
Abstract
Liquid biopsy is a novel diagnostic approach at first developed to characterize the molecular profile of solid tumors by analyzing body fluids. For cancer patients, it represents a noninvasive way to monitor the status of the solid tumor with respect to representative biomarkers. There is growing interest in the utilization of circulating tumor DNA (ctDNA) analysis also in the diagnostic and prognostic fields of lymphomas. Clonal immunoglobulin (IG) gene rearrangements are fingerprints of the respective lymphoid malignancy and thus are highly suited as specific molecular targets for minimal residual disease (MRD) detection. Tracing of the clonal IG rearrangement patterns in ctDNA pool during treatment can be used for MRD assessment in B-cell lymphomas. Here, we describe a reproducible next-generation sequencing assay to identify and characterize clonal IG gene rearrangements for MRD detection in cell-free DNA.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
- Cell-free DNA
- Plasma
- Immunoglobulin rearrangements
- Therapy monitoring
- Liquid biopsy
- Minimal residual disease
- Digital droplet PCR
- Next-generation sequencing
1 Introduction
Circulating cell-free DNA (cfDNA) is fragmented extracellular DNA, which is released from apoptotic and necrotic cells in small fragments of <200 bp [1]. cfDNA is typically isolated from the blood stream; however, it is also possible to detect cfDNA in other biological fluids such as urine or cerebrospinal fluid [2,3,4,5,6]. Interestingly, in cancer patients, a fraction of 0.01–60% of the total cfDNA consists of circulating tumor DNA (ctDNA), which originates from neoplastic lesions [7].
The fact that ctDNA shares the same biological features as the cellular DNA of the tumor, such as point mutations, gene amplifications, and immunoglobulin (IG) and T-cell receptor (TR ) gene rearrangements in lymphoma, makes utilizing ctDNA as a noninvasive biopsy in diagnostic approaches and monitoring the status of minimal residual diseases (MRD) very attractive.
The ideal markers for MRD detection in B-cell lymphomas are clonal IG gene rearrangements. The IG heavy chain (IGH ) gene rearrangements are frequently used as target due to their unique junctional regions. However, IGH rearrangements may not be that reliable because of the somatic hypermutations (SHM ) mainly in the V gene regions taking place during B-cell development and maturation. This might result in mismatches in primer binding [8]. Alternatively, incomplete IGHD-IGHJ rearrangements and IGK gene rearrangements could be used as targets for MRD. Both rearrangement types are mainly unmutated. Incomplete rearrangements in the IGH locus do not contain SHM in the majority of cases, because transcription only starts from the promoters in the V genes [9]. The finding of hypermutation in a small proportion of incomplete DJH rearrangements suggests important biological implications concerning the process of SHM . The rearrangements of the IGK genes can also be an important complementary MRD target, as in rearrangements involving the kappa deletion element (Kde), no SHM can occur after Kde recombination, since the deletion of the JK-CK introns removes the IGK enhancer that is essential for SHM [10].
PCR-based methods like allele-specific real-time quantitative PCR or digital droplet (dd) PCR targeting the clonal IG rearrangements are currently the gold standard for MRD quantification in cfDNA but are limited by a sensitivity of 1xE-05 in a polyclonal B-cell background. Since ctDNA is present at a very low overall amount in the peripheral blood, highly sensitive technologies are needed to detect MRD in cfDNA . Next-generation sequencing (NGS) of IG rearrangements (IG-NGS) is the technology that can overcome the limitation of PCR-based approaches with a potential higher sensitivity.
