and store at room temperature.
-
2.
Prepare Hv-Min Carbon Source by dissolving in 150 mL H2O: 41.7 mL 60% Sodium dl-lactate, 37.5 g Succinic acid Na2 salt·6H2O; 3.15 mL 80% Glycerol. Carefully adjust to pH 7.5 with first 5 M NaOH and then 1 M NaOH. Fill up with H2O to a final volume of 250 mL and sterilized by filtration. Aliquot in 50 mL and store at 4 °C.
-
3.
Prepare Tris–HCl pH 7.0.
-
4.
Prepare Hv-Trace elements solution by adding to 100 mL water a few drops of 37% concentrated HCl. Dissolve the following salts one by one in the following order: 36 mg MnCl2·4H2O; 44 mg ZnSO4·7H2O; 230 mg FeSO4·7H2O; 5 mg CuSO4·5H2O. Sterilize by filtration and store at 4 °C.
-
5.
Prepare Hv-minimal salts by mixing 30 mL 1 M NH4Cl; 36 mL 0.5 M CaCl2 and 6 mL Hv-Trace elements solution (see step 4). Sterilize by filtration and store at 4 °C.
-
6.
Prepare 0.5 M KPO4 buffer (pH 7.0). Mix 61.5 mL 1 M K2HPO4 and 38.5 mL 1 M KH2PO4. Check pH and adjust to pH 7.0, if required. Add an equal volume of H2O (100 mL). Autoclave and store at room temperature.
-
7.
Prepare Thiamine/Biotin mix by combining 9.5 mL Thiamine (1 mg/mL) and 1.2 mL Biotin (1 mg/mL).
-
8.
Prepare Hv-minimal medium (Hv-min). For 1 L medium mix: 600 mL 30% salt water, 330 mL H2O and 30 mL Tris–HCl pH 7.0. Autoclave. When cooled down add 25.5 mL Hv-Min Carbon Source (see step 2), 12 mL Hv-Min Salts (see step 5), 1.95 mL 0.5 M KPO4 buffer pH 7.0 (see step 6), 900 μL Thiamine/Biotin mix (see step 7), 1 mL 1,000× Uracil stock solution (see item 10 Subheading 2.2). Store at room temperature in a dark cupboard.
-
9.
Scratch the surface of an Haloferax volcanii glycerol cryo-stock with a sterile inoculation loop and inoculate 5 to 10 mL medium as a start culture. Incubate at 42 °C under agitation. Dilute with prewarmed medium or let grow until the culture has reached the desired cell density/volume (OD600nm = 0.6).
3.2 In Vivo Pulse Labeling with L-AHA
-
1.
Prepare a fresh stock solution of L-AHA (see item 1 Subheading 2.3).
-
2.
Pulse label 5 mL of cells at OD600nm = 0.6 with 0.1–1 mM L-AHA (or methionine as negative control) at the desired temperature and time (see Note 4 ).
-
3.
Fractionate cells in two 2 mL aliquots and centrifuge for 3 min at 6,000 × g.
-
4.
Discard supernatant.
-
5.
Snap-freeze cell pellet in liquid nitrogen.
3.3 Protein Extraction
-
1.
Resuspend cell pellet in 500 μL Extraction Buffer (EB) supplemented with 1% SDS (see item 1 Subheading 2.4 and Note 5 ).
-
2.
Boil the samples for 13 min at 95 °C.
-
3.
Cool for 5 min at RT.
-
4.
Centrifuge at 16,000 × g for 5–10 min RT.
-
5.
Transfer supernatant into a fresh cup.
-
6.
Optional: Freeze supernatant at −20 °C or proceed with click chemistry (see Subheading 3.4).
3.4 Click-Chemistry
3.4.1 Reduction
-
1.
Add 10 μL of β-Mercaptoethanol (final concentration 2%).
-
2.
Incubate the lysate for 1 h in the dark (RT).
3.4.2 Acetone Precipitation
-
1.
Afterward, split the 500 μL of cell lysate into two cups.
-
2.
Add 4 Volume (1 mL) of acetone precooled at −20 °C (acetone end concentration = 80%).
-
3.
Vortex and let proteins precipitate for 1 h at −20 °C.
-
4.
Centrifuge for 10 min at 16,000 × g at 4 °C.
-
5.
Discard the supernatant without disturbing the protein pellet.
