
Abstract
Although MRD monitoring by the classic polymerase chain reaction (PCR) approach is a powerful outcome predictor, about 20% of mantle cell lymphoma (MCL) and 50% of follicular lymphoma (FL) patients still lack a molecular marker and are thus resulting not eligible for MRD monitoring. Targeted locus amplification (TLA), a new NGS technology, has been revealed as a feasible marker screening approach able to identify uncommon B-cell leukemia/lymphoma 1 (BCL1) and B-cell leukemia/lymphoma 2 (BCL2) rearrangements in MCL and FL cases defined as having “no marker” by the classic PCR approach.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
- Mantle cell lymphoma
- Follicular lymphoma
- Immunoglobulin
- Translocations
- Molecular marker
- Next-generation sequencing
- Targeted locus amplification
1 Introduction
Mantle cell lymphoma (MCL) and follicular lymphoma (FL) are non-Hodgkin lymphomas with an aggressive and indolent clinical course, respectively [1]. Despite the high rate of success of modern immunotherapies in the treatment of these patients, relapsing disease at variable time from disease presentation is still the rule, and the consequent acquisition of more aggressive behavior overtime is common [2, 3]. Therefore, it is crucial to track the disease course by highly sensitive minimal residual disease (MRD) approaches, in order to assess both the effective treatment efficacy and to early identify patients at risk of relapse [4]. In the last decade several prospective clinical trials revealed MRD as a strong outcome predictor both in MCL and FL [5,6,7,8].
Chromosomal translocations, which juxtapose oncogenes to the immunoglobulin (IG) regions, are ideal molecular markers for MRD in mature B-cell lymphoproliferative diseases. In detail, MCL and FL are characterized by chromosomal translocations that transpose the B-cell leukemia/lymphoma 1 (BCL1) and B-cell leukemia/lymphoma 2 (BCL2) genes, respectively, near the IG heavy chain (IGH ) regions; t(11;14) and t(14;18) result in the overexpression of cyclin D1 (CCND1) and BCL2 proteins and to the constitutive activation of proliferative and antiapoptotic cellular pathways, respectively [9].
Actually, fluorescence in situ hybridization (FISH) revealed that almost 90% of MCL and 80% of FL harbor the translocation in the diagnostic tissues, but this technology is not sensitive enough to monitor MRD in follow-up samples [7, 10].
On the other hand, polymerase chain reaction (PCR), able to detect up to one clonal cell among 100,000 analyzed cells, can overcome this limitation. Currently, due to its high international standardization level, it represents the gold-standard approach employed for MRD monitoring purposes in MCL and FL [7, 11].
The well-known t(11;14) and t(14;18) breakpoints concern, respectively, (a) major translocation cluster (MTC), involving the BCL1 region at 11q13 and the IGH locus at 14q32; (b), major breakpoint region (MBR), and minor cluster region (mcr), occurring between BCL2 gene at 18q21 and the 14q32.3 IGH region. In FL MBR is most frequently involved (80% of the identified rearrangements), while mcr is less frequently identified (~15%) [8]; some rare (<5% of cases) “minor” breakpoints involving regions 3′ and 5′ of the MBR and mcr and named 3′MBR, 5′mcr, and distal MBR have also been described [12].
Moreover, between the juxtaposed chromosomic regions, nucleotides, also called N insertions, are randomly added, establishing the tumor “fingerprint-like sequences” essential for MRD monitoring allele-specific oligonucleotide (ASO) assay design [13].
Although classic PCR approaches for marker screening and MRD monitoring have been defined and standardized within the EuroClonality-NGS and EuroMRD working groups (/www.euroclonality.org), about 20% of MCL and 50% of FL patients still lack a molecular marker, thus resulting not eligible for MRD monitoring.
In the last few years, IGH amplicon-based next-generation sequencing (NGS) applications successfully provided new scenarios in several hematological diseases, such as acute lymphoblastic leukemia [14, 15], multiple myeloma [16, 17], and different lymphomas [18, 19] describing those NGS approaches as feasible tools for marker identification and MRD monitoring, allowing clinical correlations in large patient populations.
