
Abstract
Regeneration, the ability to restore body parts after an injury or an amputation, is a widespread property in the animal kingdom. This chapter describes methods used to study this fascinating process in the annelid Platynereis dumerilii. During most of its life, this segmented worm is able to regenerate upon amputation the posterior part of its body, including its pygidium (terminal non-segmented body region bearing the anus) and a subterminal posterior growth zone which contains stem cells required for the formation of new segments. Detailed description of Platynereis worm culture and how to obtain large quantity of regenerating worms is provided. We also describe the staging system that we established and three important methods to study regeneration: whole mount in situ hybridization to study gene expression, 5-ethynyl-2′-deoxyuridine (EdU) labeling to characterize cell proliferation, and use of pharmacological treatments to establish putative roles of defined signaling pathways and processes.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
- Regeneration
- Annelid
- Platynereis dumerilii
- Whole mount in situ hybridization
- Gene expression
- EdU
- Cell proliferation
- Pharmacological inhibitors
1 Introduction
Regeneration, the ability to restore a lost or damaged body part is a fascinating process that has intrigued scientists since the pioneering study of Hydra regeneration by A. Trembley during the 1700s [1]. While having been intensively studied during the first part of the twentieth century, reparative regeneration has been less investigated since the rise of genetic and molecular studies of development in the 70s. This is intrinsically linked to the limited regenerative potential of the main developmental biology models, with the noticeable exception of zebrafish [2], which have emerged at that time. These last years, there has been a strong revival of the interest for regeneration, in part driven by possible applications for regenerative medicine [3].
Annelida (annelids) constitute a major lineage of the Lophotrochozoa super phylum, a group of primary importance to understand animal and especially bilaterian evolution [4]. Annelids represent a quite large phylum, with over 22,000 species including ragworms, earthworms and leeches. They can live in various ecosystems, mostly in the sea, but also in fresh water and humid terrestrial environments. They present a diversity of forms and life history traits; some live in a tube, while others are burrowed deep in the sand, stuck on algae or even parasitic [4, 5].
Interestingly, annelids, with the noticeable exception of leeches, are among the Metazoa that show the most important regenerative abilities [6, 7]. Indeed, many annelids are able to regenerate, after an amputation or injury , the posterior part of their body, their anterior part (including the head), or both, as well as appendages (named parapodia) and all kind of tentacles and cirri [6]. While the capacity to regenerate their posterior parts is almost shared by all annelids, anterior or head regeneration is less widespread [6].
There is a quite long history of experimental and descriptive morphological studies of regeneration in many annelid species [8, 9]. Many of these studies notably investigated possible sources of the cells involved in regeneration [8, 10], as well as the importance of the nervous system to allow a proper regeneration [11]. More recently, cellular and molecular aspects of annelid regeneration have been studied in a couple of model species, Pristina leydyi, Capitella teleta, and Enchytraeus japonensis, all belonging to the same group of annelids, the Sedentaria (for review see [6, 12]). While these studies provided interesting information, there is, however, still a crucial need for additional annelid models that allow to address fundamental and mechanistic questions about regeneration.
One major model species that has been successfully developed for decades is the Nereididae Platynereis dumerilii , which was originally described by Audouin and Milne Edwards in 1834 (Fig. 1) [13], and belongs to the Errantia lineage. Platynereis dumerilii is a medium-sized marine annelid that is easily maintained in laboratories world-wide. Like many other marine animals, such as corals, sea urchins and fishes, Platynereis’s life cycle is synchronized with the lunar cycle [14]. Each worm will reproduce only once in its life before dying, and the timing of this reproduction is tightly regulated by this circalunar life cycle. Platynereis has emerged as an intensely studied model organism for developmental, marine, neuro, and evolutionary biology, as well as to study regeneration [15, 16]. Platynereis worms have indeed extensive regenerative capabilities: after amputation of the posterior part of their body, which leads to the removal of the pygidium (terminal non-segmented body part of the worm), the stem cell–rich subterminal growth zone (responsible for the continuous growth of the worms [17]) and several segments, Platynereis worms are able to regenerate both pygidium and growth zone which will in turn produce new segments [18]. Platynereis is also able to regenerate various body outgrowths, such as tentacles and appendages (parapodia), but not its head. Platynereis worms can thus properly regenerate both complex differentiated structures which includes different types of tissues or organs (pygidium and parapodia, for example) and stem cells (posterior growth zone) [17, 18]. In this chapter, we will describe protocols routinely used to breed and maintain Platynereis in the laboratory and prepare biological materials required for regeneration studies. We will also introduce molecular biology and functional tools used to address key questions about regeneration.

Platynereis dumerilii . Pictures of juvenile and adult (male and female) worms. Males and females harbor specific morphological features linked to sexual metamorphosis, notably their color. While juveniles are mainly brownish, females are bright yellow, as they are full of oocytes. Males are white in their anterior part, as they are full of sperm, and red in their posterior part, due to extensive accessory blood capillaries. Morphological differences between juveniles and maturing worms are not limited to their color. Indeed, during sexual maturation, the whole intestine of the worm regresses and the trunk of the animal is progressively modified to become a bag full of gametes. In addition, mature worms harbor bigger and darker eyes compared to juveniles
2 Materials
Prepare all solutions using ultrapure autoclaved water (H2O). Prepare and store all reagents at room temperature (unless indicated otherwise).
2.1 Platynereis Worms Culture and Biological Material Production
-
1.
Filtered natural fresh seawater (NFSW): filter seawater with a 0.22 μm filter.
-
2.
