
Abstract
Identification of immunoglobulin (IG) and T-cell receptor (TR) gene rearrangements in acute lymphoblastic leukemia (ALL) patients at initial presentation are crucial for monitoring of minimal residual disease (MRD) during subsequent follow-up and thereby for appropriate risk-group stratification. Here we describe how RNA-Seq data can be generated and subsequently analyzed with ARResT/Interrogate to identify possible MRD markers. In addition to the procedures, possible pitfalls will be discussed. Similar strategies can be employed for other lymphoid malignancies, such as lymphoma and myeloma.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
- Minimal residual disease
- Acute lymphoblastic leukemia
- Immunoglobulin
- T-cell receptor
- Gene rearrangements
- RNA-Seq
- Whole exome sequencing
- Whole genome sequencing
- Marker identification
1 Introduction
Most clinical protocols for patients with acute lymphoblastic leukemia (ALL) nowadays include minimal residual disease (MRD)-based stratification [1,2,3,4]. Molecular MRD analysis is, at least in Europe, most commonly used and is generally based on analysis of rearranged immunoglobulin (IG) and T-cell receptor (TR ) genes according to international guidelines [5,6,7,8]. In a diagnostic setting, IG /TR gene rearrangements are generally identified using DNA-based PCR analysis, followed by classical Sanger sequencing or next-generation sequencing (NGS) [5, 8]. In recent years, whole transcriptome RNA sequencing (RNA-Seq) is increasingly used to identify fusion genes and to assign patients into distinct molecular subgroups according to the WHO 2016 classification, or for protocol-based clinical decisions [9]. Clearly, it would be beneficial if RNA-Seq data could also be used for the identification of IG /TR gene rearrangements pertaining to the leukemic clone. A recent study already showed that RNA-Seq data allowed the identification of IG heavy chain (IGH ) gene rearrangements in approximately 90% of B-ALL patients [10]. It should however be noted that the majority of ALL rearrangements is unproductive; this is in clear contrast to rearrangements present in normal B cells, which virtually all are functional. Therefore, caution is warranted in the analysis of RNA-Seq data for IG /TR marker screening in ALL (and in other lymphoproliferative disorders requiring multiple RNA/DNA analyses) [11, 12], and applying computational methods that only focus on productive rearrangements (e.g., like for most repertoire analyses) will clearly result in incomplete interpretation of IG /TR data for marker identification [13].
In this chapter, we describe how RNA-Seq data can be obtained and subsequently evaluated using the ARResT/Interrogate immunoprofiling platform [arrest.tools/interrogate] to identify possible IG /TR markers. Similar strategies can likely be employed for other lymphoid malignancies, such as lymphoma and myeloma. Finally, comparable data analysis tools may be used for whole genome sequencing and whole exome sequencing data.
2 Materials
The following equipment, materials, and reagents (or equivalents) should be available:
2.1 RNA-Input Quality Check
-
1.
Agilent 2100 Bioanalyzer (Agilent Technologies).
-
2.
Chip priming station (supplied with the Agilent 2100 Bioanalyzer).
-
3.
IKA vortex mixer (supplied with the Agilent 2100 Bioanalyzer).
-
4.
16-pin bayonet electrode cartridge (supplied with the Agilent 2100 Bioanalyzer).
-
5.
Agilent RNA 6000 Nano Kit (Agilent Technologies).
-
6.
Microcentrifuge (≥1300 × g).
2.2 Preparation of RNA-Seq Library
-
1.
Illumina TruSeq® Stranded mRNA Library Prep Kit, 96 Sample.
-
2.
TruSeq® RNA CD Index Plate (96 Indexes, 96 Samples) (Illumina).
-
3.
96-well storage plates, round well, 0.8 ml (“midi” plate) (Thermo Fisher Scientific).
-
4.
Agencourt AMPure XP 60 ml kit (Beckman Coulter Genomics).
-
5.
Agilent DNA 1000 Kit (Agilent Technologies).
-
6.
Ethanol 200 proof (absolute) for molecular biology (500 ml) (Sigma-Aldrich).
-
7.
Microseal “B” adhesive seals (Bio-Rad).
-
8.
Nuclease-free ultrapure water.
-
9.
RNaseZap (to decontaminate surfaces).
-
10.
RNase-/DNase-free 8-tube strips and caps.
-
11.
RNase-/DNase-free multichannel reagent reservoirs, disposable (VWR).
-
12.
SuperScript II Reverse Transcriptase (1 vial per 48 reactions) (Thermo Fisher Scientific).
-
13.
Tris–HCl 10 mM, pH 8.5.
-
14.
Tween 20 (Sigma-Aldrich).
-
15.
96-well thermal cycler (with programmable heated lid).
-
16.
Magnetic stand-96 (Thermo Fisher Scientific).
