
Abstract
AIRR-seq data sets are usually large and require specialized analysis methods and software tools. A typical Illumina MiSeq sequencing run generates 20–30 million 2 × 300 bp paired-end sequence reads, which roughly corresponds to 15 GB of sequence data to be processed. Other platforms like NextSeq, which is useful in projects where the full V gene is not needed, create about 400 million 2 × 150 bp paired-end reads. Because of the size of the data sets, the analysis can be computationally expensive, particularly the early analysis steps like preprocessing and gene annotation that process the majority of the sequence data. A standard desktop PC may take 3–5 days of constant processing for a single MiSeq run, so dedicated high-performance computational resources may be required.
VDJServer provides free access to high-performance computing (HPC) at the Texas Advanced Computing Center (TACC) through a graphical user interface (Christley et al. Front Immunol 9:976, 2018). VDJServer is a cloud-based analysis portal for immune repertoire sequence data that provides access to a suite of tools for a complete analysis workflow, including modules for preprocessing and quality control of sequence reads, V(D)J gene assignment, repertoire characterization, and repertoire comparison. Furthermore, VDJServer has parallelized execution for tools such as IgBLAST, so more compute resources are utilized as the size of the input data grows. Analysis that takes days on a desktop PC might take only a few hours on VDJServer. VDJServer is a free, publicly available, and open-source licensed resource. Here, we describe the workflow for performing immune repertoire analysis on VDJServer’s high-performance computing.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others

Key words
1 Introduction
Immune repertoire sequencing produces large, highly complex data sets that require specialized analysis methods and software tools. We developed VDJServer to address critical barriers in broader adoption of immune repertoire sequencing, namely, the lack of a complete, start-to-finish analysis pipeline, the lack of a data management infrastructure, and limited access for many researchers to highperformance computing (HPC) resources. VDJServer fills these gaps, specifically providing (1) an open suite of interoperable repertoire analysis tools that allows users to upload a set of sequences and pass them through a seamless workflow that executes all steps in an analysis, (2) access to sophisticated analysis tools running in an HPC environment, (3) interactive visualization capabilities for exploratory analysis, (4) a data management infrastructure, and (5) a graphical user interface to facilitate use by experimental and clinical research groups that lack extensive bioinformatics expertise.
Here, we describe the workflow for performing immune repertoire analysis on VDJServer’s high-performance computing. The major steps of the workflow include creating a project to hold sequencing data and analysis results, uploading and preparing immune repertoire sequencing files, preprocessing the raw sequence data, performing V(D)J assignment and annotation of the processed sequences, defining study metadata and analysis comparison groups, performing repertoire characterization and comparison, and visualizing and downloading analysis results.
2 Materials
VDJServer requires a user account with a valid email address to access the system. Creating an account is free, as well as using the VDJServer resources. Accounts are used to insure data and results are private and secure. Create an account at https://vdjserver.org to get started. Contact VDJServer with any questions or concerns by using the Feedback option on the website or send email to at vjdserver@utsouthwestern.edu.
3 Methods
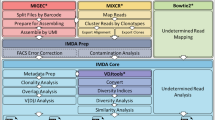
For researchers without access to high-performance computing (HPC), VDJServer provides free access to the Texas Advanced Computing Center (TACC) through a standard web browser via a graphical user interface [1]. A suite of tools for a complete analysis workflow are provided, including modules for preprocessing and quality control of sequence reads, V(D)J gene assignment, repertoire characterization, and repertoire comparison (Fig. 1). VDJServer incorporates analysis software from the Immcantation suite [2, 3], VDJPipe [4], and other interoperability tools [5, 6]. Germline gene sets for human and mouse are derived from IMGT [7], and a draft germline set for Indian origin rhesus macaque IG is also provided [8]. VDJServer provides the Community Data Portal for publicly sharing data and analysis results, and studies can be published to the AIRR Data Commons [9], which is not covered in this workflow. To publicly share data, please see the AIRR Community method chapter entitled, “Data Sharing and Re-Use.” Here, we discuss the different steps in the immune repertoire analysis workflow using VDJServer.

Workflow immune repertoire analysis on high-performance computing using VDJServer V1
3.1 Create Project
After login with a user account, click on the Add Project button to create a new project and give the project a name. Each project is a logical container for files, jobs, analysis results, and visualizations, and any number of projects may be created. All data in a project is private to the user account but can be shared with other VDJServer users by adding them on the Project Settings page (see Note 1).