Also NGS capture panels appeared to be useful in the detection of multiple molecular targets, but their limited sensitivity hampers the application to the MRD context [20, 21].
Targeted locus amplification (TLA), a NGS-based technology firstly developed in 2014 by Cergentis B.V., allows the detection of structural variants not identified by classic PCR methods. TLA protocol differs from NGS capture approaches: actually, it is based on the principle of physiological cross-linking of genome regions placed in physical proximity. Moreover, employing the targeted enrichment of short, locus-specific sequences, it results in the sequencing of all single nucleotide variants (SNVs) and structural variants, such as chromosomal translocations [22, 23].
Since its first publication, TLA approach has been employed in different contexts such as transgene detection, vector design, and novel SNVs identification, thus resulting in a promising technology also for onco-hematology [24,25,26]. In this context, the application of a multiplex TLA , as a marker screening tool, showed promising results in acute leukemia through detection of cryptic rearrangements and multiple (un)known translocated genes involved in leukemia pathogenesis [27, 28].
Recently the implementation of TLA targeting the fusion partners of the IGH enhancer described the presence of novel, uncommon BCL1 and BCL2 rearrangements in MCL and FL patients lacking a MRD molecular marker by classic PCR marker screening approach [29]. The newly identified TLA rearrangements allowed the design of highly sensitive ASO MRD assays (up to 1E-05), thus priming the potential use of this NGS technology to increase the number of lymphoma patients eligible for MRD monitoring in clinical trials.
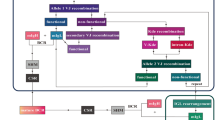
Here we provide a detailed description of the TLA protocol as marker screening tool in MCL and FL patients, followed by an ASO MRD assay based on the TLA sequence (Fig. 1).

TLA library preparation workflow
2 Materials
2.1 Reagents and Kits
-
Red blood cell lysis buffer (NH4Cl).
-
0.9% NaCl solution.
-
Maxwell® RSC Blood DNA Kit (Promega).
-
Go Taq G2 Flexi DNA Polymerase (Promega).
-
Agarose gel electrophoresis.
-
TAE 1×.
-
Targeted locus amplification gDNA library prep kit (Cergentis).
-
QuantiFluor® ONE dsDNA System (Promega).
-
AMPure XP Beads (Beckman Coulter).
-
80% ethanol.
-
MilliQ.
-
2-propanol.
-
Nextera DNA Flex Library Prep kit (Illumina).
-
Nextera DNA CD Indexes (24 Indexes, 24 sample-Illumina) (Illumina).
-
High sensitivity D1000 ScreenTape (Agilent).
-
NaOH.
-
MiSeq Illumina v3 reagent kit (600 cycles).
2.2 Instruments and Software
-
50 ml conical tubes.
-
Centrifuge with conical tube adapters and minispin centrifuge.
-
1–1.5 ml vials.
-
2 ml reaction tubes (with safe lock caps).
-
0.5 ml PCR tubes.
-
Heating block.
-
Maxwell RSC (Promega).
-
NanoDrop2000 Spectrophotometer (Thermo Scientific).
-
Thermal cycler with heated lid.
-
Electrophoresis Systems.
-
Quantus fluorometer (Promega).
-
Magnetic rack fitting with 1.5–2 ml tubes.
-
2200 Tapestation system (Agilent).
-
MiSeq sequencer (Illumina).
-
PC, software for genome alignment and primer design.
3 Methods
3.1 Mantle Cell Lymphoma and Follicular Lymphoma Cell Collection
Collect bone marrow (BM) and peripheral blood (PB) samples in EDTA vacutainers, ranging from 2 to 7 ml and from 7 to 14 ml for BM and PB, respectively. Next, the red blood cell (RBC) lysis procedure is carried out to collect total white blood cells (WBC), as follows:
-
1.
Mix 10–15 ml of BM or PB with lysis buffer 1× in 50 ml conical tubes up to a final volume of 45 ml.