MRD assessment in cfDNA by IG-NGS requires the identification of the lymphoma-associated clonotypes in diagnostic tumor tissue. Therefore, fresh or formalin-fixed paraffin-embedded (FFPE) lymph node material or diagnostic peripheral blood or bone marrow with sufficient tumor infiltration is required for the initial marker identification. The EuroClonality-NGS working group has recently shown that IGH and IGK rearrangements are highly suitable for detecting clonality in frozen and FFPE-embedded tissue specimens [11]. Due to the small fragment size of cfDNA (~166 bp) and the high frequency of SHM in the variable heavy framework region 3 (IGHV-FR3), the EuroClonality-NGS IGHV-FR3 multiplex PCR was redesigned and optimized for the specific requirements of MRD detection in cfDNA . IGHD-IGHJ and IGK (IGKV-IGKJ, IGKV-KDE, and intron RSS-KDE) primer sets remained unchanged; only the reaction conditions and primer concentrations were modified to facilitate balanced amplification of all rearrangements also in this type of material.
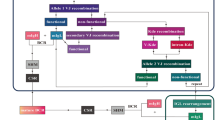
As illustrated in Fig. 1, using a one-step NGS PCR protocol, clonal IG rearrangements are amplified in cfDNA and combined with molecular barcodes and sequencing adapters. The amplicons bind the flow cell of the Illumina MiSeq through the introduced adapters and are sequenced by synthesis. A standardized bioinformatic analysis of the high-throughput sequencing data allows the verification of clonal IG rearrangements and their precise quantitation. The bioinformatic platform ARResT/Interrogate (at http://arrest.tools/interrogate/), developed within the EuroClonality-NGS working group, allows identification of clonotypes and MRD follow-up in the same workflow.

Schematic representation of the cfDNA-based NGS IG rearrangement analysis in lymphoma. Adapted from “Next Generation Sequencing (Illumina),” by BioRender.com (2021). Retrieved from https://app.biorender.com/biorender-templates
The EuroClonality-NGS “central intra-tube quality/quantification control” (cIT-QC), comprising of known copy numbers of clonal rearrangements, is added to each reaction to enable the quantification of ctDNA as fraction of cfDNA and the correction of potential amplification biases. The cIT-QC is used to calculate the coverage of each single rearrangement copy in order to calculate the read coverage per cell and to determine the MRD level. The utility of this approach has been published recently [12].
Here we provide detailed instructions on amplicon sequencing of clonal IG rearrangements in cfDNA using modified EuroClonality-NGS protocols for IGH (VJ + DJ) and IGK (VJ + intron-Kde/V-Kde) (http://www.euroclonality.org/protocols/). The process of marker identification in FFPE samples or diagnostic bone marrow or peripheral blood is not part of this chapter; for that we refer to the publication of Scheijen et al. [11].
2 Materials
Solutions must be prepared with double-distilled water (supplied as ultrapure water or purified by filtering of 18 MΩ-cm at 25 °C). All reagents should be stored at 18–25 °C unless otherwise indicated.
2.1 Sample Collection
-
1.
Blood collection tubes (S-Monovette®EDTA, PAXgene®, Cell-free DNA BCT® Blood collection tubes [Streck]).
2.2 cfDNA Extraction
-
1.
QIAamp® Circulating Nucleic Acid Kit (QIAGEN). Add 200 ml isopropanol (100%) to 300 ml buffer ACB concentrate to prepare ready-to-use buffer ACB. Add 25 ml ethanol (96–100%) to 19 ml buffer ACW1 concentrate to prepare ready-to-use buffer ACW1. Add 30 ml ethanol (96–100%) to 13 ml buffer ACW2 concentrate to prepare ready-to-use buffer ACW2.
-
2.
QIAvac 24 Plus vacuum pump (QIAGEN) or any vacuum pump capable of producing a pressure of −800 to −900 mbar. Alternatively, use the Maxwell® RSC instrument (Promega) with the corresponding Maxwell® RSC ccfDNA Plasma Kit (Promega) for automated extraction.
-
3.
Vortexer.
-
4.
Centrifuge.
-
5.
Heat block.
-
6.
Water bath.
-
7.
Sterile 50 ml conical tubes.
2.3 Digital Droplet PCR (ddPCR )
-
1.
2× ddPCR ™ Supermix (no dUTP, Bio-Rad).