3.2 Upload Files into Project
From the Upload and Browse Project Data page, click on the Upload button and select files from the local computer, from Dropbox, or from a URL (ftp/http), to be uploaded; multiple files can be selected (see Note 2). Click the Start button to start uploading. Upload FASTQ sequence read files (compress with gzip for faster upload), FASTA files with barcode and primer sequences, TSV containing metadata, or any file to be associated with the project.
3.3 Set File Attributes and Link Paired-End Read Files
VDJServer attempts to detect the file type (FASTQ, FASTA, AIRR TSV, etc.) from the file extension, but this can be changed with the File Type setting. Use Barcode or Primer for files containing those sequences. For paired-end read sequencing files, set the Read Direction on each file to either the Forward or Reverse orientation, and then link the two files together on the Link Paired Read Files page. The forward orientation refers to the V gene end of the template, and reverse orientation refers to the J gene or constant region end of the template. Correct orientation is necessary for proper matching of barcodes and primers. Linked files will show together as a pair on the Upload and Browse Project Data page.
3.4 Preprocessing with VDJPipe or pRESTO
From the Upload and Browse Project Data page, select sequence read files for preprocessing by clicking on the checkbox next to each file. Click on the Run Job button and select either VDJPipe or pRESTO; a job submission screen will be displayed. VDJPipe and pRESTO have similar capabilities; pRESTO should be used for UMI ; otherwise, VDJPipe is significantly faster (up to 20×) on larger data sets. A single workflow is available for pRESTO, while VDJPipe offers a number of customized workflows. VDJPipe’s single function workflows perform individual preprocessing steps, while the complete workflow performs all steps. If unsure about filtering parameters to use for preprocessing, such as length or quality settings, it is useful to run VDJPipe’s Sequence Statistics workflow. This will visualize length, quality, and nucleotide distributions of the read data. The job submission screen will provide parameters, with default values, that can be changed for the individual preprocessing steps (see Note 3). Finally, click the Launch Job button to submit the preprocessing job to the TACC supercomputer. The user will receive an email when the job is finished.
3.5 Review Preprocessing Statistics
When the preprocessing job is complete, the job on the View Analyses and Results page will change from an In Progress label to a View Output button. Click the button to show the View Output page, which has three main sections: job output files, analysis charts, and log files. Job output files provides a list of output files generated from the preprocessing job. Analysis Charts provides visualizations for pre- and post-filtering statistics, and log files are job error logs and workflow provenance metadata. The provided visualizations include:
-
1.
Nucleotide composition for each read position.
-
2.
GC% histogram.
-
3.
Sequence length histogram.
-
4.
Mean quality score histogram.
-
5.
Quality score distribution for each read position.
Use the Analysis Charts to review the preprocessing results; they show the pre- and post-filtering statistics to understand how preprocessing has affected the data. If preprocessing removed too many reads, or alternatively has not removed enough, then a new preprocessing job should be run with looser or more stringent parameters. Among the job, log files are summary logs that will give information about the number of reads processed during each preprocessing step.
3.6 Make Job Output Files Available in Project Data Area
Once satisfied with the preprocessing results, the appropriate job output files need to be made available in the project data area so that they can be selected as input for additional analysis jobs. This can be done in two ways. The first is on the View Analyses and Results page. Click the Job Actions button for the job and select Include Job Output; this will make all output files available. Conversely, select Exclude Job Output from the Job Actions button, which will remove all output files for the project data area. Alternatively, the second way, the user can make individual job output files available from the View Output page for the job by clicking the Make Available in Project Data Area button next to each file. Clicking that button again will remove the file from the project data area. Job output files available in the project data area will show in their own section on the Upload and Browse Project Data page, grouped together by the job with the job name as a title.
3.7 Gene Annotation with IgBLAST
Select files for IgBLAST processing, either job output files or uploaded FASTA files, on the Upload and Browse Project Data page by clicking on the checkbox next to each file (see Note 4). Click on the Run Job button and select IgBLAST; a job submission screen will be displayed. Select the organism species (human, mouse, or rhesus macaque), the strain (if appropriate), and the sequence type (IG or TR ). VDJServer maintains separate germline databases, so processing multiple sequence types and/or organism species requires running multiple IgBLAST jobs. Finally, click the Launch Job button to submit the preprocessing job to the TACC supercomputer. The user will receive an email when the job is finished.
