
Abstract
Recently, clustered regularly interspaced palindromic repeats (CRISPR)-Cas9 derived editing tools had significantly improved our ability to make desired changes in the genome. Wild-type Cas9 protein recognizes the target genomic loci and induced local double strand breaks (DSBs) in the guidance of small RNA molecule. In mammalian cells, the DSBs are mainly repaired by endogenous non-homologous end joining (NHEJ) pathway, which is error prone and results in the formation of indels. The indels can be harnessed to interrupt gene coding sequences or regulation elements. The DSBs can also be fixed by homology directed repair (HDR) pathway to introduce desired changes, such as base substitution and fragment insertion, when proper donor templates are provided, albeit in a less efficient manner. Besides making DSBs, Cas9 protein can be mutated to serve as a DNA binding platform to recruit functional modulators to the target loci, performing local transcriptional regulation, epigenetic remolding, base editing or prime editing. These Cas9 derived editing tools, especially base editors and prime editors, can introduce precise changes into the target loci at a single-base resolution and in an efficient and irreversible manner. Such features make these editing tools very promising for therapeutic applications. This review focuses on the evolution and mechanisms of CRISPR-Cas9 derived editing tools and their applications in the field of gene therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Genome editing has become a powerful tool for both basic biomedical research and translational medicine. The development of reprogrammable nucleases, especially clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 system, has revolutionized the field of genome editing [1, 2]. Since the discovery demonstrated that the wild-type (WT) Cas9 system produced double strand breaks at target region in vitro in the guidance of a pair of small RNA molecules, crRNA and tracrRNA, which were later engineered to a single guide RNA (sgRNA) [1, 2], the system soon found its applications in multiple organisms and in a wide range of scenarios [3,4,5,6,7,8,9]. In eukaryotic cells, Cas9 can be used to induce double strands breaks (DSBs) in the genome, which are frequently repaired by non-homologous end joining (NHEJ) pathway since NHEJ is more active than other DNA repair systems in repairing DSBs, leading to the formation of insertion or/and deletion (indel) mutations [5, 10].
Besides producing DSB, Cas9 could also be engineered to renounce the ability of DNA cleavage but reserve the ability of DNA binding, thereby serving as a platform for recruiting other DNA regulators or modifiers to induce localized transcriptional, epigenetic or genetic manipulations. To the best of our knowledge, up to date the platform has been utilized to develop transcriptional regulators [11,12,13,14,15,16,17], histone methylation [18,19,20] or acetylation modifiers [17, 21], DNA methylation modifiers [19], cytosine or adenine base editors [22,23,24,25,26] and primer editors [25, 27,28,29]. Among these tools, base editors and primer editors have the potential for clinical translation because both of them can produce permanent and desired changes in genomic sequences [22, 26, 27, 30, 31]. By recruiting deaminases, base editors produce targeted deamination on cytosines (Cs) or adenines (As) dependending on the types of deaminases, which in turn are converted into thymines (Ts) or guanines (Gs) respectively by endogenous DNA repair or replication mechanisms [22, 23, 32,33,34,35]. Prime editors consist of a Cas9 nickase and reverse transcriptase (RT) fusion protein and a prime editing guide RNA (pegRNA). When binding to target DNAs, the Cas9 nickase introduced a nick in the non-target strand (NTS), which is then bound by the primer binding sequences (PBS) of pegRNA [27]. RT recognized the DNA/RNA duplex and extended the ssDNA under the guidance of RT template of the pegRNA [27]. Finally, the information of the extended ssDNA is written into the genome through endogenous DNA repair mechanisms to accomplish the editing [27, 36,37,38].
These editing tools, together with Cas9 nuclease, enable virtually any desired changes in the genome, therefore have significant potential for clinical applications for disease treatment. Cas9 derived tools have achieved significant success in animal models of human genetic disease as well as cancer, either via inactivating the expression or function of specific genes [39,40,41,42,43], correcting disease relevant mutations to restore gene function [44,45,46,47,48], or reactivating the expression of functional redundant genes to compensate for mutant genes [49,50,51,52]. With the development of in vivo delivery system, it is now practicable to efficiently deliver Cas9 derived tools into multiple target organs, including liver, eye, ear and muscles etc. Up to date, more than 34 clinical trials using Cas9 tools have been recruited, among which several trials have reported therapeutic benefit for patients, highlighting the potential of Cas9 tools in clinical translation. Here, we will introduce recent advances in the evolution and mechanisms of CRISPR-Cas9 derived editing tools, focusing on the stepwise actions of these tools and the responses of endogenous DNA repair pathways. In addition, we will also introduce their applications in the field of gene therapy, with special emphases on the design of editing strategy and the in vivo delivery of these tools.