-
2.
ddH2O.
-
3.
Forward and reverse primers (see Table 1 for ddPCR primer list).
-
4.
Extracted ccfDNA samples.
-
5.
DG8 Cartridges for QX200 Droplet Generator (Bio-Rad).
-
6.
DG8 Gaskets for QX200 Droplet Generator (Bio-Rad).
-
7.
QX200™ Droplet Digital PCR system (Bio-Rad).
-
8.
Twin.tec PCR Plate 96 (Eppendorf).
-
9.
Pierceable Foil Heat Seal (Bio-Rad).
-
10.
Thermal cycler.
-
11.
PX1 PCR plate sealer (Bio-Rad).
-
12.
Centrifuge.
-
13.
Vortexer.
2.4 One-Step Next-Generation Sequencing (NGS) PCR
-
1.
FastStart™ High Fidelity reaction buffer (Roche) w/o MgCl2.
-
2.
FastStart™ High Fidelity Taq polymerase (Roche).
-
3.
MgCl2 (Roche).
-
4.
dNTPs.
-
5.
ddH2O (HPLC purified).
-
6.
Library preparation target primers (desalted) (see Table 2).
-
7.
High sensitivity dsDNA concentration measuring kit (i.e., Qubit® dsDNA HS Assay Kit (ThermoFisher Scientific) or QuantiT PicoGreen dsDNA Assay Kit (ThermoFisher Scientific)).
-
8.
Qubit assay 0.5 ml tubes.
-
9.
2× 250 MiSeq Reagent Kits (Illumina) (i.e., v2, v2-Nano Kit (500 cycles), or v3 Kit (600 cycles), depending from number of samples and sequencing depths).
-
10.
PhiX Control v3 (Illumina) (10 nM). Dilute the PhiX (10 nM) to 4 nM by mixing 2 μl 10 nM PhiX with 3 μl EBT buffer.
-
11.
HT1 (Hybridization buffer) (Illumina).
-
12.
NaOH. Prepare a fresh 0.2 N NaOH daily by mixing 20 μl 10 N NaOH with 980 μl ddH2O.
-
13.
EBT-Buffer (Illumina).
-
14.
Tween 20®.
-
15.
Thermal Cycler.
-
16.
Low binding tubes.
-
17.
Illumina MiSeq sequencer.
-
18.
Light Cycler LC480/Qubit Fluorometer.
2.5 Purification of Subpools by Gel Extraction
-
1.
Gel electrophoresis chamber.
-
2.
Gel Red (VWR).
-
3.
Gel loading dye.
-
4.
MinElute Gel extraction kit (Qiagen).
-
5.
Scalpel or X-Tracta tips.
-
6.
Agarose.
-
7.
TBE (or TAE) Buffer.
3 Methods
All experimental procedures should be carried out at room temperature unless otherwise indicated.
3.1 Sample Preparation
-
1.
Centrifuge blood collection tubes at 2000 × g for 10 min. If using S-Monovette®EDTA tubes, processing time should not be longer than 4 h after sample taking.
-
2.
Carefully move the supernatants (plasma) into a 5 ml tube with a conic bottom without damaging the buffy coat phase.
-
3.
Centrifuge the isolated plasma at 16,000 × g, 4 °C for 10 min (see Note 1).
-
4.
Move supernatants to a clean 5 ml tube. Proceed immediately to cfDNA extraction (Subheading 3.2) or store at −80 °C (see Note 2).
3.2 cfDNA Extraction
cfDNA should be extracted from 4 ml purified blood plasma (set samples to 4 ml by adding phosphate-buffered saline if the volume is less than 4 ml).
-
1.
Prior to starting the extraction the following should be done:
-
(a)
Equilibrate samples and buffers to room temperature (18–25 °C).
-
(b)
Heat a water bath or heating block to 60 °C for use with 50 ml tubes.
-
(c)
Heat a heating block to 56 °C for use with 2 ml Eppendorf tubes.