Dried adult fish food (e.g., TetraMin flakes, Tetra).
-
3.
Dried fish fry food (e.g., Micron Growth Food, Sera).
-
4.
Live Chlorophyta algae (e.g., Tetraselmis marina).
-
5.
7.5% MgCl2: 75 g MgCl2 hexahydrated powder in 1 L H2O.
-
6.
1× Phosphatase buffer saline (1× PBS): 800 mL H2O, 8 g NaCL, 200 mg KCl, 1.44 g Na2HPO4, 240 mg KH2PO4, HCl to pH 7.4, H2O until volume is 1 L. Autoclave the solution.
-
7.
2 M NaOH: 80 g NaOH in 1 L H2O (see Note 1).
-
8.
16% Paraformaldehyde (PFA) stock solution: 80 g PFA, 450 mL 1× PBS (see Note 2), stir and heat at 60 °C until dissolution of the powder. Add droplets of 2 M NaOH until solution turns quite clear. Cool at room temperature, adjust pH to 7 with HCl, add 1× PBS until volume is 500 mL, filter using a 0.20 μm filtration column, aliquot as 12 mL in 15 mL tubes and store at −20 °C. Thaw aliquots at 60 °C, leftover can be stored at 4 °C and used within 2 weeks.
-
9.
0.1% Tw 1× PBS (1× PBST): 1 mL Tween 20 in 1 L 1× PBS (see Note 3).
-
10.
Fixation solution: 1 mL 16% PFA, 3 mL 1× PBST. Prepare fresh.
-
11.
Dehydration solution: 2 mL MeOH, 2 mL 1× PBST. Prepare fresh.
2.2 Whole Mount In Situ Hybridization and EdU Labelling
-
1.
25% rehydration solution: 30 mL MeOH, 10 mL 1× PBST. Prepare fresh.
-
2.
50% rehydration solution: 20 mL MeOH, 20 mL 1× PBST. Prepare fresh.
-
3.
75% rehydration solution: 10 mL MeOH, 30 mL 1× PBST. Prepare fresh.
-
4.
Digestion buffer: 50 μL 20 μg/μL Proteinase K (PK) in 25 mL 1× PBST. Prepare fresh.
-
5.
10× glycine: 4 g glycine in 200 mL of 1× PBS, adjust pH to 7.5 with HCl. Aliquot in 15 mL tubes, store at −20 °C.
-
6.
1× glycine: 5 mL 10× glycine, 45 mL 1× PBST. Prepare fresh.
-
7.
20× SSC: 175.3 g NaCl, 88.2 g Na3C6H5O7 in H2O, adjust pH to 7.5 with HCl, add H2O until volume is 1 L. Autoclave the solution.
-
8.
Heparin: 50 mg Heparin, 1 mL H2O. Prepare fresh.
-
9.
Hybridization buffer (HB): 25 mL formamide, 12.5 mL 20× SSC, 125 μL Heparin, 250 mg Torula (yeast) RNA powder, 50 μL Tween 20. Adjust to 50 mL with H2O. Store at −20 °C.
-
10.
Working solution of RNA probes: 1000 to 1500 ng of probes in 1 mL of HB. Store at −20 °C.
-
11.
4× wash buffer: 10 mL 20× SSC, 25 mL formamide, 50 μL Tween 20, 15 mL H2O. Prepare fresh.
-
12.
2× wash buffer: 5 mL 20× SSC, 50 μL Tween 20, 45 mL H2O. Prepare fresh.
-
13.
0.2× wash buffer: 500 μL 20× SSC, 50 μL Tween 20, 49.5 mL H2O. Prepare fresh.
-
14.
Blocking solution: 50 μL sheep serum, 1 mL 1× PBST. Prepare fresh.
-
15.
Anti-digoxigenin-alkaline phosphatase (AP) conjugate solution: 1 μL Anti-Digoxigenin-AP antibody, 3999 μL 1× PBST. Prepare fresh.
-
16.
1 M Tris: 121.14 g Tris–HCl, pH 9.5, 1 L H2O. Autoclave the solution.
-
17.
3 M NaCl: 87.75 g NaCl, 500 mL H2O. Autoclave the solution.
-
18.
1 M MgCl2: 101.75 g MgCl2, 500 mL H2O. Autoclave the solution.
-
19.
Staining buffer: 10 mL 1 M Tris–HCl pH 9.5, 10 mL 3 M NaCl, 5 mL 1 M MgCl2, 100 μL Tween 20, 75 mL H2O. Prepare fresh.
-
20.
1× Coloration solution: 1 μL nitro blue tetrazolium chloride (NBT), 3.5 μL 5-brom-4-chloro-3′-indolyphosphate p-toluidine salt (BCIP), 1 mL staining buffer. Prepare fresh.
-
21.
Stop solution: 100 mL 1 M Tris-HCl pH 7.5, 100 mL 3 M NaCl, 1 mL Tween 20, 799 mL H2O.
-
22.
Mounting solution: 90 mL of glycerol, 10 mL 1× PBST.
-
23.
10 mM EdU stock solution: 5 mg EdU powder, 2 mL DMSO. Mix well. Aliquot in 1 mL and store at −20 °C.
-
24.
5 μM EdU incorporation solution: 1 μL EdU stock solution, 1999 μL NFSW. Prepare fresh (see Note 4).
-
25.
EdU reaction solution: Follow specific manufacturer’s instructions (e.g., Invitrogen Click-iT® EdU Imaging Kits). Prepare fresh every time.
-
26.