-
2.
Invert to mix and incubate at room temperature (RT) for 10–15 min.
-
3.
Centrifuge at 226 × g for 15 min at RT.
-
4.
Discard the supernatant and add lysis buffer up to a final volume of 20 ml.
-
5.
Centrifuge at 226 × g for 10 min at RT.
-
6.
Decant the supernatant and resuspend the pellet with 0.9%NaCl solution.
-
7.
Aliquot 5–10 × 106 cells in 1–1.5 ml vials.
-
8.
Centrifuge at 6440 × g for 1 min and discard the supernatant.
-
9.
Store the samples at −80 °C.
3.2 gDNA Extraction and Quality Control
gDNA is extracted from 5–10 × 106 BM and PB dry cell pellets (see Note 1). High purity gDNA is obtained using semiautomated or automated DNA extraction procedures, avoiding DNA cross-contamination among the samples. Here we describe the Maxwell® Rapid Sample Concentrator (RSC) Blood protocol.
-
1.
Set the heating block at 56 °C.
-
2.
Resuspend 5–10 × 106 dry cell pellet using 300 μl of 0.9% NaCl solution or PBS.
-
3.
Add 300 μl of Lysis Buffer and vortex for 10 s.
-
4.
Add 30 μl of Proteinase K Solution and vortex.
-
5.
Incubate at 56 °C for 20 min.
-
6.
During sample incubation, prepare cartridges as described in the instrument operating manual.
-
7.
Load the samples in the cartridge and proceed with the gDNA extraction program.
After extraction, gDNA quantity (ng/μl) and quality (OD ratio A260/A280 and A260/A230) are evaluated by the NanoDrop2000 Spectrophotometer (Thermo Scientific, Waltham, MA, USA). gDNA is stored at 4 °C or −20 °C until library preparation.
Next, a control gene (P53 exon 8) amplification is performed to further qualitatively check the gDNA [30].
-
1.
Prepare the PCR mix and set the thermal cycler as described in Tables 1 and 2, respectively.
-
2.
Prepare the reaction mix on ice.
-
3.
Add 100–500 ng as input gDNA.
-
4.
To each run add a positive gDNA from a healthy subject and a no-template control (NTC).
Run the PCR products on a 2% agarose gel. The P53 exon 8 amplification signal should appear as a 150 bp band; samples without any signal should not be considered for TLA library preparation.
3.3 Targeted Locus Amplification (TLA) Library Preparation
TLA consists of different steps that allow gDNA cross-linking and circularization, followed by IGH target enrichment. The protocol outline takes 4 workdays; for more detailed information and technical support, please refer to Cergentis (www.Cergentis.com).
Day 1:
-
Assembly and fixation: gDNA is assembled and fixed to fold, to cross-link and to connect regions placed in a very close proximity through the genome.
-
First enzymatic digestion, ligation, and reverse cross-linking: long cross-linked genomic templates originating from the same locus are obtained using restriction enzymes, ligase, and proteinase.
Day 2:
-
Second enzymatic digestion and ligation: the large gDNA fragments are digested to obtain molecules suited for PCR amplification and then circularized.
Day 3:
-
TLA PCR: the circularized gDNA molecules are amplified using the IGH enhancer complementary primer.
Day 4:
-
TLA library indexing: the TLA PCR products are fragmented and tagged with adapter sequences using the Bead-Linked Transposomes (BLT) kit.
-
TLA library sequencing on an Illumina platform .
3.3.1 Assembly and Fixation
At least 5 μg of gDNA (see Note 2) is required for TLA library preparation
-
1.
Measure the gDNA concentration using Quantus fluorometer (see Note 3) and dilute the sample in the concentration range of 50–100 ng/μl (see Note 4).
-
2.
Add, in two separate 2 ml tubes, 290 μl Assembly Buffer (AB), 2.5 μg DNA, and dilution buffer (DB) until a 900 μl final volume.
-
3.
Add 100ul Assembly Mix (AM) and incubate 15 min at 37 °C.