-
17.
Microplate centrifuge.
-
18.
Vortex.
The following supplies are specifically required for the “HS” workflow described in this protocol (see Note 1).
-
1.
96-well Hard-Shell 0.3 ml PCR plate (Bio-Rad).
-
2.
Microseal “A” film (Bio-Rad).
-
3.
High-Speed Microplate Shaker (VWR).
-
4.
Midi plate insert for heating system (Illumina) (two inserts recommended for successive heating procedures).
-
5.
Stroboscope.
-
6.
SciGene TruTemp Heating System (Illumina) (115 V) or SC-60-504 (220 V).
-
7.
Hybex Microsample Incubator (SciGene) 1057-30-0 (115 V) or 1057-30-2 (230 V) (two systems recommended for successive heating procedures).
3 Methods
Be careful when handling RNA samples (see Note 2).
Here we describe the workflow for RNA-Seq using the Illumina® TruSeq Stranded mRNA Kit (Illumina® Document # 1000000040498 v00) Library Kit chemistries and workflows [https://support.illumina.com//sequencing/sequencing_kits/truseq-stranded-mrna/documentation.html]. Before you proceed, please check carefully for any changes issued by the manufacturer regarding the kit or protocol (see Note 3).
3.1 RNA Isolation and Quality Assessment
Input RNA quality and quantity is essential to transcriptome sequencing. Illumina True Seq mRNA library Kit requires 0.1–1 μg total RNA as input (see Note 4). To assess the RNA quality, use the Agilent RNA 6000 Nano Kit:
-
1.
Ensure a correct setup of the chip priming station (see Note 5).
-
2.
The following kit components need to be prepared before first time use according to the manufacturer’s instructions:
-
(a)
RNA ladder aliquots, stable at −70 °C for extended time periods.
-
(b)
Agilent RNA 6000 Nano gel matrix aliquots (65 μl), can be stored at 4 °C for 1 month (protect from light during use).
-
(a)
-
3.
To prepare the gel-dye matrix, allow one aliquot Agilent RNA 6000 Nano gel matrix (65 μl) and RNA 6000 Nano dye concentrate (blue cap) to come to room temperature for 30 min.
-
4.
Vortex the dye concentrate for 10 s and spin down.
-
5.
Pipette 1 μl of dye concentrate to 65 μl of gel matrix (one aliquot) in a microcentrifuge tube.
-
6.
Vortex thoroughly and check for proper mixing of gel and dye.
-
7.
Spin at room temperature for 10 min at 13,000 × g, protect from light, and use within 1 day. Store at 4 °C if not used immediately.
-
8.
To load the gel-dye matrix, check proper setup of the chip priming station, and place an unused RNA 6000 Nano chip on the chip priming station.
-
9.
Pipette 9.0 μl of the gel-dye mix to the bottom of the well “G” with black background.
-
10.
Position the plunger of the syringe in the chip priming station to 1 ml; then close the chip priming station and pressurize quickly by pressing down the plunger. Keep for exactly 30 s.
-
11.
Release the plunger and pull back gently to the 1 ml mark.
-
12.
Open the chip priming station and pipette 9.0 μl gel-dye matrix to the bottom of the two wells marked “G” without background.
-
13.
To load ladder and sample to the chip, pipette 5 μl of RNA marker (green cap) to all sample wells and the well that is marked with the ladder symbol.
-
14.
Add 1 μl of ladder (prepared in step 2, Subheading 3.1) to the well that is marked with the ladder symbol.
-
15.
Add 1 μl of RNA of interest to all sample wells (see Note 6). If there are less than 12 samples to measure, put 1 μl of RNA marker (green cap) to wells that are not used.
-
16.
Vortex the chip horizontally in the IKA vortex (1 min, 2400 rpm), and insert to the Bioanalyzer 2100 station within 5 min to perform analysis using 2100 Expert Software.
Analysis will deliver a microcapillary electropherogram together with the RNA concentration measured and the RNA integrity number (RIN). The presence of a marker peak and two ribosomal RNA peaks (18S and 28S) will indicate a successful measurement of RNA with at least intermediate quality. Integrity of RNA is quantified on a scale from 1 (poor) to 10 (best) by RIN, based on a proprietary algorithm developed by Agilent © [14]. Figure 1 shows a good and a poor RIN example. In the poor RIN example, the ribosomal peaks are hardly detectable and RNA degradation is observed as a smear of RNA with decreasing size. Illumina True Seq protocols recommend a RIN of 8.0 or higher to be used for library preps (see Note 7).