Counterstaining solution: 1 μL of fluorescent nuclear-specific dye (e.g., Hoechst or DAPI); 999 μL of 1× PBST.
-
27.
DABCO antiphotobleaching solution: 625 mg N2(C2H4)3 (1,4-Diazabicyclo[2.2.2]octane, DABCO), 225 mL glycerol, 25 mL H2O. Stir for several hours until complete dissolution. Protect from light with aluminum foil. Store at 4 °C for months.
3 Methods
3.1 Platynereis Worms Culture
Platynereis dumerilii is a marine worm found worldwide in temperate seas [19]. Since decades researchers have no longer taken animals directly from the sea (except if information related to environmental cues are needed), as Platynereis’s full life cycle is completed easily and successfully in laboratory settings (see Note 5) [20]. To raise Platynereis worms, always rinse glassware with distilled water and never use detergents.
-
1.
Prepare a thermostatic room at 18 °C, with light control and equipped with shelves.
-
2.
Set up a daily illumination regime in the room to 8 h of full darkness and 16 h of full light.
-
3.
Place worms in a Tupperware-like box of middle size (30 × 20 × 10 cm) filled with 800 mL of NFSW.
-
4.
Switch on a low-light lamp 7 nights per month (on a 28-days monthly cycle) in the worm room during the whole night to artificially reproduce the moon illumination (see Note 6).
-
5.
3 days after the end of the moon illumination regime, collect sexually mature worms. Maturing males become red and white (full of sperm), while females become yellow (full of eggs) (Fig. 1). Both males and females display an increase of their eyes size as compared to juvenile worms.
-
6.
Transfer the collected male and female worms in separate boxes supplied in air by a pump, using a large pipette (see Note 7).
-
7.
Do not feed them as mature worms do not eat anymore (sexual maturation leads to a complete regression of the gut).
-
8.
Collect sexually mature worms following steps 5–7 every 2 days for 2 weeks (see Note 8).
-
9.
Monitor maturation boxes daily to identify mature animals ready to reproduce. They can easily be recognized as they start to swim.
-
10.
Collect one swimming sexually mature male and one swimming sexually mature female and put them in a 300 mL beaker filled with NFSW (see Note 9).
-
11.
Wait 5 to 10 min for the elegant nuptial dance of the worms to complete, several thousands of gametes to be released and fertilization to occur (see Note 10).
-
12.
When the fertilization is done, the female body looks empty and it is not swimming anymore, remove both the female and the male (see Note 11).
-
13.
Put the beaker in a thermostatic room at 18 °C.
-
14.
15 min after fertilization, check the beaker for a substantial egg jelly that covers the whole developing egg batch. This is a clear indicator of a successful fertilization.
-
15.
Discard unfertilized eggs, if any, from the content of the beakers using a pipette.
-
16.
24 h postfertilization (hpf), pour the content of the beaker through a 100 μm sieve. The net will retain small ciliated larvae that have developed from fertilized eggs.
-
17.
Rinse larvae three times with 1 L NFSW to carefully remove all the jelly.
-
18.
Transfer cleaned larvae back to a clean beaker by rinsing the sieve upside-down.
-
19.
Check them every 24 h during the three following days and remove dead individuals if needed.
-
20.
5 days postfertilization (dpf), start feeding larvae with 1 mL of live algae. Algae will form a mat at the bottom of the beaker embedding the larvae.
-
21.
Feed again larvae with 1 mL of live algae at 7 dpf and 9 dpf.
-
22.
At 10 dpf, using a pencil, carefully shift the algae mat containing the larvae to a box filled with 1 L of NFSW, 25 mL of algae and supplied in air by a pump.
-
23.
Until 30 dpf, fed twice a week with 5 mL of algae per box. Do not change the water during this period of time.
-
24.
Until 60 dpf, fed twice a week with a mix of 5 mL of algae and 0.2 mg of fry food per box. Do not change the water during this period of time.
-
25.
At 60 dpf, transfer up to 40 small worms per new box, filled with 800 mL of NFSW (see Notes 12 and 13). Change seawater every 2 weeks.
-
26.
Feed the worms three times per week, alternating between adult fish food (twice per week) and mashed spinach (once per week) (see Notes 14 and 15).
3.2 Production and Fixation of Samples at Specific Stages of Regeneration
To minimize, as much as possible, variability, notably due to the age and the size of the animals, strict procedures for worm selection and amputation should be followed.
-
1.
Select worms long of 30–40 segments and 3–4 month-old (see Note 16).
-
2.
Transfer selected worms to a beaker filled with 100 mL of 7.5% MgCl2.
-
3.
Wait 20 min until worms are anesthetized (see Note 17).
-
4.
Transfer anesthetized worms to a glass plate.
-
5.
Using a pencil, spread a worm on a glass plate (see Note 18).
-
6.
Under a dissecting scope, perform a sharp amputation to remove the 6 posterior-most segments of worms using a microknife (see Note 19, Fig. 2).
-
7.
Transfer to a clean box filled with NFSW at 18 °C and fed them normally three times per week.
-
8.
Monitor amputated worms daily under a dissecting scope to determine the current stage of regeneration (see Note 20, Fig. 2).
-
9.
Stage 1 (1 dpa): wound healing is achieved but no posterior outgrowth is present.
-
10.
Stage 2 (2 dpa): a small regenerated region (blastema) is visible with a notch, corresponding to the anus, in its central part.
-
11.
Stage 3 (3 dpa): presence of a larger blastema and two small anal cirri.
-
12.
Stage 4 (4 dpa): large blastema and long anal cirri are present.
-
13.