-
(a)
-
2.
Pipet 400 μl QIAGEN Proteinase K into a pre-labeled 50 ml tube.
-
3.
Add 4 ml plasma to the tube.
-
4.
Add 3.2 ml buffer ACL to the tube. Mix well by vortexing for 30 s.
-
5.
Incubate for 30 min at 60 °C.
-
6.
Add 7.2 ml buffer ACB, mix well by vortexing for 15–30 s.
-
7.
Incubate the mixture for 5 min on ice.
-
8.
Insert the QIAamp Mini column into the VacConnector on the QIAvac 24 Plus. Insert a 20 ml tube extender into the open QIAamp Mini column. Make sure that the tube extender is firmly inserted into the QIAamp Mini column to avoid leakage of the sample.
-
9.
Carefully pour the mixture from step 6 into the tube extender of the QIAamp Mini column. Set the vacuum pump to produce a vacuum of −800 mbar to −900 mbar until all lysates are drawn through (takes up to 15 min) (see Note 3).
-
10.
Release the pressure to 0 mbar, discard the tube extenders carefully and leave the QIAamp Mini columns attached to the VacConnector on the QIAvac 24 Plus.
-
11.
Add 600 μl washing buffer ACW1 to the QIAamp Mini column. Switch on the vacuum pump (−800 mbar to −900 mbar) while the lid is open (see Note 3). After the entire washing buffer has been drawn through the column, switch the vacuum pump off and release the pressure to 0 mbar.
-
12.
Add 750 μl washing buffer ACW2 to the QIAamp Mini column. Switch on the vacuum pump (−800 mbar to −900 mbar) while the lid is open (see Note 3). After the entire washing buffer has been drawn through the column, switch the vacuum pump off and release the pressure to 0 mbar.
-
13.
Add 750 μl ethanol (96–100%) to the QIAamp Mini column. Switch on the vacuum pump (−800 mbar to −900 mbar) while the lid is open (see Note 3). After the entire washing buffer has been drawn through the column, switch the vacuum pump off and release the pressure to 0 mbar.
-
14.
Close the lid of the QIAamp Mini column. Remove it from the vacuum manifold and discard the VacConnector. Move the QIAamp Mini column to a clean 2 ml collection tube and centrifuge at full speed (16,000 × g) for 3 min.
-
15.
Move the QIAamp Mini column to a new 2 ml collection tube, open the lid, and incubate at 56 °C for 10 min to dry the membrane completely.
-
16.
Move the QIAamp Mini column to a clean 1.5 ml elution tube. Carefully pipet 20–150 μl of Buffer AVE onto the filter of the QIAamp Mini column. Close the lid and incubate at room temperature for 3 min (see Note 4).
-
17.
Centrifuge for 1 min at 16,000 × g. Discard columns.
-
18.
For downstream processing, isolated cfDNA can be stored at 4 °C for up to 24 h. For longer storage freeze at <−30 °C.
-
19.
Use a fragment analyzer to accurately size and qualify the extracted cfDNA (see Note 5).
3.3 Digital Droplet PCR (ddPCR)-Mediated Copy Number Quantification
-
1.
Prepare the reaction mixture by adding 10 μl 2× ddPCR Supermix, 0.3 μl forward primer (20 μM), 0.3 μl reverse primer (20 μM), and 0.1 μl probe (20 μM) to 1 μl DNA and fill with ddH2O up to 20 μl.
-
2.
Generate the droplets by pipetting 20 μl sample into the sample wells and 70 μl QX200 Droplet generation oil into the Oil well in the DG8 Cartridge, cover using the DG8 Gaskets and place into the QX200 Droplet generator.
-
3.
Carefully transfer 40 μl of the generated droplets to the Twin.tec PCR Plate by slowly pipetting using a multichannel pipette. Seal plate using a pierceable Foil Heat Seal and a PX1™ PCR Plate sealer.
-
4.