Cas9 and its derivates
CRISPR system
Reprogrammable nucleases that target DNA molecules are the fundamental players in genome editing, which introduce targeted cleavage of specific DNA sequences. To date, four classes of DNA nucleases that possess genome editing potential have been discovered, which were meganucleases, zinc finger nucleases (ZFNs) [53, 54], transcription activator–like effector nucleases (TALENs) [55,56,57] and CRISPR-associated nucleases [58]. Distinct from the former three nucleases that recognize DNA through sequence-specific protein-DNA interaction, Cas9s utilized a bi-interaction modes to recognize their target DNA, i.e., protein-DNA and RNA–DNA interactions [1, 2].
CRISPR system was first discovered in bacteria genome, where regular sequences consisting of repeat sequences and spacer sequences were found [59]. Later, it was demonstrated to function as a bacterial defense system against invaded foreign DNAs [60]. Upon the invasion of foreign DNAs, such as plasmids and phages, the CRISPR system is activated to cut a small piece of invaded DNA and insert it into the CRISPR locus of bacterial genome [60] The inserted foreign DNAs are then transcribed, together with the sequences from the host genomes, to form fusion RNAs that was named CRISPR repeat RNA (crRNA), which guides CRISPR proteins to cleave the invaded DNAs [2, 61,62,63,64,65].
Cas9 nuclease
In 2012, Jenniffer Doundna and Emmanuelle Charpentier labs provided the first in vitro data demonstrating that Cas9 system derived from Streptococcus pyogenes can be re-engineered to recognize new target DNA [1]. They showed that changing the protospacer sequences of crRNA can guide the Streptococcus pyogenes Cas9 (SpCas9) to recognize target DNA that contains the spacer sequences in front of a motif called protospacer adjacent motif (PAM). By recognizing the target DNA, SpCas9 cleaved both DNA strands 3 nucleotides upstream the PAM, leading to double strand break (DSB). Several months later, Virginijus Siksnys lab reported another Cas9 system, from Streptococcus thermophilus, also capable of introducing in vitro DSB in a reprogrammable manner (Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria) [2]. Soon after these pioneer works, Feng Zhang and George Church labs independently demonstrated that this system can be functionally introduced into mammalian cells to target a large variety of genomic loci [4, 5]. In mammalian cells, DSBs produced by Cas9, or other nucleases are mainly repaired by an error-prone DNA repair mechanism, named non-homologous end joining (NHEJ) pathway, resulting in the formation of insertion and deletion (indel) mutations [4, 5, 66].
Following these works, Cas9s from different microorganisms were investigated. These efforts generated a growing list of Cas9 systems that were capable of editing eukaryotic genomes, including SaCas9 [39, 67,68,69], CjCas9 [70,71,72,73], NmCas9 [74,75,76,77], FnCas9 [78,79,80,81] and SauriCas9 [82, 83] etc. Importantly, these Cas9s recognize different PAMs, which increases the targeting scope of Cas9 based editing strategies (Table 1). Structurally, all Cas9 proteins studied so far exhibit bilobed architecture that consist of one recognition (REC) and one nuclease (NUC) lobes [84]. Three major functional domains were identified, including two nuclease domains, RuvC and HNH, and one PAM-interacting domain (PI domain) (Fig. 1). The PI domain, usually locating in the C-terminus of Cas9 proteins, recognizes the PAM sequences. Upon recognizing PAM, Cas9 sharply bends and undertwists DNA to flip DNA nucleotides out of the duplex and toward the sgRNA. Once the spacer region of the target DNA matched the sgRNA guide sequences, the sgRNA invades into the DNA duplex to partially detached the nontarget strand (NTS), thereby forming an “R-loop” structure [85,86,87,88,89]. The binding of Cas9 protein to the target DNA induced conformational changes in Cas9 structure, which triggers its cleavage activity [84, 87]. The HNH and RuvC nuclease domains of Cas9 protein are responsible for cleavage of the complementary and noncomplementary strands of the target DNA, respectively [84]. Noteworthy, the cleavage of DNA strand is independent to each other [84]. Therefore, Cas9 proteins can be mutated into nickases that nicked either TS or NTS or dead proteins without any cleavage activity [90].