Stage 5 (5 dpa): presence of an indentation separating the differentiating pygidium from the anterior part of the regenerated region, faint segmental boundaries can be seen (see Note 21).
-
14.
To collect regenerating tissue, identify a worm in the desired stage of regeneration following steps 9 to 13.
-
15.
Amputate the worm two segments more anterior than the primary amputation plane following steps 2 to 7.
-
16.
Transfer the sample using a pencil in a 5 mL tube containing 4 mL of fixation solution.
-
17.
Incubate for 1.5 h while agitating on a rotating wheel.
-
18.
Rinse twice with PBST.
-
19.
Dehydrate for 20 min in dehydration solution at room temperature (RT) while agitating on a rotating wheel.
-
20.
Replace the solution by 4 mL of 100% MeOH for 1 h while agitating on a rotating wheel.
-
21.
Transfer sample to a 2 mL tube with 1 mL of 4 °C MeOH.
-
22.
Store at −20 °C.

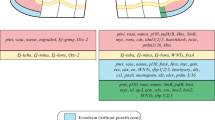
Regeneration stages. On the top of the figure is drawn a growing juvenile worms with its posterior growth zone (orange line and arrowhead) and, anterior to the growth zone, developing segments (purple asterisks) and, posterior to the growth zone, the pygidium characterized by the presence of two specific outgrowths, anal cirri (green arrowheads) and two large glands (gray circles). Amputation plane is represented by red dotted lines. The five stages of regeneration are depicted. At stage 1, wound healing is achieved. At stage 2, a small blastema composed of proliferating cells is formed and its size increases during subsequent stages. At stage 3, small anal cirri can be observed. They strongly extend at stage 4 and some signs of pygidium differentiation become obvious (e.g., presence of glands). At stage 5, pygidium differentiation has pursued and a few segments delimited by faint segmental boundaries are observed. Growth continues and an increasing number of differentiating segments (with obvious segmental boundaries and developing parapodia) can subsequently be observed
3.3 Whole Mount In Situ Hybridization
Whole mount in situ hybridization (WMISH) is the specific annealing of a labeled RNA probe to complementary sequence of a target mRNA in a fixed specimen, followed by detection and visualization of the nucleic acid hybrids [21] (see Note 22) (Fig. 3).
-
1.
Transfer fixed samples to large baskets placed in a box containing 40 mL of 100% MeOH, under a fume hood and with orbital agitation (see Note 23).
-
2.
Move baskets to a box containing 40 mL of the 25% rehydration solution.
-
3.
Incubate for 5 min at RT.
-
4.
Move baskets to a box containing 40 mL of the 50% rehydration solution.
-
5.
Incubate for 5 min at RT.
-
6.
Move baskets to a box containing 40 mL of the 75% rehydration solution.
-
7.
Incubate for 5 min at RT.
-
8.
Move baskets to a box containing 40 mL of 1× PBST.
-
9.
Incubate for 5 min at RT.
-
10.
Repeat steps 8 and 9.
-
11.
Move baskets to a box containing 25 mL of digestion buffer.
-
12.
Incubate for 10 min at RT without agitation.
-
13.
Move baskets to a box containing 50 mL of 1× glycine.
-
14.
Incubate for 1 min.
-
15.
Move baskets to a box containing 50 mL of fixation solution.
-
16.
Incubate for 20 min.
-
17.
Repeat steps 8 and 9, five times.
-
18.
Transfer samples from a large basket to a pillbox with 2 mL 1× PBST.
-
19.
Using a pipette, place up to 10 samples per small basket (one basket per probe) under a dissecting scope (see Notes 24 and 25).
-
20.
Transfer small baskets to 2 mL tubes containing 1 mL of HB using forceps.
-
21.
Incubate at 65 °C for 1 h.
-
22.
Denature working solution of RNA probes at 80 °C during 10 min in a wet bath.
-
23.
Transfer small baskets to 2 mL flat bottom tubes containing 300 μL of denaturated probe.
-
24.
Place in an oven at 65 °C with orbital agitation (100 rpm) for 16 h (see Notes 26 and 27).
-
25.
Transfer small baskets to 2 mL tubes containing 1 mL of 4× wash solution.
-
26.
Incubate for 30 min at 65 °C (see Note 28).
-
27.
Similarly perform in 2 mL tubes a second wash of 30 min at 65 °C in new 4× wash buffer.
-
28.
Similarly perform in 2 mL tubes another two 15 min washes in 2× wash buffer at 65 °C, followed by two more 30 min washes in 0.2× wash buffer.
-
29.
Transfer small baskets to 2 mL tubes containing 800 μL of blocking solution.
-
30.
Incubate for 1 h at RT with orbital agitation.
-
31.
Transfer small baskets to 2 mL tubes containing 800 μL of AP conjugate solution.
-
32.
Incubate for 1 h at RT with orbital agitation (see Note 29).
-
33.
Transfer the samples to 12 wells plates with 1 mL of staining buffer.
-
34.
Add 1 mL of 1× coloration solution to each well (see Note 30).
-
35.
Allow blue staining to appear at RT (see Note 31), which can take from few hours to few days depending on probes (see Note 32).
-
36.
Check coloration every 30 min under a dissecting scope.
-
37.
Transfer samples to a 2 mL tube containing 2 mL of stop solution to arrest the reaction.
-
38.
Replace stop solution with 2 mL of 1× PBST.
-
39.
Incubate 5 min while agitating on a rotating wheel.
-
40.
Repeat 4 times steps 36 and 37.
-
41.