-
4.
Add 90 μl Fixation Buffer (FB) and incubate exactly 10 min at 37 °C.
-
5.
Add 90 μl Quencher Buffer (QB) and 800 μl thoroughly mixed AMPure XP Beads and incubate 15 min at RT.
-
6.
Place the tubes on a magnetic rack until the beads are separated.
-
7.
Discard the supernatant and add 1800 μl of fresh 80% ethanol to each tube and wait until the beads are completely separated.
-
8.
Repeat step 7.
-
9.
Remove residual ethanol and air dry the beads for 5 min.
-
10.
Resuspend the beads in 520 μl 1× Restriction Buffer (RB), pool the samples in one 2 ml tube, and incubate 15 min at 55 °C (see Note 5).
3.3.2 First Enzymatic Digestion and Ligation
-
1.
Incubate the samples on ice for 5 min.
-
2.
Add 5 μl of Restriction enzyme 1 (RE1) and incubate for 2 h at 37 °C.
-
3.
Inactivate RE1 by incubating the samples 25 min at 65 °C and then keeping them on ice for 5 min.
-
4.
Add 125 μl 10× Ligation Buffer (10×LB) and 125 μl MilliQ (see Note 5).
-
5.
Add 5 μl Ligase (LIG) and incubate 1 h at RT.
-
6.
Reverse cross-linking by using 5 μl Proteinase K (ProtK) and incubating the samples overnight at 65 °C.
3.3.3 Second Enzymatic Digestion and Ligation
-
1.
Mix 150 μl 10× Restriction Buffer (10×RB), 135 μl MilliQ, and 5 μl Restriction Enzyme 2 (RE2) and incubate for 1 h at 37 °C (see Note 5).
-
2.
Inactivate RE2 for 20 min at 65 °C and then keep the samples on ice for 5 min.
-
3.
Add 175 μl 10×LB, 70 μl MilliQ, and 5 μl LIG; then incubate the samples with the ligation mix for 1 h at RT.
-
4.
Divide the samples in two 2 ml tubes, add 875 μl 2-propanol and 10 μl Magnetic Bead (MB), and incubate 1 h at RT.
-
5.
Place tubes on the magnetic rack and remove supernatant when the beads are completely separated.
-
6.
Wash the beads, with 1800 and 900 μl 80% ethanol. Between these two beads-wash steps, place the samples on the magnet, wait till the beads are separated, and then discard the supernatant. After the second wash, leave the beads to air dry for 15 min.
-
7.
Resuspend the beads with 105 μl Elution Buffer (EB), place the samples on the magnet, and when the beads are completely separated, transfer 100 μl of the eluted sample in a 1.5 ml tube.
-
8.
Measure the TLA template concentration using the Quantus fluorometer.
3.3.4 TLA PCR
-
1.
Prepare 600 ng of TLA template and add MilliQ until a 90 μl final volume.
-
2.
Set up the TLA PCR mix and thermal profile as described in Table 3.
-
3.
Divide the PCR mix in three PCR tubes and set up the thermal profile as described in Table 4.
-
4.
Pool all the three PCR tubes into a 1.5 ml tube.
-
5.
Add 200 μl of AMPure XP Beads and incubate 15 min at RT.
-
6.
Place the samples on the magnet and remove the supernatant when the beads are completely separated.
-
7.
Wash twice with 900 μl 80% ethanol and air dry the beads for 5 min.
-
8.
Resuspend the beads with 55 μl EB and place on the magnet for 1 min.
-
9.
Transfer 50 μl of the eluted TLA PCR products into a clean 1.5 tube and measure the concentration using Quantus fluorometer (see Note 5).
3.3.5 TLA Library Indexing
TLA library indexing is performed through Nextera DNA Flex Library Prep kit (Illumina), using a Bead-Linked Transposomes protocol to fragment and tag the TLA PCR products with adapter sequences, according to Illumina manual protocol procedures.
-
1.
TLA library preparation is performed using at least 50–100 ng of TLA PCR products as starting material.