RNA quality assessment by Bioanalyzer. RNA from bone marrow samples of patients with first diagnosis of ALL was isolated by silica columns (Qiagen, AllPrep) and subjected to microcapillary electrophoresis on the Agilent 2100 Bioanalyzer using the Agilent RNA 6000 Nano Kit as described. (a) Electropherogram representing a high-quality RNA sample (RIN 9.4). (b) Electropherogram representing a low-quality RNA sample with ongoing RNA degradation (RIN 4.2)
3.2 Library Preparation
The following protocol will describe how to obtain a sequencing-ready RNA-Seq library using the Illumina TruSeq® Stranded mRNA library prep kit (October 2017) [https://support.illumina.com//sequencing/sequencing_kits/truseq-stranded-mrna/documentation.html]. Sequencing kit chemistries and protocols are subject to change by the manufacturer. Before beginning, please check with the current version of the Kit and protocol you are using.
The original protocol includes two workflow options based on the number of samples to be processed. We are here describing the “HS” option for >48 samples (see Note 8).
3.2.1 Purification and Fragmentation of mRNA
In this step, polyadenylated mRNA is pulled down from the total RNA sample using oligo dT-attached magnetic beads (see Note 9). The isolate is then purified and fragmented.
-
1.
Bring 0.1–1.0 μg of total RNA to 50 μl volume using nuclease-free ultrapure water and transfer to the individual wells of the RNA bead plate.
-
2.
Vortex RNA purification beads and transfer 50 μl to each well.
-
3.
Seal the plate with Microseal “B” and shake plate (1 min, 1000 rpm).
-
4.
Heat the plate in the microheating system (5 min, 65 °C, lid closed), and cool down on ice (1 min).
-
5.
Incubate the plate at room temperature (5 min). Meanwhile bring the microheating system to 80 °C.
-
6.
For magnetic pull-down, remove the seal and put the plate on the magnetic stand. Inspect visually until the liquid is clear (ca. 5 min).
-
7.
Remove the supernatant from all wells. Then remove the plate from the stand.
-
8.
Pipette 200 μl Bead Washing Buffer to all wells; seal the plate and mix by shaking (1 min, 1000 rpm).
-
9.
Repeat steps 6 and 7 of Subheading 3.2.1.
-
10.
Pipette 50 μl Elution Buffer to all wells; seal the plate and mix by shaking (1 min, 1000 rpm).
-
11.
Heat the plate in the microheating system (2 min, 80 °C, lid closed), and cool down on ice (1 min).
-
12.
Place the plate at the bench and remove the seal.
-
13.
To prepare for RNA fragmentation, pipette 50 μl Bead Binding Buffer to all wells; seal the plate and mix by shaking (1 min, 1000 rpm).
-
14.
Incubate the plate at room temperature (5 min).
-
15.
Repeat steps 6–9 of Subheading 3.2.1 (magnetic pull-down, bead washing, another magnetic pull-down).
-
16.
Pipette 19.5 μl Fragment, Prime, Finish Mix to all wells, seal the plate, and mix by shaking (1 min, 1000 rpm).
-
17.
Remove the seal and transfer the samples well by well to the RNA fragmentation plate.
-
18.
Seal the plate and run the following program on the thermocycler with preheated lid:
-
(a)
94 °C for 8 min
-
(b)
Hold at 4 °C.
-
(a)
-
19.
Quickly spin down.
3.2.2 First Strand cDNA Synthesis
Purified RNA fragments are reverse transcribed to first strand cDNA using random hexamer primers.
-
1.
For magnetic pull-down, put the plate on the magnetic stand. Inspect visually until the liquid is clear (ca. 5 min).
-
2.
Remove the seal and transfer 17 μl supernatant well by well to the cDNA plate.
-
3.
Spin down the First Strand Synthesis Act D Mix (5 s, 600 × g).
-
4.
Mix SuperScript II reverse transcriptase and the First Strand Synthesis Act D Mix at a 1:10 ratio (1 μl SuperScript II reverse transcriptase plus 9 μl First Strand Synthesis Act D Mix (can be stored for extended time periods at −20 °C).
-
5.
Pipette 8 μl of this mix to each well of the cDNA plate and mix by shaking (20 s, 1600 rpm).
-
6.
Spin down by centrifugation (1 min, 280 × g).
-
7.
Run the following program on the thermocycler with the lid preheated to 100 °C:
-
(a)
25 °C for 10 min.
-
(b)
42 °C for 15 min.
-
(c)
70 °C for 15 min.
-
(d)
Hold at 4 °C.
-
(a)
3.2.3 Second Strand cDNA Synthesis
To maintain strand specificity during cDNA synthesis and to remove the mRNA template, dUTP is replaced by dTTP in second strand cDNA synthesis. Second strand cDNA synthesis results in blunt-end double-stranded cDNA, which can be stored for 1 week (first safe stopping point).
-
1.
Add 5 μl Resuspension buffer to each well.
-
2.