Perform the PCR Reaction on a thermal cycler by incubating the sample for 10 min at 95 °C for denaturation followed by 40 cycles of 30 s incubation at 94 °C for initial denaturation and 1 min incubation at 60 °C for annealing. Apply a final extension step by incubating for 10 min at 98 °C and store the sample at 4 °C.
-
5.
Move the plates to the QX200 Droplet Reader to measure the amplified Droplets (see Note 6).
-
6.
To measure the amplified fragments, start a new experiment in the QuantaLife™ Software, adjust Supermix to ddPCR ™ Supermix w/o dUTP for the desired wells, name targets and select the appropriate channels as used in the probe (FAM, HEX, etc.).
-
7.
Place the plate in the reader and run the experiment, choose DyeSet: FAM or FAM/HEX.
-
8.
Save the analysis and load the exported file into the QuantaSoft™ Pro software.
-
9.
Set the threshold in the 1D-Amplitude to the positive fraction (see Notes 7 and 8).
-
10.
Export the results to Excel or any other spreadsheet application.
-
11.
Calculate the mean of the copies/20 μl reads and divide it by 2 to determine the copy number/cell.
-
12.
Divide the copy number/cell by 150 to calculate the concentration (ng/μl). Calculate the cfDNA concentration per 1 ml of plasma.
3.4 NGS Library Preparation
For library preparation, keep the reagents on ice until use. Use precooled racks for preparing the reaction mixture. Alternatively, the reaction could be prepared by placing the tubes on ice. Use the EuroClonality-NGS cIT-QC with known copy numbers of clonal rearrangements, in order to calculate the read coverage per cell (see Note 9 and [12]).
-
1.
Prepare the target-specific master mixes according to Table 3. Vortex and centrifuge the tubes.
-
2.
Add 40 μl target-specific master mix to 1500 cell equivalents of a pre-quantified cfDNA . Adjust the volume to 50 μl with ddH2O. Consider preparing one reaction for buffy coat (positive control) and another without added DNA (negative control).
-
3.
Perform the targeted PCR Reactions on a thermal cycler as indicated in Table 4.
-
4.
Verify the amplification of the target genes by agarose gel electrophoresis by loading 7 μl PCR product and 3 μl DNA loading dye into a 2% agarose gel (Fig. 2). Estimate the concentration of the amplified products by comparing the bands’ intensity with these of the DNA ladder. To prepare a 2% agarose gel, weigh 8 g agarose and add it to 400 ml TBE buffer in a 1 l glass container. Dissolve the agarose by heating for intervals of 15–20 s in a microwave at 800 W, swirl the container gently, and repeat until the agarose completely dissolves. Allow the mixture to cool to <60 °C. Add 1:10,000 Gel-Red, mix gently and pour the dissolved agarose into the casting form, place the comb in place, and wait until the gel sets (around 30 min).
-
5.
Pool 2–10 μl PCR products of the same target (i.e., cfFR3, IGHDJ, IGK ). Consider the differences in the bands’ intensities to pool equal amounts of the samples. Load the mixture on a new 2% agarose gel for separation by electrophoresis. After clear separation of the bands, extract the corresponding DNA fragments by excising the bands out of the gel using a clean scalpel or X-tracta tips. Using a gel extraction kit, extract the DNA from the agarose gel and elute in 10–20 μl elution buffer. Alternatively, pools could be purified using a magnetic beads-based purification kit.
-
6.
Quantify the extracted DNA fragments using a Qubit® assay and use a fragment analyzer to identify the length of the extracted fragments. Fragment length should vary from 280 bp to 290 bp for IGH-VJ (cfFR3), 250–270 bp for IGH-DJ, and around 300 bp for IGK .
-
7.
Using the fragment length and the DNA concentration (ng/μl), convert the DNA concentration (ng/μl) to nM using the following formula:
$$ \mathrm{Concentration}\left(\mathrm{ng}/\upmu \mathrm{l}\right)/\left(\mathrm{DNA}\ \mathrm{fragment}\ \mathrm{size}\times 650\right)\times 1,000,000 $$ -
8.