As with all analysis jobs on VDJServer, job status is shown on the View Analyses and Results page, and the job output is available with the View Output button. Multiple output formats are provided including VDJServer’s custom RepSum TSV, VDML, Change-O TSV, and AIRR TSV. Individual files can be downloaded by clicking on the filename, or all output files can be downloaded by clicking on Archive of Output Files in the log file section. It is recommended that AIRR TSV files are used for any custom analysis as they contain the most comprehensive annotations, and they are interoperable with many AIRR-seq tools.
3.8 Define MiAIRR Study Metadata and Repertoire Comparison Groups
By this point in the workflow, raw sequence data has been preprocessed, and sequences have been annotated. However, to achieve the greatest utility of repertoire analysis, it is recommended that metadata is entered and comparison groups are defined, though it is not strictly necessary as individual files can be analyzed in isolation. Entering metadata also has the benefit of providing MiAIRR compliance when it’s time to publish the study. Metadata is entered on the Metadata Entry page and consists of the six MiAIRR components: study, subject, diagnosis, sample, cell processing, and nucleic acid processing. VDJServer adds a seventh component with sample groups for doing group comparisons. Metadata can be manually entered on the page, but it is typically more efficient to prepare the metadata in a separate spreadsheet file, then import that spreadsheet into VDJServer (see Note 5). To do this, go to the appropriate section on the Metadata Entry page, click on the Metadata Actions button, and select Export to File. Open the spreadsheet file in Excel or another program, use one row for each entry, and fill in the values for each column. Save the file as Tab-delimited Text and upload the file into the project. Finally, on the Metadata Entry page, click on the Metadata Actions button and select Import From File. A panel will be shown where the user can pick the file to import and choose to either replace or append the current metadata.
Sample groups are a specialized feature of VDJServer that allows sample repertoires to be grouped together for performing intragroup and intergroup comparisons. Sample groups are defined by using one or more grouping operations. These grouping operations include:
-
1.
Grouping by the values of a study metadata field. The Group By option provides a popup list of all the possible fields. VDJServer will determine all of the values for that field among the study metadata and create a separate group for each value.
-
2.
Grouping by a logical operation. The Logical option defines a simple Boolean expression. Sample repertoires where that expression evaluates as true will be included in the sample group, while those for which the expression evaluates as false will be excluded. Pick the study metadata field from the popup list of available fields, pick the comparison operator, and provide a value.
-
3.
Individual samples can be picked. The Repertoires option provides a popup list of sample repertoires. By default, when no sample repertoires are selected, then all sample repertoires are included in the sample group. Click on specific sample repertoires in the list to include them in the sample group.
-
4.
The three grouping operations can be combined together. A sample repertoire needs to satisfy all applicable grouping operations to be included in the sample group.
3.9 Repertoire Characterization and Comparison with RepCalc
RepCalc performs a wide variety of analysis functions including clonal assignment, gene usage, gene combination usage, CDR3 length distribution and amino acid properties, CDR3 and clonal sharing and uniqueness, clonal abundance, diversity profile, and B cell–specific mutation analysis and clonal lineage. RepCalc uses a combination of tools to perform the analyses including VDJServer’s custom repertoire summarization and Change-O, Alakazam, and SHazaM from the Immcantation suite.
To run RepCalc, no files need to be selected on the Upload and Browse Project Data page; instead, RepCalc will directly access the appropriate output files from a previous IgBLAST job. Click the Run Job button and select RepCalc; a job submission screen will be displayed. Pick the IgBLAST job to use as input. If study metadata was defined, the screen will indicate its availability and automatically perform group comparison; otherwise, RepCalc will only perform analysis on individual files. Change the default values to include or exclude specific analysis functions. Finally, click the Launch Job button to submit the job to the TACC supercomputer. The user will receive an email when the job is finished.
3.10 Visualize Analysis Results and Download Data
When the RepCalc job has completed successfully, click the View Output button on the View Analyses and Results page to display the analysis results. For RepCalc jobs, the View Output page has three main sections: job output files, analysis charts, and log files. Job output files provides a list of clonal assignment output files, Analysis charts provide analysis visualizations, and log files are job error logs and workflow provenance metadata. RepCalc produces a set of interactive analysis charts:
-
1.
Absolute and relative gene usage.