Spin down Second Strand Marking Mix (5 s, 600 × g), and pipette 20 μl to each well, and mix by shaking (20 s, 1600 rpm).
-
3.
Spin down the plate (1 min, 280 × g).
-
4.
Place the plate on the thermocycler with lid preheated to 30 °C and run at 16 °C for 60 min. Allow to come to room temperature afterwards.
-
5.
Pipette 90 μl of AMPure XP beads to new cDNA Clean-Up Plate, and transfer the content of the cDNA plate well by well to cDNA Clean-Up Plate. Mix by shaking (2 min, 1800 rpm).
-
6.
Incubate at room temperature (15 min), and then spin down (1 min, 280 × g).
-
7.
Put the plate on the magnetic stand. Inspect visually until the liquid is clear (ca. 5 min). Remove 135 μl supernatant from each well.
-
8.
Wash the beads with the plate retained on the magnetic stand by adding 200 μl ethanol 80% to all wells (see Note 10). After 30 s, remove all supernatant from each well (see Note 11).
-
9.
Repeat step 8 of Subheading 3.2.3 and carefully remove all remaining ethanol (using a small-volume pipette). Let the samples air dry on the magnetic stand (15 min).
-
10.
Remove the plate from the stand and pipette 17.5 μl Resuspension buffer to all wells. Mix by shaking (2 min, 1800 rpm). Let stand at room temperature (2 min).
-
11.
Spin down (1 min, 280 × g).
-
12.
Put the plate on the magnetic stand. Inspect visually until the liquid is clear (ca. 5 min). Transfer 15 μl supernatant to the new plate (adapter ligation plate). This is the first safe stopping point, where the sealed plate can be stored at −20 °C for 1 week.
3.2.4 3′ End Adenylation and Adapter Ligation
During these steps, single adenine nucleotides are added to the 3′ fragment ends to prevent fragment ligation during addition of adapters. Next, indexing adapters are ligated to 3′ fragment ends. These will later hybridize fragments to the flow cell. The ligated fragments can be stored for 1 week (second safe stopping point).
-
1.
Add 2.5 μl Resuspension buffer to all wells of the adapter ligation plate and spin down (5 s, 600 × g).
-
2.
Pipette 12.5 μl A-Tailing Mix to each well and mix by shaking (2 min, 1800 rpm).
-
3.
Cover plate with Microseal “B” and spin down (1 min, 280 × g).
-
4.
Incubate on 37 °C microheating system (30 min), then transfer to 70 °C microheating system (5 min, lid closed), and cool down on ice (1 min).
-
5.
Spin down TruSeq® RNA CD Index Plate (1 min, 280 × g).
-
6.
Transfer to all wells of the adapter ligation plates in this order:
-
(a)
2.5 μl Resuspension buffer
-
(b)
2.5 μl Ligation mix
-
(c)
2.5 μl RNA Adapters from the Index adapter Plate (to each corresponding well).
And mix by shaking (2 min, 1800 rpm).
-
(a)
-
7.
Spin down (1 min, 280 × g) plate and transfer to the microheating system (10 min, 30 °C, lid closed), and then cool down on ice.
-
8.
Spin down the Stop Ligation Buffer (5 s, 600 × g) and add 5 μl to every well. Mix by shaking (2 min, 1800 rpm).
-
9.
Spin down the plate (1 min, 280 × g).
-
10.
Add 42 μl AMPure XP beads to every well. Mix by shaking (2 min, 1800 rpm).
-
11.
Incubate at room temperature (15 min), and then spin down the plate (1 min, 280 × g).
-
12.
Put the plate on the magnetic stand. Inspect visually until the liquid is clear (ca. 5 min). Remove all supernatant.
-
13.
Wash the beads with the plate retained on the magnetic stand by adding 200 μl ethanol 80% to all wells. After 30 s remove ethanol.
-
14.
Repeat step 13 of Subheading 3.2.4 and carefully remove all remaining ethanol (using a low-volume pipette). Let samples air dry on the magnetic stand (15 min).
-
15.
Remove plate form stand and pipette 52.5 μl of Resuspension buffer to every well, and mix by shaking (2 min, 1800 rpm).
-
16.
Incubate at room temperature (2 min).
-
17.
Spin down the plate (1 min, 280 × g) and place it on the magnetic stand. Inspect visually until the liquid is clear (ca. 5 Min).
-
18.
Transfer 50 μl supernatant well by well to a novel plate (Clean Up ALP Plate).
-
19.
Repeat steps 10–17 of Subheading 3.2.4, but use 50 μl AMPure XP beads in step 10 and 22.5 μl Resuspension buffer in step 15.
-
20.
Transfer 20 μl supernatant well by well to a novel plate (PCR plate). Sealed plate can be kept at −20 °C for 1 week (this is the second safe stopping point).