Structure of spCas9/sgRNA/DNA complex. a Overall structure of spCas9/sgRNA/DNA complex, with PI, HNH and RuvC domains shown in yellow, green and blue respectively. b and c Detailed structure showing the interaction between PI domain and PAM (b), with key amino acids highlighted in pink (c). d Detailed structure showing the HNH domain, RuvC domain and target DNA, with key amino acids (D10 and H840) shown in sphere. Mutation of Asparticacid 10 (D10) or Histidine 10 (H840) to Alanine (A) silences the RuvC or the HNH domain respectively, resulting in TS nickase or NTS nickase respectively. Mutation of both amino acids silences both domains, resulting in catalytic dead Cas9. e Detailed structure showing the key amino acids interaction with target strand in HNH domain. f Detailed structure showing the key amino acids interaction with target strand in RuvC domain
Cas12 nuclease
In addition to Cas9, other CRISPR systems were also found to be capable of editing eukaryotic genomes. Up to date, three major types of CRISPR systems were identified, type I-III. They were classified according to their components and action mechanisms [106]. Among these systems, Cas12a (also named Cpf1, hereafter referred as to Cpf1) proteins from type II CRISPR were also frequently used in genome editing because of its simplicity of action and easiness to be reprogrammed, since this system consists of a single nuclease protein and a single RNA component [107,108,109,110]. Like Cas9, Cpf1 recognizes its target DNA also through the two types of interactions, i.e., RNA/DNA and protein/DNA [111]. However, compared with Cas9, Cpf1 protein can only be mutated to form NTS nickase or dead Cpf1 but not TS nickase, because the activation of TS cleavage is triggered by the cleavage of NTS [111]. Therefore, Cpf1 protein is less versatile than Cas9.
Engineered Cas9 variants with expanded targeting scope
As PAM recognition is a key limitation for the targeting scope of Cas9 proteins, many efforts have been made to engineer them to relax the limitation. As mentioned above, structural investigations have revealed that PI domain is responsible for sequence-specific binding of PAM [84, 112] (Fig. 1a, b, c). Under the guidance of structural information, several Cas9 variants had been designed harboring mutations within PI domain to change the PAM preference. Most efforts have been put on SpCas9, leading to the discovery of a panel of SpCas9 PI variant, including Cas9-VQR [91], Cas9-EQR [91], and Cas9-VRER [91], Cas9-NG [93], Non-G SpCas9s [113], SpG and SpRY [94]. Among these variants, Cas9-VQR, Cas9-EQR, and Cas9-VRER changed the PAM specificity from NGG to NGA, NGAG, or NGCG, respectively [91]. And Non-G SpCas9s recognize NRNH PAMs (where R is A or G and H is A, C or T) [113]. Other variants, including Cas9-NG, SpG and SpRY, relaxed the PAM restriction from NGG to NGN (Cas9-NG and SpG) and NRN or NYN (SpRY, NRN > NYN) respectively [93, 94]. Interestingly, another variant, xCas9, also significantly relaxed the PAM restriction by recognizing NGN, NNG, GAA, GAT, and CAA PAMs. However, the majority of mutated amino acids of xCas9 did not locate in the PI domain (six out of seven), suggesting that other structures beyond PI domain also contribute to PAM recognition. Interestingly, coupling non-PI domain mutations of xCas9 to PI domains of Cas9-NG or Non-G Cas9 variants also relaxed the PAM restriction of the latter. Collectively, these variants significantly expanded the targeting scope of Cas9 systems [92].
Genome editing produced by wild-type Cas9
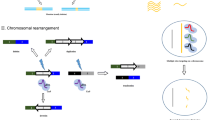
As mentioned above, in eukaryotic cells, DSBs tend to be repaired by error-prone NHEJ pathway, leading to the formation of uncontrolled indels [10] (Fig. 2). Such feature can be utilized to disrupt functional elements of the genome to manipulate gene functions. For example, introducing indels into gene coding regions can change the codon or even shift the reading frame, thereby disrupting gene function [39, 43, 114, 115]. Indels can also be introduced to the boundary regions between exons and introns to interrupt RNA splicing or to the enhancers or suppressors to change the expression pattern of specific genes [6, 116, 117]. In addition, when supplied with donor DNAs harboring sequences homologous to each end of DSBs, they can also be repaired by homology dependent mechanisms, such as homology directed repair (HDR) pathway [44, 118,119,120,121] or MMEJ [122,123,124,125,126] (Fig. 2). However, the efficiency of HDR pathway is much lower than that of NHEJ [10]. Because NHEJ is very active in the whole cell cycle and can be observed in various cell types, including cells in division and cells after mitosis. In contrast, HDR mainly plays a role in the S/G2 stage, so it is mainly limited to actively dividing cells, limiting the treatment that requires precise genomic modification of mitotic cells [127, 128].