-
6.
Wash the pellet with 1 mL of acetone precooled at −20 °C.
-
7.
Centrifuge for 10 min at 16,000 × g at 4 °C.
-
8.
Discard the supernatant and air-dry the protein pellet (see Note 6 ).
3.4.3 Alkylation
-
1.
Resuspend pellet in 25 μL of EB + 1% SDS (see item 1 Subheading 2.7).
-
2.
Add 225 μL EB without detergent (see item 2 Subheading 2.7).
-
3.
Vortex and denature proteins for 5 min at 95 °C.
-
4.
Let cool down at RT.
-
5.
Merge the corresponding supernatant together in a fresh cup.
-
6.
Measure protein concentration with a NanoDrop or equivalent (vortex samples before measuring).
-
7.
Prepare 200 μL of extracted protein at a protein concentration 1–2 μg/μL.
-
8.
Optional: use black tubes to keep the samples in the dark.
-
9.
Depending on protein concentration and the L-AHA pulse, aim for 200–400 μg of proteins.
-
10.
Equalize the volumes of added protein with EB without detergent (see item 2 Subheading 2.7).
-
11.
Add 20 μL of freshly prepared 10× concentrated alkylation solution (10× PBS, 2 M 2-chloroacetamide) (see item 3 Subheading 2.7 and Note 7 ).
-
12.
Incubate for 1 h on a rotating wheel in the dark.
-
13.
Afterward, precipitate the proteins via acetone precipitation as described above (see Subheading 3.4.2).
3.4.4 Click-Chemistry : Strain Promoted Alkyne–Azide Cycloaddition (SPAAC )
-
1.
Resuspend the pellet in 20 μL of EB containing 1% SDS (item 1 Subheading 2.8).
-
2.
Add 180 μL of EB without detergent (item 2 Subheading 2.8).
-
3.
Denature samples for 5 min at 95 °C.
-
4.
Let cool down to room temperature.
-
5.
Add 2 μL of 100 μM solution DBCO reagent (1 μM final concentration) to allow for strain-promoted cycloaddition to the L-AHA azide group (e.g., DBCO-Cy5, DBCO-Cy7, DBCO-PEG4-Biotin) (see Note 8 ).
-
6.
Incubate on a rotating wheel for 30 min at room temperature in the dark.
-
7.
Eliminate unreacted DBCO reagent by adding excess of L-AHA to 0.1–1 mM.
3.5 Methanol–Chloroform Extraction
-
1.
Add 480 μL methanol, 160 μL chloroform to the sample (~200 μL).
-
2.
Vortex vigorously and add 640 μL H2O.
-
3.
Vortex and centrifuge for 5 min at 16,000 × g RT.
-
4.
Suck off and discard the aqueous phase (upper phase).
-
5.
Add 300 μL methanol.
-
6.
Vortex and centrifuge for 30 min at 16,000 × g 4 °C.
-
7.
Discard supernatant and shortly air-dry the protein pellet.
-
8.
Proceed with the desired step.
3.6 Affinity Purification of Biotinylated Protein
3.6.1 Sample Preparation
-
1.
Performed PEG4-Biotin cycloaddition of 400 μg protein as described in Subheading 3.4.4.
-
2.
After the final methanol–chloroform extraction (see Subheading 3.5) resuspend protein pellet in 100 μL of AP buffer + 2% SDS (see item 2 Subheading 2.10).
-
3.
Denature for 5 min at 95 °C.
-
4.
Add 900 μL AP buffer without SDS (see item 1 Subheading 2.10).
-
5.
Take and store an aliquot as Input (5% of total volume).
3.6.2 Streptavidin Beads Preparation
-
1.
For each purification prepare 100 μL of slurry streptavidin-bound agarose beads. Centrifuge for 1 min at 100 × g to discard the supernatant.
-
2.
Wash the beads twice with 1 mL AP buffer + 1% SDS (see item 3 Subheading 2.10) for 5 min on a rotating wheel. Centrifuge for 1 min at 100 × g to discard the supernatant.
-
3.
Block the beads twice with 500 μL AP buffer + 1% SDS (see item 3 Subheading 2.10) and 1 mg/mL BSA for 5 min on a rotating wheel. Centrifuge for 1 min at 100 × g to discard the supernatant.
-
4.