3.2.5 DNA Fragment Enrichment
PCR is used to amplify the library and to select for DNA fragments with successful adapter ligation.
-
1.
On ice, pipette 5 μl PCR Primer Cocktail and 25 μl PCR Master Mix to each well, mix by shaking (20 s, 1600 rpm), and spin down (1 min, 280 × g).
-
2.
Transfer to thermocycler with the preheated lid set to 100 °C and run the program:
-
(a)
98 °C for 30 s.
-
(b)
15 cycles of:
-
98 °C for 10 s
-
60 °C for 30 s
-
72 °C for 30 s.
-
-
(c)
72 °C for 5 min.
-
(d)
Hold at 4 °C.
-
(a)
-
3.
Spin down plate (1 min, 280 × g) and pipette 47.5 μl AMPure XP beads to every well. Mix by shaking (2 min, 1800 rpm).
-
4.
Incubate at room temperature (15 min), and then spin down (1 min, 280 × g).
-
5.
Put the plate on the magnetic stand. Inspect visually until the liquid is clear (ca. 5 min). Remove all supernatant.
-
6.
Wash the beads with the plate retained on the magnetic stand by adding 200 μl ethanol 80% to all wells. After 30 s, remove ethanol.
-
7.
Repeat step 6 of Subheading 3.2.5 and carefully remove all remaining ethanol (using low-volume pipette). Let the samples air-dry on magnetic stand (15 min).
-
8.
Resuspend the beads in 32.5 μl Resuspension buffer. Mix by shaking (2 min, 1800 rpm).
-
9.
Incubate at room temperature (2 min), and then spin down (1 min, 280 × g).
-
10.
Put plate on magnetic stand. Inspect visually until the liquid is clear (ca. 5 min), and transfer 30 μl supernatant to a novel plate (Target sample plate 1). Libraries can be kept at −20 °C for 1 week (this the third safe stopping point).
3.2.6 Library Quality Check, Normalization, and Pooling
Library quantity and fragment size are determined using the Bioanalyzer. Indexed libraries are pooled prior to sequencing (see Note 12).
-
1.
Quantify library concentration by qPCR as outlined in manufacturers protocol (lluminaSequencing Library qPCR Quantification Guide (document # 11322363) [https://support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/qpcr/sequencing-library-qpcr-quantification-guide-11322363-c.pdf].
-
2.
Run 1 μl of each sample on the Agilent Bioanalyzer 2100 using the DNA 1000 chip. The principle follows the outline given for RNA quality control (Subheading 3.1). Average fragment size of a typical library will be ca. 260 bp.
-
3.
Pipette 10 μl library well by well to a novel plate (diluted cluster template plate), and adjust the concentration of the libraries to 10 nM using Tris–HCl 10 mM, pH 8.5 with 0.1% Tween 20. Mix by shaking (2 min, 1000 rpm) and spin down (1 min, 280 × g).
-
4.
Pool the libraries according to the system guide of the Illumina sequencing platform that is used. Pooling and normalization should be performed immediately before sequencing to avoid index hopping.
3.2.7 Sequencing
Sequencing of the libraries is performed on an Illumina® sequencing system. Multiple configurations exist which provide the targeted output of 30 million reads per sample (e.g., Illumina’s NextSeq 550, 1000 & 2000 and NovaSeq 6000 systems). Depending on the selected sequencing system, flow cell, and read length, between 4 and 132 samples (maximum: two S4 flow cells on NovaSeq 6000 with 2x100 bp sequencing) can be multiplexed in one sequencing run (see also Notes 12 and 13). For further details, please refer to standard protocol of the manufacturer for the preferred sequencing system.
3.2.8 Raw Data Processing
When sequencing multiple samples on the same sequencing run, outputs have to be demultiplexed to create individual FASTQ files for each respective sample. This is most commonly done using bcl2fastq, a tool developed by Illumina® and pre-installed on most Illumina® sequencers (https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html). Note that run outputs usually include already demultiplexed reads using default bcl2fastq options (mainly allowing one mismatch in the barcode sequence). Alternatively, bcl2fastq can be run on a Linux system using different (usually more stringent, to avoid in silico contamination) options.
3.2.9 Quality Control
FASTQ files include quality information per sequence base, which can be visualized either using the Illumina graphical software (“Sequencing Analysis Viewer”) that is pre-installed on the sequencing machines or by external software that takes the demultiplexed FASTQ files as input. The most commonly used are FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), which outputs an HTML file per sample, and multiqc, which processes multiple FastQC-outputs into a single HTML file to analyze batches [15]. FastQC was developed for DNA applications and might output QC failures for some parameters when used for RNA-Seq reads. Nonetheless, it can be used to quickly and easily check per base sequencing quality, the number of input reads, and adapter content in the reads.