Transfer samples to a 2 mL tube containing 2 mL of mounting solution.
-
42.
Agitate on a rotating wheel at 4 °C overnight.
-
43.
Select samples to be mounted under a dissecting scope.
-
44.
Place 2 or 3 samples on a slide with 20 μL of mounting solution.
-
45.
Place a small piece of clay under each corner of a coverslip and cover samples with it.
-
46.
Press the corners of coverslip to flatten them (see Note 33).
-
47.
Image the samples under a bright field microscope (Fig. 3).

Whole mount in situ hybridization. Whole mount in situ hybridization for the genes whose name is indicated are shown for two posterior regeneration stages, stage 3 (a–c) and stage 5 (d–f). All panels are ventral views (anterior is up). Amputation plane is represented by yellow dotted lines. In a and d, blue arrowheads point to expression of Pdum-pax6 in two longitudinal rows of neuroectodermal cells which will give rise to ventral neurons of the ventral nerve cord. In b is shown the expression of Pdum-neurogenin in a large number of neuronal precursor cells of the both the central and peripheral nervous system. In c, brown arrows point to the expression of Pdum-cdki1a in internal cells located in the anal region. In e, green arrowheads point to segmental ectodermal stripes of cells expressing Pdum-prdm3/16 which is also expressed in cells of the posterior growth zone (red arrowheads). These cells also express Pdum-evx (red arrowheads in f)
3.4 EdU Labelling for Investigating Cell Proliferation During Platynereis Regeneration
5-ethynyl-2′-deoxyuridine (EdU) is a nucleoside analog that is widely used to detect cells that are in the S-phase of their cell cycle in various species (see Note 34). In Platynereis, EdU labeling has been used to study cell proliferation in whole mount animals during development , postembryonic growth, and regeneration (e.g., [17, 18, 22]) (Fig. 4).
-
1.
Select live worms at desired regeneration stages.
-
2.
Place one worm per well in 12-well plastic plates filled with 2 mL of EdU incorporation solution.
-
3.
Incubate for 1 h at RT (see Note 35).
-
4.
Discard EdU incorporation solution.
-
5.
Rinse each well with 10 mL of NFSW.
-
6.
Repeat 2 times step 5 (see Note 36).
-
7.
Follow the aforementioned fixation procedure (Subheading 3.2, steps 16 to 22) (see Note 37).
-
8.
Follow the aforementioned rehydration and digestion procedures (Subheading 3.3, steps 1 to 17).
-
9.
Place up to 10 samples in the bottom of a 1.5 mL tube filled with 1 mL 1× PBST.
-
10.
Remove the 1× PBST using a pipette.
-
11.
Add 300 μL of EdU reaction solution in the tube.
-
12.
Incubate 1 h in the dark at RT.
-
13.
Remove EdU reaction solution.
-
14.
Add 1 mL of 1× PBST in the tube.
-
15.
Incubate for 5 min at RT in the dark under orbital agitation.
-
16.
Remove 1× PBST.
-
17.
Repeat 2 times steps 13 to 15.
-
18.
Add 800 μL of counterstaining solution to the tube.
-
19.
Incubate overnight at 4 °C in the dark under orbital agitation.
-
20.
Remove counterstaining solution.
-
21.
Perform 5 times steps 14 to 16.
-
22.
Add 2 mL of DABCO anti-photobleaching solution.
-
23.
Incubate overnight at 4 °C in the dark while agitating on a rotating wheel.
-
24.
Follow the aforementioned mounting procedure (Subheading 3.3, steps 42 to 45).
-
25.
Observe your samples under epifluorescent or confocal microscope (Fig. 4).

EdU Labeling. Confocal image of the posterior part of a stage 3 worm (3 dpa) incubated in 5 μM EdU for 1 h before fixation. Hoechst counterstaining has been performed and allows to visualize all nuclei (in blue). EdU labeling is shown in red. Amputation plane is represented by yellow dotted lines. Anterior is up
3.5 Pharmacological Treatments for Functional Studies During Platynereis Regeneration
Performing functional studies during postembryonic developments used to be challenging for many organisms in which genetic tools are not easily or fully mastered. One way to alter or modify various molecular signaling pathways or cellular mechanisms is to soak regenerating animals in specific pharmacological inhibitors or activators (see Notes 38 and 39). An initial and crucial step consists in defining the efficient concentration that induces defects in the regeneration process (e.g., morphological abnormalities) and/or in its timing, without (or with minimal) toxic effects (i.e., with a minimal number of dead or autotomized animals (see Note 40)).
-
1.
Define three to four different inhibitor/activator concentrations to be tested (see Note 41).
-
2.
Prepare fresh inhibitor/activator solutions and control solution (see Note 42).
-
3.
Define your negative control condition (see Note 43).
-
4.
Follow the aforementioned procedure for worm amputations (Subheading 3.2, steps 1 to 6).
-
5.
Place amputated worms in 12-wells plates, one worm per well filled with 2 mL of solution.
-
6.
Use at least 12 worms per concentrations and 12 worms as controls.
-
7.
Incubate the worms for 24 h.
-
8.
Remove the solution from each well.
-
9.
Add 2 mL of 7.5% MgCl2 per well.
-
10.
Observe individually each worm under a dissecting scope.
-
11.
Record their regeneration score according to the regeneration stage that has been reached (stages 1–5) following the procedure describe previously (Subheading 3.2, steps 9 to 13) (see Notes 44 and 45).
-
12.
Remove the 7.5% MgCl2 solution in each well.
-
13.
Add 2 mL of fresh inhibitor / activator / control solution.