Dilute the amplified libraries to 2 nM or 4 nM when using a V3 Kit. According to the desired total reads, choose the corresponding MiSeq Reagent Kit (V2, V3, nano V2, etc.) (see Note 10).
-
9.
Define the desired read count of each target and pool all targets in the corresponding ratio.

PCR products analyzed by agarose gel electrophoresis. The target-specific band size is around 250 bp in IGH-VJ, 300 bp in IGH-DJ, and 300 bp in IGK . Amplification reactions using buffy coat and distilled water served as positive and negative controls, respectively
3.5 Next-Generation Sequencing
The Illumina MiSeq instrument must be set up correctly before use. Post-run and maintenance wash procedures must be performed after each run.
-
1.
Before starting, thaw the HT1 Hybridization buffer on ice and the cartridge in water for at least 1 h. After the cartridge thaws, invert it 10× to mix the thawed reagents. Inspect that all positions are thawed. If air bubbles are present, gently tap the cartridge on the bench. Keep cool until use.
-
2.
After preparing and pooling the sequencing libraries, denature the PhiX control using 0.2 N NaOH by mixing 5 μl 4 nM PhiX with 5 μl 0.2 N NaOH, centrifuge for 1 min at 300 × g, and incubate for 5 min at room temperature. Stop the denaturation by adding 990 μl precooled HT1 buffer. The PhiX end concentration of the mixture is 20 pM (see Note 11).
-
3.
Denature a 2 nM library by mixing 5 μl 2 nM library with 5 μl 0.2 N NaOH, centrifuge for 1 min at 300 × g, and incubate for 5 min at room temperature. Stop the denaturation by adding 990 μl precooled HT1 buffer. The end concentration of the library is 10 pM (see Note 12).
-
4.
Dilute both denatured PhiX and denatured library pool to the same concentration using precooled HT1 buffer. Combine 10% denatured PhiX (60 μl) to the denatured library pool (540 μl) for a low-diversity library (see Note 13). Mix well by inverting the tube and centrifuge for 1 min at 300 × g. Keep the mixture on ice until loading into the cartridge.
-
5.
Create a new sample sheet using the Illumina Experiment Manager software by choosing the corresponding Illumina device and selecting “other analysis followed by FastQ Only.” Define cycle reads for forward and reverse to 200 each. Select the desired Index primers and save the sheet.
-
6.
Load 600 μl library pool mixture into the sample position in the cartridge. Load the cartridge and the flow cell into the MiSeq device and start the run according to the manufacturer’s instructions (see Note 14) for monitoring data quality.
-
7.
Export the data for demultiplexing and processing.
3.6 Bioinformatic Analysis
Output data are analyzed using the previously described bioinformatic platform ARResT/Interrogate [12, 13]; see below.
-
1.
Sequencing output data are demultiplexed using the bcl2fastq tool (Illumina) with 0 mismatches allowed in the barcode sequence (“—mismatches 0”).
-
2.
ARResT/Interrogate can be accessed at http://arrest.tools/interrogate and requires an account which can be created by emailing contact@arrest.tools.
-
3.
Switch to the “processing” panel, create a new analysis, and then upload the samples in compressed FASTQ format (the extension should be “.fastq.gz”).
-
4.
Make sure to select the “ARResT.cfDNA ” pipeline scenario for IGHfcFR3 samples and “ARResT.Routine” for IGH-DJ and IGK samples.
-
5.
Click on the blue “test it” button. If the test was OK, one should be able to click on the green “process” button. If not, check the “process output” tab for feedback, and email contact@arrest.tools if necessary.
-
6.
Progress of the bioinformatic pipeline can be followed in the “process output” tab. There is no need to wait; one may even close the browser; either log in later or better make sure to provide an email address to receive email notifications.
-
7.