3.2.10 Adapter Trimming
When read length exceeds DNA insert size, a run can sequence beyond the DNA insert and read bases from the sequencing adapter. To prevent these bases from appearing in FASTQ files, the adapter sequence is trimmed from the 3′ ends of reads. Trimming the adapter sequence improves alignment accuracy and performance in Illumina FASTQ generation pipelines [https://support-docs.illumina.com/SHARE/AdapterSeq/DNAandRNACDIndexes.html].
3.3 Bioinformatic Analysis of the Sequencing Data Using ARResT/Interrogate
We use the ARRest/Interrogate immunoprofiling platform for data analysis, which has been developed and validated within EuroClonality-NGS [16,17,18].
-
1.
ARResT/Interrogate can be accessed at http://arrest.tools/interrogate and requires an account that can be created by emailing contact@arrest.tools (see Note 14).
-
2.
Once logged in, select the “Interrogate.WholeMark” user mode at the top-left of the user interface.
-
3.
Switch to the “processing” panel, and follow the instructions to upload the samples in compressed FASTQ format (the extension should be “.fastq.gz”) (see Note 15).
-
4.
Click on the blue “test it” button. If the test was OK, one should be able to click on the green “process” button. If not, check the “process output” tab for feedback; email contact@arrest.tools if necessary.
-
5.
When the run is complete (see Note 16), follow the instructions on the user interface to retrieve the result files.
-
6.
The main result file is a table in both tab-delimited and Microsoft Excel formats.
-
7.
The table contains information on the analyzed samples and on the reported rearrangements. The “usable” column refers to the sum of fragments with rearrangements and should be in the 1000s. Other columns currently include the rearranged genes and their approximate genomic coordinates; the junction class (rearrangement type), amino acid and nucleotide sequence, and its segmentation; the absolute and relative abundances, in fragments and in reads; technical comments; and the consensus nucleotide sequence of the rearrangement. Column descriptions are provided.
-
8.
The sorting and filtering to obtain a meaningful set of rearrangements depends on the context (see Notes 17 and 18). We will be releasing further guidelines for interpretation over time; please stay updated via ARResT/Interrogate [arrest.tools/interrogate] and EuroClonality-NGS [euroclonalityngs.org] (see Note 19).
4 Notes
-
1.
Alternatively, an “LS” workflow (for <48 samples) is also described in the manufacturer’s protocol which requires no additional technical equipment.
-
2.
RNA degrades at room temperature. Keep RNA on ice at all times. On warm days, ensure that the ice does not melt. Always wear gloves. Use RNAse-free materials and reagents. Clean surfaces of bench and flow cabinets with RNaseZap before use.
-
3.
If you prepare an RNA-Seq library for the first time, we strongly advise to take note of the manufacturer’s protocol, which includes further systematic considerations.
-
4.
We have successfully performed transcriptome sequencing using RNA isolated by silica columns (Qiagen©), guanidinium thiocyanate-phenol-chloroform extraction (TRIzol©), or automated RNA-purification (Maxwell ©).
-
5.
Note that syringes have to be replaced with each reagent kit. Make sure that the electrode cartridge is inserted to the Bioanalyzer and that the vortex mixer is adjusted to 2400 rpm. It is recommended to follow a daily electrode decontamination to avoid decomposition of the RNA samples.
-
6.
Heat samples (70 °C, 2 min) before loading to minimize secondary structure.
-
7.
Illumina® TruSeq Stranded mRNA Kit protocol requires a RIN of 8.0 or higher. RNA from primary patient samples is a precious and often irreplaceable material. Therefore, we have also used samples with RINs between 6.0 and 8.0 to allocate ALL samples to molecular disease subtypes based on gene expression profiling and calling of driver gene fusions. However, the frequency of samples which could not be allocated to a specific leukemia driver subtype increased substantially with decreasing RIN. While still more than half of the samples could be classified with a RIN between 6.00 and 8.00, this rate was markedly lower in a small sample set with RINs <6.0. This should be kept in mind when processing samples with low RINs. However, PCR-based approaches might still be successfully performed in samples with reduced RNA quality.
-
8.
Safe stopping points are indicated throughout the protocol. Before proceeding to the next step, carefully read the protocol, and make sure that you have all reagents required brought to the indicated temperatures, that heating systems are preheated to the temperatures required, and that thermal cyclers have been programmed according to the given programs.
-
9.
Magnetic bead wash steps are critical for nucleic acid and sequencing quality. Make sure to vortex the beads thoroughly before use and then pipette immediately, to avoid settling of the beads within the container. It might be helpful to prepare aliquots in smaller tubes and vortex more often. When using multistep pipettes with beads, choose smaller volumes, and pipette quickly to avoid settling of the beads (concentration gradient) within the pipette tip.
-
10.