-
14.
Repeat steps 7 to 12 for 3 more days (see Note 46).
-
15.
On day 5, repeat steps 7 to 10 and discard worms.
-
16.
Based on the results obtained, choose concentration with the greatest regeneration defects and the lowest toxicity.
-
17.
Following the aforementioned procedure of pharmacological treatment and scoring (steps 2 to 14), repeat at least twice the regeneration experiment with the selected concentration using at least 24 worms per experiment (see Note 47).
-
18.
Perform statistical analysis of the obtained data (see Note 48).
4 Notes
-
1.
Pay attention to the exothermic reaction induced by mixing sodium hydroxide and water. Use borosilicate glasses free of any scratches, chips or breaks.
-
2.
PFA is highly toxic. PFA solution must be prepared under a fume hood. It is very important to wear lab coat, spectacles and gloves during the whole procedure.
-
3.
Tween 20 is a highly viscous product. Cut the end of the tip before pipetting and mix well.
-
4.
Let EdU stock solution thaw at room temperature and be aware that this can last about 2 h.
-
5.
If you want to establish your own culture, worm larvae are easily shared by the Platynereis community.
-
6.
Be extremely careful to respect illumination and moonlight regimes, as any change in these regimes have long term negative effects on the worm culture; prepare a worm room schedule and follow it meticulously.
-
7.
Some worms in the course of maturation may harbor an orange color and it may be difficult to morphologically categorized them as females or males yet. In such a case, place the “undetermined” worms either in a separated box (with an air supply) or in the “males” box. Do not place them in the “females” box as if one of them turns to be a male, it will release gametes and induce reproduction of most or all of the females of this box.
-
8.
Illumination regime triggers sexual maturation for 2 weeks per month. The other 2 weeks of the month, there is no sexually mature worms available. Set up two culture rooms with alternate illumination regime in order to have mature animals all month long.
-
9.
Every month, perform fertilization to replenish the worm culture after the sexual maturation period, which leads to the death of many animals.
-
10.
When unfertilized eggs are released by a female, they have to be fertilized almost immediately to induce proper embryogenesis. If males are late to release sperm, you can press their body to manually induce release of sperm. Do that directly in the beaker, trap the male against the beaker wall with your finger and press anywhere on the male body until sperm is ejected. This procedure is not possible for females.
-
11.
Males are able to release sperm a couple of times, and thus to fertilize several batch of eggs before dying.
-
12.
Avoid putting more than 40 worms in a box, otherwise they will not grow enough before maturation and few gametes will be produced per animal.
-
13.
While fertilization should be done in NFSW, adults/juveniles worms (>2-month-old) can be raised in a mix of 50% NFSW and 50% artificial seawater.
-
14.
Use organic spinach as Platynereis worms are very sensitive to pesticides.
-
15.
Alternative feeding using powdered spirulina (1.0 g/L) is possible [23].
-
16.
Exclude worms that are already in the process of regeneration due to accidental injury as they may regenerate differently if amputated a second time.
-
17.
Properly anesthetized worms are not moving anymore (while contractions of the gut are still present), even when put under the light of the dissecting scope. If this is not the case, increase anesthesia incubation time by a couple of minutes.
-
18.
There is no need to hold them, they will stay in place if well anesthetized.
-
19.
Pay attention to perform amputation between two segments and perpendicularly to the body axis. Avoid amputating parapodia from one side and not the other, as this will affect posterior regeneration timing.
-
20.
More than 90% of amputated worms follow this timeline, but some worms from a specific box/batch are occasionally delayed for yet undetermined reason.
-
21.
The regeneration process is finished at this stage; the next steps are considered as posterior elongation [17], that is, production of new segments from the regenerated posterior growth zone.
-
22.
As an extension of the WMISH procedure, a technique named whole-body gene expression pattern registration allows the rigorous comparison of expression patterns, at a specific developmental stage, of an animal which harbors invariant development . It requires a standardized reference template at the desired developmental stage and allows the production of a standardized digital anatomical atlases of gene expression patterns, at near-cellular resolution [24].
-
23.
Home-made large baskets are made of a plastic cylinder (diameter: 4 cm) with a heat-sealed 100 μM mesh at its bottom. This allows to incubate samples in solution while still contained in the basket and to move them easily from one solution to another, by moving the basket itself using forceps. Place all samples of the same stage in a same basket. Annotate each basket to avoid confusion. Consider to use five samples per stage per gene.
-
24.
Home-made small baskets are composed of the largest part of a P1000 tip (8 mm long) with a heat-sealed 100 μM mesh at its bottom.
-
25.
You can mix up to 2 stages within a same small basket (5 samples per stage, same probe). You can perform easily a WMISH experiment with up to 30 small baskets.
-
26.
Hybridization temperatures as well as probe concentrations can be modulated to optimize coloration intensity and signal to background ratio.
-
27.
After use, small baskets and flat bottom tubes are treated with 0.5 M NaOH overnight with agitation then carefully washed and rinsed with H2O, so they can be reused. Regular tubes are washed with H2O only and also reused.
-
28.
Pay attention to perform the first four washes at 65 °C.
-
29.
Alternatively, steps 29 and 31 can be done at 4 °C overnight.
-
30.
Pay attention that staining solution should be pH 9.5, otherwise the enzymatic reaction will not work.
-
31.
Staining can be slowed down by putting plates at 18 °C and even 4 °C.
-
32.