When the run is complete, switch to the “file” panel and select it from the drop-down menu, and click on “load results;” when looking for clonotypes of very low abundance, load the “not prefiltered” results.
-
8.
Switch to the “questions” panel.
-
9.
To access normalized values (i.e., number of cells instead of number of reads), it is also necessary to provide the number of cells (derived from the DNA amount) in the sample, e.g., ~15,000 cells from 100 ng of DNA (also see Note 15)—this will be used as the denominator for the percentage calculation. There is a widget in the “processing” panel and the same in the “questions” panel (Fig. 3), which sets the value for all samples; if one needs to set different values for different samples, this should be done in a sample sheet and its “cells” column. Do not include spike-ins in those numbers.
-
10.
One should be able to see extra relevant widgets and messages in “questions” (and remember to hover over the “?” tooltip anchors) – to see normalized abundances, check the “use” box (Fig. 3).

Messages and widgets related to cIT-QC (spike-ins)
4 Notes
-
1.
Precool the centrifuges to 4 °C prior sample preparation.
-
2.
Avoid freeze-thaw cycles of plasma as this may lead to DNA degradation.
-
3.
In case of clogged QIAamp Mini column, close the VacValve, carefully remove the whole assembly, and transfer the remaining sample from the tube extender to a new 50 ml tube. Move the QIAamp Mini column from the assembly into a clean 2 ml collection tube and centrifuge it at full speed for 1 min or until the sample has completely passed through the membrane. Reassemble the QIAamp Mini column with Tube extender, VacConnector, and VacValve. Transfer the remaining lysate into the Tube extender, and proceed as described previously.
-
4.
The recovered eluate volume will be up to 5 μl less than the elution volume applied to the column.
-
5.
The expected fragment length is 130–200 bp.
-
6.
The droplet measurements must be carried out within 3–4 days.
-
7.
The threshold must be defined manually and set closely as possible above the fraction of negative droplets of the positive control.
-
8.
In the case of positive droplets in the negative controls, channel 2 should be also checked for positive droplets. Positive droplets in channel 2 refer to nonspecific amplifications, and these should not be considered in the analysis.
-
9.
Central intra-tube control (cIT-QC), also known as spike-in DNA, is a mixture of 9 cell lines, comprising 46 clonal rearrangements with known copy numbers. Spike-in DNA is added to each reaction for the quantification of ctDNA as fraction of cfDNA and for correction of potential amplification biases [12].
-
10.
MiSeq Reagent V2 Kit provides up to 1.5 × 107 Reads, V3 Kit provides up to 2.5 × 107 Reads, V2 Micro up to 4 × 106 Reads, and V2 Nano up to 1 × 106 Reads.
-
11.
Denatured PhiX is stable for up to 3 weeks at −20 °C.
-
12.
If using a V3 kit, start with a 4 nM library to end with 20 pM library pool.
-
13.
PhiX concentration varies according to the level of diversity. For high diversity panels, use 1% PhiX.
-
14.
Use the Illumina Sequencing analysis viewer (SAV) software for monitoring the quality parameters of the running assay. Check the cluster optimization overview for optimal control on cluster density.
-
15.
Input copy numbers of cfDNA might be low due to availability. For retrieval of clonal IG rearrangements in cfDNA , we recommend using 1500 cfDNA copy numbers. Define the level of detection (lod) by dividing 1 on the amplified copy number (in this case 1500 copies). For MRD detection during treatment, the LOD has to be determined for each sample according to the variability of input copy numbers.
-
16.
Because of the nature of the cfDNA primers that result in short amplicons and thus compromise the rearranged gene annotations, the presented clonotypes only feature the junction amino acid sequence. There are more feature types that can be accessed, including the rearranged genes and the full nt sequences themselves (click on any clonotype and check the “minitable” or through the “forensics” panel and its “sequences” tab) (Fig. 4).