Wash the beads with 1 mL AP buffer + 1% SDS (see item 3 Subheading 2.10) for 5 min on a rotating wheel. Centrifuge for 1 min at 100 × g to discard the supernatant.
3.6.3 Affinity Purification
-
1.
Mix the sample (see step 4 Subheading 3.6.1) with the prepared streptavidin beads (see Subheading 3.6.2).
-
2.
Incubate for 30 min on a rotating wheel at room temperature.
-
3.
Transfer the material onto a gravity flow column. Collect and store the flow-through (= unbound fraction) (5% of total volume).
-
4.
Wash the beads 3 times with 2 mL HiAP buffer + 1% SDS (see item 4 Subheading 2.10). Collect all the wash in a single 15 mL tube and store 10% of the resulting pooled wash fractions.
-
5.
Wash the beads twice 1 mL AP buffer + 0.1% SDS (see item 5 Subheading 2.10).
-
6.
Beads were collected with 1 mL AP buffer + 0.1% SDS (see item 5 Subheading 2.10) and transfer to a fresh cup.
-
7.
Centrifuge for 1 min at 100 × g and discard as much of the supernatant as possible.
3.6.4 Elution (See Note 9 ) (an Exemplary Result Is Provided in Fig. 3)
3.6.4.1 Fast-Elution for Gel Electrophoresis
-
1.
Add 50–100 μL 2× HU buffer + DTT (see item 12 Subheading 2.10) to the beads.
-
2.
Incubate for 10 min at 95 °C.
-
3.
Collect supernatant.
3.6.4.2 Competitive Elution for Downstream Processing
-
1.
Wash twice the beads with 0.1 M NH4OAc, 0.1 mM MgCl2 solution.
-
2.
Centrifuge for 1 min at 100 × g and discard as much of the supernatant as possible.
-
3.
Elute bound material twice with 500 μL Competitive elution buffer (see item 14 Subheading 2.10) for 10 min at 95 °C.
-
4.
Collect and pool the supernatants.
-
5.
Split the collected supernatant into 2 unequal fractions (80–90%) and (10–20%).
-
6.
Fill up the splitter eluate with competitive elution buffer.
-
7.
Lyophilized overnight using a speed-vac.
-
8.
Resuspend the 10–20% eluate fraction in 40 μL 2× HU buffer + DTT (see item 12 Subheading 2.10). Denature sample and check pull-down efficiency (see Subheading 3.9).
-
9.
If desired, proceed with the 80–90% eluate fraction with desired downstream analysis.
3.7 Gel Electrophoresis and Detection
3.7.1 SDS-PAGE
-
1.
Resuspend pellet in 2× HU buffer + DTT (see item 12 Subheading 2.10).
-
2.
Denature samples for 5 min at 60 °C.
-
3.
Vortex firmly before loading 15–25 μL on an SDS-polyacrylamide gradient gel.
-
4.
Separate samples for around 1 h at constant 170–190 V.
3.7.2 2D Gel Electrophoresis (an Exemplary Result Is Provided in Fig. 4)
3.7.2.1 Isoelectric Focusing (See Note 2 )
-
1.
Sample (see Subheading 3.5) are solubilized in sample buffer containing 7 M urea, 2 M thiourea, 4% CHAPS, 40 mM DTT, and 0.4% ampholytes (pH 3–10) as suggested by the manufacturer.
-
2.
Isoelectric focusing is performed overnight using an immobilized pH gradient strip (pH 3–6) using the run parameters described below (Table 1).
3.7.2.2 Second Dimension SDS-PAGE
-
1.
Immobilized pH gradient strip is incubated in equilibration solution 1 (6 M urea, 30% glycerol, 2% SDS , and 0.05 M Tris–HCl pH 8.5) for 10 min.
-
2.
The strip is incubated for 10 min in equilibration solution base mix.
-
3.
The strip is shortly immersed in SDS-PAGE running buffer.
-
4.
Load the strip on a 4–12% polyacrylamide gradient gel (keep the strip in place by adding 40 °C warm low-melting agarose).
-
5.
The gel was run for 15 min at 80 V followed by 1 h at 160 V.
-
6.
Proceed with gel fixation (see Subheading 3.8).
3.8 In-Gel Detection (Exemplary Results Are Provided in Figs. 2 and 4)
-
1.
Incubate the gel in gel fixation solution (see item 4 Subheading 2.11) for at least 20 min under agitation.