-
2.
After adapter ligation, quantify the libraries using Quantus fluorometer and check the quality using High sensitivity D1000 ScreenTape (Agilent). Figure 2 shows typical TLA library profiles with an average fragment size of 300–450 bp when analyzed with a size range of 150–1500 bp.

TLA library profiles obtained using High sensitivity D1000 ScreenTape (Agilent)
3.3.6 Sequencing
-
1.
Pool 20 ng of each of the libraries and check the quantity and quality to obtain the concentration (nM) of the final TLA pool.
-
2.
Dilute the final TLA pool to 4 nM and follow the Illumina’s MiSeq sequencing protocol to denature the library.
-
3.
Sequence the TLA final pool spiked with 1% PhIX on a MiSeq platform using a v3 chemistry (600 cycles, paired end-PE).
3.4 Bioinformatics Analysis
Bioinformatics analysis is performed by Cergentis. To identify break-spanning reads, FASTQ files are mapped against the human genome version hg19 using BWA-SW, which is a Smith-Waterman alignment tool. Then, the Integrated Genomic Viewer (IGV; http://software.broadinstitute.org/software/igv/) tool is used to confirm rearrangements, translocations, to identify TLA-BCL1 or TLA-BCL2 defined as breakpoint sequences mapping on chromosome 11 (for MCL), chromosome 18 (for FL), and the mate chromosome 14 (IGH locus).
3.5 TLA Sequence Validation by Allele-Specific Oligonucleotide (ASO) Approach
Once bioinformatically defined, TLA-BCL1 or TLA-BCL2 is further investigated to detect the breakpoint nucleotide sequences and to develop the ASO MRD assay based on the TLA sequence.
The free websource BlastN (https://blast.ncbi.nlm.nih.gov/Blast.cgi) aligns the query to the human genome version hg38, thus defining the t(11;14) and t(14;18) translocated regions and the N insertion nucleotides.
Then, the ASO MRD assay based on the TLA sequence is designed as follows:
-
1.
An ASO forward primer is designed on the N insertions, which are randomly inserted between the translocated regions and which are not mapped with the Blast alignment.
-
2.
The consensus probe and reverse primer are set to anneal the joining IGH region (JH) involved in the translocation .
-
3.
The primer annealing temperature (Tm) is established both manually and using a primer design tool such as Integrated DNA Technologies (IDT free websource available at https://eu.idtdna.com/pages) following these criteria:
-
(a)
Primer Tm ranges between 58 °C and 62 °C.
-
(b)
Probe Tm is 10 °C higher than primer Tm.
-
(c)
It is recommended that the primer GC content is 40–60%.
-
(a)
Tables 5 and 6 show the list of JH primer available for TLA validation, while the ASO forward MRD assay design is detailed in Fig. 3.

ASO forward MRD assay design on TLA-BCL1 sequence
TLA-BCL1 or TLA-BCL2 assay validation is performed using highly sensitive quantification approaches as quantitative PCR (ASO qPCR) [13], setting a tenfold standard curve starting from 500 ng BM and or PB diagnostic sample serially diluted in pooled polyclonal healthy gDNAs or gDNA from a cell line not featuring any of the t(11;14) and t(18;14) translocations.
TLA-BCL1 or TLA-BCL2 is confirmed as MRD molecular markers if the validation experiment achieves a sensitivity level that allows the identification of 1 clonal cell within 100,000 analyzed cells, defined according to EuroMRD guidelines for qPCR data interpretation [11].
4 Notes
-
1.
High cell amounts lead to beads carryover, which is affecting TLA library quality. On the other hand, the minimum gDNA yield requested by TLA library preparation is not obtained when starting from a too low cell amount. gDNA samples should have an OD 260/280 range 1.8–1.9 and OD 260/230 > 1.5.
-
2.
gDNA quantification is performed using a fluorometer. Nanodrop could overestimate gDNA yield, thus affecting TLA library quality.
-
3.
Verify the gDNA concentration after dilution and before starting TLA library preparation.