-
2.
Nucleotide and amino acid CDR3 length distribution.
-
3.
Clonal abundance and cumulative abundance.
-
4.
Diversity profile curves.
-
5.
Selection pressure quantification.
Each chart provides three pop-up lists for selecting files, sample repertoires, or sample groups to be displayed on the chart. Chart figures can be downloaded by clicking on the Download Chart button, which will generate a figure identical to the chart being displayed in the browser, and the data for the chart can be downloaded by clicking on the Download Data button. Not all analysis output has an associated visualization but can be downloaded by clicking on Archive of Output Files in the log file section, with the data provided in TSV format for easy import into Excel and other tools.
4 Notes
-
1.
Processing on shared facilities external to the user’s institution raises privacy concerns. We recommend that data is fully anonymized before analysis begins.
-
2.
Uploading a large number (hundreds) of files at one time is susceptible to network errors and timeouts that may prevent a file or two from not being uploaded. Be sure to check the total file count to insure it matches. If files are missing, use the Search field to narrow the list and verify. Another technique is to upload the files in batches, e.g., 20 files at a time, and check after each batch that all the files got uploaded.
-
3.
Steps that will not be used, as indicated by a red warning box, should be removed by clicking on the red X button.
-
4.
With many files, use the Search field to restrict the files shown to just the desired files, and then click Select All to select them all at once.
-
5.
While VDJServer V1 collects MiAIRR study metadata, it does not yet utilize the AIRR Standards Repertoire metadata format for import/export interoperability. However, VDJServer V2 will directly utilize the AIRR Data Model and AIRR Standards data formats. Conversion scripts exist for converting VDJServer V1 into VDJServer V2 metadata so no data will be lost or require reentry.
References
Christley S, Scarborough W, Salinas E, Rounds WH, Toby IT, Fonner JM et al (2018) VDJServer: a cloud-based analysis portal and data commons for immune repertoire sequences and rearrangements. Front Immunol 9:976
Gupta NT, Vander Heiden JA, Uduman M, Gadala-Maria D, Yaari G, Kleinstein SH (2015) Change-O: a toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics 31:3356–3358
Vander Heiden JA, Yaari G, Uduman M, Stern JN, O'Connor KC, Hafler DA et al (2014) pRESTO: a toolkit for processing high-throughput sequencing raw reads of lymphocyte receptor repertoires. Bioinformatics 30:1930–1932
Christley S, Levin MK, Toby IT, Fonner JM, Monson NL, Rounds WH et al (2017) VDJPipe: a pipelined tool for pre-processing immune repertoire sequencing data. BMC Bioinformatics 18:448
Toby IT, Levin MK, Salinas EA, Christley S, Bhattacharya S, Breden F et al (2016) VDJML: a file format with tools for capturing the results of inferring immune receptor rearrangements. BMC Bioinformatics 17:333
Vander Heiden JA, Marquez S, Marthandan N, Bukhari SAC, Busse CE, Corrie B et al (2018) AIRR community standardized representations for annotated immune repertoires. Front Immunol 9:2206
Giudicelli V, Brochet X, Lefranc MP (2011) IMGT/V-QUEST: IMGT standardized analysis of the immunoglobulin (IG) and T cell receptor (TR) nucleotide sequences. Cold Spring Harb Protoc 2011:695–715
Cottrell CA, van Schooten J, Bowman CA, Yuan M, Oyen D, Shin M et al (2020) Mapping the immunogenic landscape of near-native HIV-1 envelope trimers in non-human primates. PLoS Pathog 16:e1008753
Christley S, Aguiar A, Blanck G, Breden F, Bukhari SAC, Busse CE et al (2020) The ADC API: a web API for the programmatic query of the AIRR data commons. Frontiers Big Data 3:22
Author information
Authors and Affiliations
Consortia
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this protocol

Cite this protocol
Christley, S., Stervbo, U., Cowell, L.G., on behalf of the AIRR Community. (2022). Immune Repertoire Analysis on High-Performance Computing Using VDJServer V1: A Method by the AIRR Community. In: Langerak, A.W. (eds) Immunogenetics. Methods in Molecular Biology, vol 2453. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-2115-8_22
Download citation
DOI: https://doi.org/10.1007/978-1-0716-2115-8_22
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-2114-1
Online ISBN: 978-1-0716-2115-8
eBook Packages: Springer Protocols