-
2.
Wash the gel 3 times with H2O for 5 min under agitation.
-
3.
Scan the gel using a compatible fluorescence imager.
3.9 Detection of Affinity Purified L-AHA-Labeled Proteins
(an exemplary result is provided in Fig. 3).
-
1.
Proceed with gel electrophoresis as described in Subheading 3.7.1.
-
2.
Transfer and immobilized proteins on Nylon-Immobilon membrane using your favorite transfer protocol.
-
3.
Incubate the membrane for 20 min in blocking buffer + 10% SDS (see item 5 Subheading 2.11) under agitation.
-
4.
Incubate with 0.1 μg/mL IRDye-conjugated Streptavidin in blocking buffer + 10% SDS for 20 min under agitation (see Note 10 ).
-
5.
Wash twice the membrane with blocking buffer + 10% SDS (see item 5 Subheading 2.11) for 20 min under agitation.
-
6.
Wash twice the membrane with blocking buffer + 1% SDS (see item 6 Subheading 2.11) for 20 min under agitation.
-
7.
Wash twice the membrane with blocking buffer + 0.1% SDS (see item 7 Subheading 2.11) for 20 min under agitation.
-
8.
Use compatible imaging system to record fluorescent (chemiluminescent) signals (see Note 10 ).
4 Notes
-
1.
Culture in methionine-free medium is critical for the efficiency of L-AHA labeling.
-
2.
There is a multitude of DBCO reagents available which offer various advantages for downstream analysis. The DBCO reagents should be wisely selected depending on the experimental goals.
-
3.
Use commercial immobilized strips and follow the protocol recommended by the manufacturer.
-
4.
Depending of the doubling time of your experimental system and the time of labeling use concentration of 0.1–1 mM L-AHA . At this point 15NH4-salt or SILAC based labeling (see [24, 27], respectively, for procedure suitable for H. volcanii ) can be simultaneously performed to improve confidence of downstream identification of neosynthesized proteins by mass spectrometry.
-
5.
No reducing agent, such as dithiothreitol (DTT) or β-mercaptoethanol, should be added to avoid reduction of the L-AHA azide group.
-
6.
Do not overdry pellets, otherwise the proteins will be difficult to solubilize.
-
7.
Various amounts of 2-chloroacetamide powder can be fractionated into 1.5 mL black cups and stored at RT. Prior to use add the necessary volume of 10× PBS to such aliquot and mix well (make sure the reagent is completely dissolved). Use only freshly solubilized 2-chloroacetamide.
-
8.
The end concentration of DBCO derivate may be optimized empirically. For H. volcanii we typically used 1 mM DBCO reagents as it provided the best signal–noise ratio in our hands.
-
9.
Streptavidin–biotin complexes are extremely stable. The elution procedure should be selected according to the downstream analytical procedure. The experimenter should be aware about the pros and contras of the different elution procedures. The following procedures are exemplary elution procedures that were qualitatively well performing in our hands. Alternatively, on beads protease digest can be directly performed and further processed for mass spectrometry. However, note that in this condition biotinylated-AHA peptides will remain attached to the affinity purification matrix. Therefore additional labeling, using SILAC or 15N, is essential to distinguish neosynthesized proteins from background (e.g., [4, 5, 8]).
-
10.
As alternative HRP-conjugated streptavidin can be used and detected by chemiluminescence. Please refer to manufacturer manual for experimental conditions.