-
4.
To check the quality of TLA library preparation, “undigested control,” “digested control,” and “ligation control” samples can be collected and processed with ProtK for 1 h at 65 °C. After AMPure XP beads purification, separate the samples on a 1% agarose gel. The “undigested control” is expected as a >10 kb signal; a smear between 0.3 and 2 kb and >5 kb are reported for the “digested control” and “ligation control.”
-
5.
Check TLA PCR products by separating the sample on a 1.2% agarose gel. Samples with a smear between 0.3 and 5 kb can proceed to the indexing phase.
References
Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R et al (2016) The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 127:2375–2390
Dreyling M, Ferrero S, Hermine O (2014) How to manage mantle cell lymphoma. Leukemia 28:2117–2130
Freedman A (2014) Follicular lymphoma: 2014 update on diagnosis and management. Am J Hematol 89:429–436
Ferrero S, Dreyling M (2017) Minimal residual disease in mantle cell lymphoma: are we ready for a personalized treatment approach? Haematologica 102:1133–1136
Pott C, Hoster E, Delfau-Larue M-H, Beldjord K, Böttcher S, Asnafi V et al (2010) Molecular remission is an independent predictor of clinical outcome in patients with mantle cell lymphoma after combined immunochemotherapy: a European MCL intergroup study. Blood 115:3215–3223
Ladetto M, Lobetti-Bodoni C, Mantoan B, Ceccarelli M, Boccomini C, Genuardi E et al (2013) Persistence of minimal residual disease in bone marrow predicts outcome in follicular lymphomas treated with a rituximab-intensive program. Blood 122:3759–3766
Grimaldi D, Genuardi E, Ferrante M, Ferrero S, Ladetto M (2018) Minimal residual disease in indolent lymphomas: a critical assessment. Curr Treat Options in Oncol 19:71
Pott C, Brüggemann M, Ritgen M, van der Velden VHJ, van Dongen JJM, Kneba M (2019) MRD detection in B-cell non-Hodgkin lymphomas using Ig gene rearrangements and chromosomal translocations as targets for real-time quantitative PCR. Methods Mol Biol 1956:199–228
Seto M (2002) Molecular mechanisms of lymphomagenesis through transcriptional disregulation by chromosome translocation. Int J Hematol 76:323–326
Pott C, Sehn LH, Belada D, Gribben J, Hoster E, Kahl B et al (2020) MRD response in relapsed/refractory FL after obinutuzumab plus bendamustine or bendamustine alone in the GADOLIN trial. Leukemia 34:522–532
van der Velden VHJ, Cazzaniga G, Schrauder A, Hancock J, Bader P, Panzer-Grumayer ER et al (2007) Analysis of minimal residual disease by Ig/TCR gene rearrangements: guidelines for interpretation of real-time quantitative PCR data. Leukemia 21:604–611
van Dongen JJM, Langerak AW, Brüggemann M, Evans PAS, Hummel M, Lavender FL et al (2003) Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 17:2257–2317
Donovan JW, Ladetto M, Zou G, Neuberg D, Poor C, Bowers D et al (2000) Immunoglobulin heavy-chain consensus probes for real-time PCR quantification of residual disease in acute lymphoblastic leukemia. Blood 95:2651–2658
Faham M, Zheng J, Moorhead M, Carlton VEH, Stow P, Coustan-Smith E et al (2012) Deep-sequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood 120:5173–5180
Brüggemann M, Kotrová M, Knecht H, Bartram J, Boudjogrha M, Bystry V et al (2019) Standardized next-generation sequencing of immunoglobulin and T-cell receptor gene recombinations for MRD marker identification in acute lymphoblastic leukaemia; a EuroClonality-NGS validation study. Leukemia 33:2241–2253
Oliva S, Genuardi E, Belotti A, Frascione PMM, Galli M, Capra A et al (2019) Minimal residual disease evaluation by multiparameter flow cytometry and next generation sequencing in the forte trial for newly diagnosed multiple myeloma patients. Blood 134:4322