It is possible to stop coloration for the night or the week-end and to resume it later. To do that, remove all the 1× coloration solution, rinse with agitation with 1 mL of stop solution, then replace by another 1 mL of stop solution and leave in the dark at 4 °C. To resume coloration, make a wash with staining buffer before adding 1× coloration solution and resuming on step 35.
-
33.
Depending of the thickness of the sample (notably the nonamputated anterior part), you will have to press more or less on the coverslip. Carefully check flattening of the sample in order not to smash it.
-
34.
Unlike other labeling methods of S-phase cells using modified nucleosides, such as Bromodeoxyuridine (BrdU), EdU labeling has the advantage not to be antibody-based and therefore does not require DNA denaturation (usually achieved by treatment with a strong acid, often HCl) for its detection, which is harmful for tissue integrity and may therefore hamper further microscopic observations.
-
35.
Longer incubation times can be envisioned to obtained more labeled (EdU+) cells. For regenerating worms, we used incubation times up to 5 h.
-
36.
Fixation can be done immediately after incorporation or worms can be kept for defined period of time in NFSW (without EdU) before fixation. In the latter case, a so-called pulse and chase experiment is performed.
-
37.
Samples can be stored at −20 °C or directly used for the subsequent steps.
-
38.
Cell permeable small molecule inhibitors of various signaling pathways were also successfully used during Platynereis embryonic development [22, 25, 26]. One advantage to use pharmacological inhibitors relies on the fact they allow to specifically target the period of inhibition while avoiding interferences with earlier functions. Always try to use inhibitors at the lowest concentration producing significant and reproducible effects, to minimize off-target effects and toxicity.
-
39.
To further characterize pharmacological treatment effects on regeneration, you can perform EdU labeling to assess cell proliferation and WMISH using specific markers (for example of particular tissue or cell types) on treated and control worms at different time points after amputation, as described in Subheadings 3.3 and 3.4.
-
40.
Autotomy or self-amputation, is the discarding of a portion of the body containing several segments. This process usually occurs when animals are facing stressful conditions (or are mechanically damaged). Autotomy can happen in normal culture conditions, at a low rate. When scoring worms during treatments, autotomy is easily visible as worms are separated in at least two fragments, the one bearing the head may start to regenerate again, while the other will not.
-
41.
This is done based on previous experiments during regeneration or on larvae, or using information from the literature on other organisms.
-
42.
Depending on the type of chemical used, stock solutions have to be made by dissolving it in a specific chemical product. Dimethylsulfoxide (DMSO) is widely used to dissolve various drugs. Working solutions are then made from this stock solution diluted in NFSW. Control solution is composed of NFSW supplemented by the chemical product used to initially dissolve the chemical to produce the stock solution.
-
43.
Negative control worm condition consists in amputated worms soaked in control solution that contains the chemical used to dissolve the drug (typically DMSO) at a concentration corresponding to that of the solution with the highest concentration. In general, DMSO is not affecting regeneration nor development in most organisms, but this needs to be confirmed. Perform an initial experiment with a batch of animals soaked in NFSW and a batch of animals in NFSW with DMSO, follow them individually every day and compare regeneration scores obtained from the two series of animals.
-
44.
Some worms may show a morphology that is intermediate between that of two successive stages of regeneration and therefore they can be scored as 1.5, 2.5, 3.5, and 4.5.
-
45.
List any morphological abnormalities that may be visible on treated worms. Pay attention and count every day the number of worms that may have died or performed autotomy during the treatment, as these are indicators of toxicity and therefore important parameters to help choosing the most efficient concentration. To further test a possible toxicity of the inhibitors, you can also perform additional experiments: (1) treat nonamputated worms 5 days with the inhibitor (at the different tested conditions) and count the number of autotomized or dead worms, if any; (2) treat amputated worms with the inhibitors for 5 days, then place them in NFSW and check for potential regeneration recovery at 7, 10, and 15 dpa.
-
46.
Drug solutions have to be replaced every 24 h to maintain their activity for the whole experiment.
-
47.
When possible, use at least two different drugs to confirm effects on regeneration and its timing.
-
48.
For inhibitor treatments, perform two-way ANOVA multiple comparisons between control versus treated worms per scoring day. Comparisons between different inhibitor concentrations are also interesting to perform.