Representation of the “minitable” with the selected clonotypes and full nt sequences
References
Volckmar A-L, Sültmann H, Riediger A, Fioretos T, Schirmacher P, Endris V et al (2018) A field guide for cancer diagnostics using cell-free DNA: from principles to practice and clinical applications. Genes Chromosom Cancer 57:123–139
Li Y, Pan W, Connolly ID, Reddy S, Nagpal S, Quake S et al (2016) Tumor DNA in cerebral spinal fluid reflects clinical course in a patient with melanoma leptomeningeal brain metastases. J Neuro-Oncol 128:93–100
Pan W, Gu W, Nagpal S, Hayden Gephart M, Quake SR (2015) Brain tumor mutations detected in cerebral spinal fluid. Clin Chem 61:514–522
Swinkels DW, de Kok JB, Hanselaar A, Lamers K, Boerman RH (2000) Early detection of leptomeningeal metastasis by PCR examination of tumor-derived K-ras DNA in cerebrospinal fluid. Clin Chem 46:132–133
Salvi S, Gurioli G, Martignano F, Foca F, Gunelli R, Cicchetti G et al (2015) Urine cell-free DNA integrity analysis for early detection of prostate cancer patients. Dis Markers 2015:574120
Xia Y, Huang CC, Dittmar R, Du M, Wang Y, Liu H et al (2016) Copy number variations in urine cell free DNA as biomarkers in advanced prostate cancer. Oncotarget 7:35818–35831
Leon SA, Shapiro B, Sklaroff DM, Yaros MJ (1977) Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res 37:646–650
van Dongen JJM, Langerak AW, Brüggemann M, Evans PA, Hummel M, Lavender FL et al (2003) Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 concerted action BMH4-CT98-3936. Leukemia 17:2257–2317
González D, González M, Alonso ME, Lopez-Perez R, Balanzategui A, Chillon MC et al (2003) Incomplete DJH rearrangements as a novel tumor target for minimal residual disease quantitation in multiple myeloma using real-time PCR. Leukemia 17:1051–1057
Langerak AW, Nadel B, de Torbal A, Wolvers-Tettero ILM, Van Gastel-Mol EJ, Verhaaf B et al (2004) Unraveling the consecutive recombination events in the human IGK locus. J Immunol 173:3878–3888
Scheijen B, Meijers RWJ, Rijntjes J, van der Klift MY, Möbs M, Steinhilber J et al (2019) Next-generation sequencing of immunoglobulin gene rearrangements for clonality assessment: a technical feasibility study by EuroClonality-NGS. Leukemia 33:2227–2240
Knecht H, Reigl T, Kotrová M, Appelt F, Stewart P, Bystry V et al (2019) Quality control and quantification in IG/TR next-generation sequencing marker identification: protocols and bioinformatic functionalities by EuroClonality-NGS. Leukemia 33:2254–2265
Bystry V, Reigl T, Krejci A, Demko M, Hanakova B, Grioni A et al (2017) ARResT/interrogate: an interactive immunoprofiler for IG/TR NGS data. Bioinformatics 33:435–437
Devonshire AS, Whale AS, Gutteridge A, Jones G, Cowen S, Foy CA et al (2014) Towards standardisation of cell-free DNA measurement in plasma: controls for extraction efficiency, fragment size bias and quantification. Anal Bioanal Chem 406:6499–6512
Acknowledgments
FKZ 01KT1807 TRANSCAN V-NOVEL by BMBF.
Author information
Authors and Affiliations
Consortia
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this protocol

Cite this protocol
Pott, C., Kotrova, M., Darzentas, N., Brüggemann, M., Khouja, M., on behalf of the EuroClonality-NGS Working Group. (2022). cfDNA-Based NGS IG Analysis in Lymphoma. In: Langerak, A.W. (eds) Immunogenetics. Methods in Molecular Biology, vol 2453. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-2115-8_7
Download citation
DOI: https://doi.org/10.1007/978-1-0716-2115-8_7
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-2114-1
Online ISBN: 978-1-0716-2115-8
eBook Packages: Springer Protocols