Martinez-Lopez J, Lahuerta JJ, Pepin F, González M, Barrio S, Ayala R et al (2014) Prognostic value of deep sequencing method for minimal residual disease detection in multiple myeloma. Blood 123:3073–3079
Ladetto M, Brüggemann M, Monitillo L, Ferrero S, Pepin F, Drandi D et al (2014) Next-generation sequencing and real-time quantitative PCR for minimal residual disease detection in B-cell disorders. Leukemia 28:1299–1307
Kurtz DM, Green MR, Bratman SV, Scherer F, Liu CL, Kunder CA et al (2015) Noninvasive monitoring of diffuse large B-cell lymphoma by immunoglobulin high-throughput sequencing. Blood 125:3679–3687
Wren D, Walker BA, Brüggemann M, Catherwood MA, Pott C, Stamatopoulos K et al (2017) Comprehensive translocation and clonality detection in lymphoproliferative disorders by next-generation sequencing. Haematologica 102:57–60
Stewart P, Gazdova J, Darzentas N, Wren D, Proszek P, Fazio G et al (2019) Euroclonality-NGS DNA capture panel for integrated analysis of IG/TR rearrangements, translocations, copy number and sequence variation in lymphoproliferative disorders. Blood 134:888
de Vree PJP, de Wit E, Yilmaz M, van de Heijning M, Klous P, Verstegen MJAM et al (2014) Targeted sequencing by proximity ligation for comprehensive variant detection and local haplotyping. Nat Biotechnol 32:1019–1025
Hottentot QP, van Min M, Splinter E, White SJ (2017) Targeted locus amplification and next-generation sequencing. Methods Mol Biol 1492:185–196
Godet I, Shin YJ, Ju JA, Ye IC, Wang G, Gilkes DM (2019) Fate-mapping post-hypoxic tumor cells reveals a ROS-resistant phenotype that promotes metastasis. Nat Commun 10:4862
Chen YH, Pallant C, Sampson CJ, Boiti A, Johnson S, Brazauskas P et al (2020) Rapid lentiviral vector producer cell line generation using a single DNA construct. Mol Ther Method Clin Dev 19:47–57
Aeschlimann SH, Graf C, Mayilo D, Lindecker H, Urda L, Kappes N et al (2019) Enhanced CHO clone screening: application of targeted locus amplification and next-generation sequencing technologies for cell line development. Biotechnol J 14:e1800371
Alimohamed MZ, Johansson LF, De Boer EN, Splinter E, Klous P, Yilmaz M et al (2018) Genetic screening test to detect translocations in acute leukemias by use of targeted locus amplification. Clin Chem 64:1096–1103
Kuiper R, van Duin M, van Vliet MH, Broijl A, van der Holt B, el Jarari L et al (2015) Prediction of high- and low-risk multiple myeloma based on gene expression and the International Staging System. Blood 126:1996–2004
Genuardi E, Klous P, Mantoan B, Drandi D, Ferrante M, Cavallo F et al (2021) Targeted locus amplification to detect molecular markers in mantle cell and follicular lymphoma. Hematol Oncol 39(3):293–303
Gaidano G, Ballerini P, Gong JZ, Inghirami G, Neri A, Newcomb EW et al (1991) p53 mutations in human lymphoid malignancies: association with Burkitt lymphoma and chronic lymphocytic leukemia. Proc Natl Acad Sci 88:5413–5417
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this protocol

Cite this protocol
Genuardi, E., Alessandria, B., Civita, A.M., Ferrero, S. (2022). Targeted Locus Amplification as Marker Screening Approach to Detect Immunoglobulin (IG) Translocations in B-Cell Non-Hodgkin Lymphomas. In: Langerak, A.W. (eds) Immunogenetics. Methods in Molecular Biology, vol 2453. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-2115-8_8
Download citation
DOI: https://doi.org/10.1007/978-1-0716-2115-8_8
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-2114-1
Online ISBN: 978-1-0716-2115-8
eBook Packages: Springer Protocols