References
Dermit M, Dodel M, Mardakheh FK (2017) Methods for monitoring and measurement of protein translation in time and space. Mol BioSyst 13:2477–2488. https://doi.org/10.1039/c7mb00476a
Ingolia NT, Hussmann JA, Weissman JS (2019) Ribosome profiling: global views of translation. Cold Spring Harb Perspect Biol 11:a032698. https://doi.org/10.1101/cshperspect.a032698
Zhao J, Qin B, Nikolay R et al (2019) Translatomics: the global view of translation. Int J Mol Sci 20:212. https://doi.org/10.3390/ijms20010212
Bagert JD, Xie YJ, Sweredoski MJ et al (2014) Quantitative, time-resolved proteomic analysis by combining bioorthogonal noncanonical amino acid tagging and pulsed stable isotope labeling by amino acids in cell culture. Mol Cell Proteomics 13:1352–1358. https://doi.org/10.1074/mcp.M113.031914
Bagert JD, van Kessel JC, Sweredoski MJ et al (2016) Time-resolved proteomic analysis of quorum sensing in Vibrio harveyi †electronic supplementary information (ESI) available: figures and tables. See DOI: 10.1039/c5sc03340c click here for additional data file. Chem Sci 7:1797–1806. https://doi.org/10.1039/c5sc03340c
Cox J, Mann M (2011) Quantitative, high-resolution proteomics for data-driven systems biology. Annu Rev Biochem 80:273–299. https://doi.org/10.1146/annurev-biochem-061308-093216
Gygi SP, Rist B, Aebersold R (2000) Measuring gene expression by quantitative p roteome analysis. Curr Opin Biotechnol 11:396–401. https://doi.org/10.1016/S0958-1669(00)00116-6
Rothenberg DA, Taliaferro JM, Huber SM et al (2018) A proteomics approach to profiling the temporal translational response to stress and growth. iScience 9:367–381. https://doi.org/10.1016/j.isci.2018.11.004
Chen X, Wei S, Ji Y et al (2015) Quantitative proteomics using SILAC: principles, applications, and developments. Proteomics 15:3175–3192. https://doi.org/10.1002/pmic.201500108
Ong S-E, Blagoev B, Kratchmarova I et al (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics 1:376. https://doi.org/10.1074/mcp.M200025-MCP200
Ong S-E, Mann M (2006) A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC). Nat Protoc 1:2650–2660. https://doi.org/10.1038/nprot.2006.427
Dieterich DC, Link AJ, Graumann J et al (2006) Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). Proc Natl Acad Sci U S A 103:9482–9487. https://doi.org/10.1073/pnas.0601637103
Landgraf P, Antileo ER, Schuman EM, Dieterich DC (2015) BONCAT: metabolic labeling, click chemistry, and affinity purification of newly synthesized proteomes. In: Gautier A, Hinner MJ (eds) Site-specific protein labeling: methods and protocols. Springer, New York, New York, NY, pp 199–215
Glenn WS, Stone SE, Ho SH et al (2017) Bioorthogonal noncanonical amino acid tagging (BONCAT) enables time-resolved analysis of protein synthesis in native plant tissue. Plant Physiol 173:1543–1553. https://doi.org/10.1104/pp.16.01762
Hatzenpichler R, Scheller S, Tavormina PL et al (2014) In situ visualization of newly synthesized proteins in environmental microbes using amino acid tagging and click chemistry. Environ Microbiol 16:2568–2590. https://doi.org/10.1111/1462-2920.12436
Hatzenpichler R, Connon SA, Goudeau D et al (2016) Visualizing in situ translational activity for identifying and sorting slow-growing archaeal−bacterial consortia. Proc Natl Acad Sci U S A 113:E4069–E4078. https://doi.org/10.1073/pnas.1603757113
Saleh AM, Wilding KM, Calve S et al (2019) Non-canonical amino acid labeling in proteomics and biotechnology. J Biol Eng 13:43–43. https://doi.org/10.1186/s13036-019-0166-3
Shin J, Rhim J, Kwon Y et al (2019) Comparative analysis of differentially secreted proteins in serum-free and serum-containing media by using BONCAT and pulsed SILAC. Sci Rep 9:3096–3096. https://doi.org/10.1038/s41598-019-39650-z
Kiick KL, Saxon E, Tirrell DA, Bertozzi CR (2002) Incorporation of azides into recombinant proteins for chemoselective modification by the Staudinger ligation. Proc Natl Acad Sci U S A 99:19–24. https://doi.org/10.1073/pnas.012583299
Agard NJ, Baskin JM, Prescher JA et al (2006) A comparative study of bioorthogonal reactions with Azides. ACS Chem Biol 1:644–648. https://doi.org/10.1021/cb6003228