Ethanol is hygroscopic (attracts and holds water from environment). Fresh aliquots of absolute alcohol should be used for every library preparation and dilutions, for example, 80% ethanol in water should be made on the day of use to ensure correct concentrations. Incorrect ethanol concentrations can decrease yield of DNA/RNA.
-
11.
Remnants of wash buffer (typically including alcohol) can disrupt further library preparation steps. On the other hand, overdried beads can crack and destroy the bound nucleic acids. If unsure whether beads are dry, fanning and smelling the tubes can be an indicator for remaining ethanol.
-
12.
We use RNA-Seq to simultaneously analyze IG /TR gene rearrangements in ALL samples and to perform molecular subgroup allocation based on gene expression profiles and driver gene fusions. For this approach, we have successfully used 75 bp or 100 bp paired end sequencing on Illumina HiSeq2000, NextSeq, and NovaSeq systems, aiming for 30 million reads per sample on all sequencers. Currently, we pool 66 samples on a NovaSeq S4 flow cell for 100 bp paired end sequencing, which yields on average 30–40 million reads per sample.
-
13.
Sequencing systems offer different read lengths for RNA-Seq, typically ranging from 50 bp to 150 bp. Longer reads (100–150 bp) provide a better coverage of transcripts and splice sites, while shorter reads (50–75 bp) are typically used when gene expression profiling is the only intended use. In a small patient subset, molecular subgroup allocation and driver fusion calling was equally effective when the sequencing read length was 75 bp, compared to our standard of 100 bp. Also based on general considerations, we would recommend a read length of 100 bp or more for marker identification from transcriptome data sets. We typically aim for a sequencing depth of 30 million reads, which is suitable for gene expression profiling, fusion calling, and marker identification. Less sequencing depth (5–25 million reads) is required for gene expression profiling only. Increasing sequencing depth to >30 million reads will improve the detection of subclonal and less covered markers.
-
14.
Details for accessing the bioinformatics pipeline and its results may change. In any case, the latest information will always be available either through the user interface of ARResT/Interrogate at http://arrest.tools/interrogate or through the authors.
-
15.
Dependent on the number of samples and quality of internet connection, uploading data can take time.
-
16.
Progress of the bioinformatic pipeline can be followed in the “process output” tab. The user does not have to wait; one may even close the browser and either log in later or better make sure to provide an email address to receive email notifications.
-
17.
In ALL patients, many rearrangements are incomplete or nonproductive, and therefore it is crucial to include such rearrangements in the analysis and not to filter on productive rearrangements only. In mature lymphoid malignancies, by definition, at least one productive IGH rearrangement should be present.
-
18.
Incomplete and nonproductive rearrangements may not be transcribed or only at very low levels (Fig. 2). It is not yet known to what extent transcription levels of cross-lineage TR gene rearrangements in leukemic rearrangements differ from transcription levels in reactive T cells. Higher read depth may facilitate identification of lowly expressed IG /TR mRNA.
-
19.
Of note, IG /TR rearrangements may also be derived from whole exome sequencing (WES) or whole genome sequencing (WGS) data sets that, in contrast to RNA-Seq data, do not depend on the transcriptional level of rearrangements. This creates a clear advantage as was recently showcased in work introducing IgCaller for WGS-derived IGH data [19].

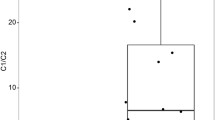
Comparison between IG /TR rearrangements detected by RNA-Seq and amplicon-based assays (“DNAamp”) in 165 ALL patients [13]. Average number of rearrangements detected for the various IG /TR loci per case
References
Pieters R, de Groot-Kruseman H, Van der Velden V, Fiocco M, van den Berg H, de Bont E et al (2016) Successful therapy reduction and intensification for childhood acute lymphoblastic leukemia based on minimal residual disease monitoring: study ALL10 from the Dutch childhood oncology group. J Clin Oncol 34:2591–2601
Stutterheim J, van der Sluis IM, de Lorenzo P, Alten J, Ancliffe P, Attarbaschi A et al (2021) Clinical implications of minimal residual disease detection in infants with KMT2A-rearranged acute lymphoblastic leukemia treated on the Interfant-06 protocol. J Clin Oncol 39:652–662