References
Galliot B (2012) Hydra, a fruitful model system for 270 years. Int J Dev Biol 56(6-8):411–423. https://doi.org/10.1387/ijdb.120086bg
Marques IJ, Lupi E, Mercader N (2019) Model systems for regeneration: zebrafish. Development 146(18):dev167692. https://doi.org/10.1242/dev.167692
Gazave E, Rottinger E (2021) 7th Euro Evo devo meeting: report on the “evolution of regeneration in Metazoa” symposium. J Exp Zool B Mol Dev Evol 336(2):89–93. https://doi.org/10.1002/jez.b.22897
Ferrier DE (2012) Evolutionary crossroads in developmental biology: annelids. Development 139(15):2643–2653. https://doi.org/10.1242/dev.074724
Brusca RC, Brusca GJ (eds) (2003) Invertebrates, 2nd edn. Sinauer Associates, Sunderland
Ozpolat BD, Bely AE (2016) Developmental and molecular biology of annelid regeneration: a comparative review of recent studies. Curr Opin Genet Dev 40:144–153. https://doi.org/10.1016/j.gde.2016.07.010
Bely AE (2014) Early events in annelid regeneration: a cellular perspective. Integr Comp Biol 54(4):688–699. https://doi.org/10.1093/icb/icu109
Boilly B (1969) Sur l’origine des cellules régénératrices chez les annélides polychètes. Archives de Zoologie expérimentale et générale 110(1):127–143
Hyman LH (1940) Aspects of regeneration in annelids. Am Nat 74:513–527
Boilly B (1969) Experimental study of the localization, by relation to the amputation plan, of the source of mesodermal regeneration cells in an annelid polychaete (Syllis amica Quatrefages). J Embryol Exp Morphol 21(1):193–206
Boilly B, Boilly-Marer Y, Bely AE (2017) Regulation of dorso-ventral polarity by the nerve cord during annelid regeneration: a review of experimental evidence. Regeneration (Oxf) 4(2):54–68. https://doi.org/10.1002/reg2.78
Bely AE, Zattara EE, Sikes JM (2014) Regeneration in spiralians: evolutionary patterns and developmental processes. Int J Dev Biol 58(6–8):623–634. https://doi.org/10.1387/ijdb.140142ab
Audouin JV, Milne Edwards H (1833) Classification des annélides, et description de celles qui habitent les côtes de la France. Ann Sci Nat 29:195–269
Zantke J, Ishikawa-Fujiwara T, Arboleda E, Lohs C, Schipany K, Hallay N, Straw AD, Todo T, Tessmar-Raible K (2013) Circadian and circalunar clock interactions in a marine annelid. Cell Rep 5(1):99–113. https://doi.org/10.1016/j.celrep.2013.08.031
Raible F, Tessmar-Raible K (2014) Platynereis dumerilii. Curr Biol 24(15):R676–R677. https://doi.org/10.1016/j.cub.2014.06.032
Williams EA, Jekely G (2016) Towards a systems-level understanding of development in the marine annelid Platynereis dumerilii. Curr Opin Genet Dev 39:175–181. https://doi.org/10.1016/j.gde.2016.07.005
Gazave E, Behague J, Laplane L, Guillou A, Preau L, Demilly A, Balavoine G, Vervoort M (2013) Posterior elongation in the annelid Platynereis dumerilii involves stem cells molecularly related to primordial germ cells. Dev Biol 382(1):246–267. https://doi.org/10.1016/j.ydbio.2013.07.013
Planques A, Malem J, Parapar J, Vervoort M, Gazave E (2019) Morphological, cellular and molecular characterization of posterior regeneration in the marine annelid Platynereis dumerilii. Dev Biol 445(2):189–210. https://doi.org/10.1016/j.ydbio.2018.11.004
Simakov O, Larsson TA, Arendt D (2013) Linking micro- and macro-evolution at the cell type level: a view from the lophotrochozoan Platynereis dumerilii. Brief Funct Genomics 12(5):430–439. https://doi.org/10.1093/bfgp/els049
Fischer A, Dorresteijn A (2004) The polychaete Platynereis dumerilii (Annelida): a laboratory animal with spiralian cleavage, lifelong segment proliferation and a mixed benthic/pelagic life cycle. Bioessays 26(3):314–325. https://doi.org/10.1002/bies.10409
Baumgart E, Schad A, Grabenbauer M (2001) In situ hybridization: general principles and application of Digoxigenin-labeled cRNA for the detection of mRNAs. In: Beesley JE (ed) Immunocytochemistry and in situ hybridization in the biomedical sciences. Birkhäuser, Boston, MA, pp 108–137. https://doi.org/10.1007/978-1-4612-0139-7_6
Demilly A, Steinmetz P, Gazave E, Marchand L, Vervoort M (2013) Involvement of the Wnt/beta-catenin pathway in neurectoderm architecture in Platynereis dumerilii. Nat Commun 4:1915. https://doi.org/10.1038/ncomms2915
Kuehn E, Stockinger AW, Girard J, Raible F, Ozpolat BD (2019) A scalable culturing system for the marine annelid Platynereis dumerilii. PLoS One 14(12):e0226156. https://doi.org/10.1371/journal.pone.0226156
Asadulina A, Panzera A, Veraszto C, Liebig C, Jekely G (2012) Whole-body gene expression pattern registration in Platynereis larvae. EvoDevo 3(1):27. https://doi.org/10.1186/2041-9139-3-27
Gazave E, Lemaître Q, Balavoine G (2017) The notch pathway in the annelid Platynereis: insights into chaetogenesis and neurogenesis processes. Open Biol 7(2):160242
Zantke J, Bannister S, Rajan VB, Raible F, Tessmar-Raible K (2014) Genetic and genomic tools for the marine annelid Platynereis dumerilii. Genetics 197(1):19–31. https://doi.org/10.1534/genetics.112.148254
Acknowledgments
Work in our team is supported by funding from Labex “Who Am I” laboratory of excellence (No. ANR-11-LABX-0071) funded by the French Government through its “Investments for the Future” program operated by the Agence Nationale de la Recherche under grant No. ANR-11-IDEX-0005-01, Centre National de la Recherche Scientifique, INSB (grant Diversity of Biological Mechanisms), Agence Nationale de la Recherche (grant TELOBLAST no. ANR-16-CE91-0007). The authors warmly thank all current and past members of the “Stem cells, Development and Evolution” team at the Institut Jacques Monod, Paris, France.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this protocol

Cite this protocol
Vervoort, M., Gazave, E. (2022). Studying Annelida Regeneration Using Platynereis dumerilii . In: Blanchoud, S., Galliot, B. (eds) Whole-Body Regeneration. Methods in Molecular Biology, vol 2450. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-2172-1_11
Download citation
DOI: https://doi.org/10.1007/978-1-0716-2172-1_11
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-2171-4
Online ISBN: 978-1-0716-2172-1
eBook Packages: Springer Protocols