Leigh JA, Albers S-V, Atomi H, Allers T (2011) Model organisms for genetics in the domain archaea: methanogens, halophiles, Thermococcales and Sulfolobales. FEMS Microbiol Rev 35:577–608. https://doi.org/10.1111/j.1574-6976.2011.00265.x
Pohlschroder M, Schulze S (2019) Haloferax volcanii. Trends Microbiol 27:86–87. https://doi.org/10.1016/j.tim.2018.10.004
Cerletti M, Paggi RA, Guevara CR et al (2015) Global role of the membrane protease LonB in archaea: potential protease targets revealed by quantitative proteome analysis of a lonB mutant in Haloferax volcanii. J Proteome 121:1–14. https://doi.org/10.1016/j.jprot.2015.03.016
Cerletti M, Paggi R, Troetschel C et al (2018) LonB protease is a novel regulator of Carotenogenesis controlling degradation of phytoene synthase in Haloferax volcanii. J Proteome Res 17:1158–1171. https://doi.org/10.1021/acs.jproteome.7b00809
Costa MI, Cerletti M, Paggi RA et al (2018) Haloferax volcanii proteome response to deletion of a rhomboid protease gene. J Proteome Res 17:961–977. https://doi.org/10.1021/acs.jproteome.7b00530
Kirkland PA, Humbard MA, Daniels CJ, Maupin-Furlow JA (2008) Shotgun proteomics of the haloarchaeon Haloferax volcanii. J Proteome Res 7:5033–5039. https://doi.org/10.1021/pr800517a
McMillan LJ, Hwang S, Farah RE et al (2018) Multiplex quantitative SILAC for analysis of archaeal proteomes: a case study of oxidative stress responses. Environ Microbiol 20:385–401. https://doi.org/10.1111/1462-2920.14014
Schulze S, Adams Z, Cerletti M et al (2020) The archaeal proteome project advances knowledge about archaeal cell biology through comprehensive proteomics. Nat Commun 11:3145–3145. https://doi.org/10.1038/s41467-020-16784-7
Allers T, Ngo H-P, Mevarech M, Lloyd RG (2004) Development of additional selectable markers for the halophilic archaeon Haloferax volcanii based on the leuB and trpA genes. Appl Environ Microbiol 70:943–953. https://doi.org/10.1128/AEM.70.2.943-953.2004
Blattner FR, Plunkett G, Bloch CA et al (1997) The complete genome sequence of Escherichia coli K-12. Science 277:1453. https://doi.org/10.1126/science.277.5331.1453
Hummel H, Böck A (1987) Thiostrepton resistance mutations in the gene for 23S ribosomal RNA of Halobacteria. Biochimie 69:857–861. https://doi.org/10.1016/0300-9084(87)90212-4
Schindelin J, Arganda-Carreras I, Frise E et al (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676
Knüppel R, Trahan C, Kern M et al (2021) Insights into synthesis and function of KsgA/Dim1-dependent rRNA modifications in archaea. Nucleic Acids Res 49(3):1662–1687. https://doi.org/10.1093/nar/gkaa1268
Acknowledgments
We are indebted of the scientists who have pioneered click chemistry, BONCAT , and other methodological aspects which have strongly inspired the establishment of this protocol. We are grateful to Prof. Dr. Karl-Dieter Entian (University of Frankfurt) for comments and suggestions. We would like to thank Dr. Robert Knüppel, Michael Jüttner and our colleagues from the chair of Biochemistry III and Biochemistry I for sharing protocols, materials, equipment, and discussion. Dr. Astrid Bruckmann (University of Regensburg) for assistance with 2D SDS-PAGE. Thanks to Prof. Dr. Sonja-Albers (University of Freiburg), Prof. Dr. Thorsten Allers (University of Nottingham), and Prof. Dr. Anita Marchfelder (University of Ulm) for kindly sharing strains and protocols. Work in the Ferreira-Cerca laboratory is supported by the chair of Biochemistry III “House of the Ribosome” – University of Regensburg, by the DFG-funded collaborative research center CRC/SFB960 “RNP biogenesis: assembly of ribosomes and nonribosomal RNPs and control of their function” (project AP1/B13) and by an individual DFG grant to S.F.-C. (FE1622/2-1; Project Nr. 409198929).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this protocol

Cite this protocol
Kern, M., Ferreira-Cerca, S. (2022). Differential Translation Activity Analysis Using Bioorthogonal Noncanonical Amino Acid Tagging (BONCAT) in Archaea. In: Entian, KD. (eds) Ribosome Biogenesis. Methods in Molecular Biology, vol 2533. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-2501-9_14
Download citation
DOI: https://doi.org/10.1007/978-1-0716-2501-9_14
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-2500-2
Online ISBN: 978-1-0716-2501-9
eBook Packages: Springer Protocols