Flohr T, Schrauder A, Cazzaniga G, Panzer-Grumayer R, van der Velden V, Fischer S et al (2008) Minimal residual disease-directed risk stratification using real-time quantitative PCR analysis of immunoglobulin and T-cell receptor gene rearrangements in the international multicenter trial AIEOP-BFM ALL 2000 for childhood acute lymphoblastic leukemia. Leukemia 22:771–782
Van Dongen JJM, van der Velden VHJ, Brüggemann M, Orfao A (2015) Minimal residual disease diagnostics in acute lymphoblastic leukemia: need for sensitive, fast, and standardized technologies. Blood 125:3996–4009
Van der Velden VHJ, Cazzaniga G, Schrauder A, Hancock J, Bader P, Panzer-Grümayer ER et al (2007) Analysis of minimal residual disease by Ig/TCR gene rearrangements: guidelines for interpretation of real-time quantitative PCR data. Leukemia 21:604–611
Van der Velden VHJ, Panzer-Grümayer ER, Cazzaniga G, Flohr T, Sutton R, Schrauder A et al (2007) Optimization of PCR-based minimal residual disease diagnostics for childhood acute lymphoblastic leukemia in a multi-center setting. Leukemia 21:706–713
van der Velden VHJ, van Dongen JJM (2009) MRD detection in acute lymphoblastic leukemia patients using Ig/TCR gene rearrangements as targets for real-time quantitative PCR. Methods Mol Biol 538:115–150
Brüggemann M, Kotrova M, Knecht H, Bartram J, Boudjogrha M, Bystry V et al (2019) Standardized next-generation sequencing of immunoglobulin and T-cell receptor gene recombinations for MRD marker identification in acute lymphoblastic leukaemia; a EuroClonality-NGS validation study. Leukemia 33:2241–2253
Bastian L, Schroeder MP, Eckert C, Schlee C, Tanchez JO, Kampf S et al (2019) PAX5 biallelic genomic alterations define a novel subgroup of B-cell precursor acute lymphoblastic leukemia. Leukemia 33:1895–1909
Li Z, Jiang N, Lim EH, Chin WHN, Lu Y, Chiew KH et al (2020) Identifying IGH disease clones for MRD monitoring in childhood B-cell acute lymphoblastic leukemia using RNA-Seq. Leukemia 34:2418–2429
Bueno C, Ballerini P, Varela I, Menendez P, Bashford-Rogers R (2020) Shared D-J rearrangements reveal cell of origin of TCF3-ZNF384 and PTPN11 mutations in monozygotic twins with concordant BCP-ALL. Blood 136:1108–1111
Abdo C, Thonier F, Simonin M, Kaltenbach S, Valduga J, Petit A et al (2020) Caution encouraged in next-generation sequencing immunogenetic analyses in acute lymphoblastic leukemia. Blood 136:1105–1107
Van der Velden VHJ, Brüggemann M, Cazzaniga G, Scheijen B, Tops B, Trka J et al (2021) Potential and pitfalls of whole transcriptome-based immunogenetic marker identification in acute lymphoblastic leukemia; a EuroMRD and EuroClonality-NGS working group study. Leukemia 35:924–928
Schroeder A, Mueller O, Stocker S, Salowsky R, Leiber M, Gassmann M et al (2006) The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol 7:3
Ewels P, Magnusson M, Lundin S, Kaller M (2016) MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32:3047–3048
Bystry V, Reigl T, Krejci A, Demko M, Hanakova B, Grioni A et al (2017) ARResT/interrogate: an interactive immunoprofiler for IG/TR NGS data. Bioinformatics 33:435–437
Knecht H, Reigl T, Kotrova M, Appelt F, Stewart P, Bystry V et al (2019) Quality control and quantification in IG/TR next-generation sequencing marker identification: protocols and bioinformatic functionalities by EuroClonality-NGS. Leukemia 33:2254–2265
Scheijen B, Meijers RWJ, Rijntjes J, Van der Klift MY, Mobs M, Steinhilber J et al (2019) Next-generation sequencing of immunoglobulin gene rearrangements for clonality assessment: a technical feasibility study by EuroClonality-NGS. Leukemia 33:2227–2240
Nadeu F, Mas-de-Les-Valls R, Navarro A, Royo R, Martin S, Villamor N et al (2020) IgCaller for reconstructing immunoglobulin gene rearrangements and oncogenic translocations from whole-genome sequencing in lymphoid neoplasms. Nat Commun 11:3390
Acknowledgments
We thank the members of EuroClonality and EuroMRD for their support, especially the participants of the WholeMark work package (Blanca Scheijen, Bastiaan Tops, Jan Trka, Karol Pál, Sonja Hänzelmann, Gianni Cazzaniga, Grazia Fazio, Simona Songia, and Anton W. Langerak).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this protocol

Cite this protocol
van der Velden, V.H.J., Bastian, L., Brüggemann, M., Hartmann, A.M., Darzentas, N. (2022). Immunoglobulin/T-Cell Receptor Gene Rearrangement Analysis Using RNA-Seq. In: Langerak, A.W. (eds) Immunogenetics. Methods in Molecular Biology, vol 2453. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-2115-8_4
Download citation
DOI: https://doi.org/10.1007/978-1-0716-2115-8_4
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-2114-1
Online ISBN: 978-1-0716-2115-8
eBook Packages: Springer Protocols