Editing processes outcomes of Cas9 nuclease, base editor and prime editor
Base editor
Besides functioning as nuclease, Cas9 proteins can also be rewired to function as DNA binding platforms to recruit other functional modulators. By linking with modulators such as transcription regulators and histone or DNA modifiers, Cas9s can be engineered to regulate transcription, histone and DNA modifications [11, 12, 21]. Among these derivates, base editors, in which dead or nickase Cas9 proteins were fused to cytosine (C) or adenine (A) deaminase (CBE or ABE), are the most interesting ones in that they can efficiently and precisely convert one target DNA base pair into another [22, 30] (Fig. 2).
As above mentioned, Cas9 proteins bind to their target DNAs to form an “R-Loop” structure, in which the NTS was partially detached and exposed outside the Cas9 complex [88]. The exposed NTS is a favorite target for C or A deaminase that prefers single-strand DNA [129]. Usually, the deaminases are fused to the N-terminus of Cas9 protein, thereby deaminating Cs or As that locate in a small window of NTS, called editing window [22, 30]. Typically, SpCas9 derived N-terminal base editors function in a window ranging from ~ 4 to ~ 8 bases, counting NGG PAM as 21–23. These Cs or As in the window were deaminated into urines (Us) or inosines (Is), which in turn are transformed into thymines (Ts) or guanines (Gs) by endogenous DNA repair or replication mechanisms [22, 30]. Reserving the cleavage activity of Cas9 for TS significantly increased the transformation efficiency, which is possibly due to the nick within TS induce endogenous mechanisms to fix the lesion according to the unbroken NTS [130].
Editing window of base editor
The width and position of the editing window varied with the types and locations of deaminases. For example, deaminases with decreased enzyme activity tend to narrow the editing window [131]. And inlaying or tethering deaminases to different locations of the Cas9 complex shift the position of the editing window [132,133,134,135,136]. By contrast, tethering multiple copies of deaminases to base editor enlarges the editing window [137]. By adjusting these parameters, now we have a large variety of editors with different editing windows [138,139,140,141]. In addition, reducing the length of sgRNA spacer has also been shown to narrow the editing windows in some cases [142,143,144]. It is also noteworthy that when equipped with both deaminases, base editors can achieve simultaneous conversion of both cytosine and adenine, which is particularly useful for saturated mutation screening [132, 145,146,147].
Off-target editing of base editor
Although base editors are efficient in on-target editing, they also produce undesired off-target editing on both DNA and RNA molecules [148,149,150,151,152]. Basically, the off-target effects of base editors can be classified into sequence (or Cas9) dependent [153,154,155,156,157] and independent [150, 152] effects. Sequence dependent off-target editing mainly occurred at the genomic DNAs, including Cas9 off-target genomic loci or the on-target locus but outside the editing window [148, 158]. Sequence independent off-target editing took place in both DNA and RNA molecules [149, 150, 152]. Since the deaminases prefer single strand nucleic acids, RNAs and occasionally occurred single strand DNAs can be their substrates [149]. These off-target effects are suspected to lead to pathologic conditions, raising the concern of safety issue. To resolve this issue, many efforts have been put to reduce the off-target base editing. Firstly, deaminases with reduced enzyme activity have been shown to significantly decrease the off-target efficiency while has little effects on the on-target efficiency [134, 148, 159, 160]. Secondly, inlaying deaminases into Cas9 protein specifically reduced the sequence independent RNA and DNA off-target editing [136, 161]. Moreover, splitting the deaminases to timely control their activity also helps reduce off-target editing, which is particularly useful in long-term delivery of base editors, such as AAV delivery [162, 163].
C to G base editing
It is noteworthy that base editors, especially CBEs, frequently produce impure editing outcomes, including indels and C to R conversions [30] (R = A or G). This is possibly because U within DNA molecules tend to be removed by endogenous uracil-N-glycosylase (UNG), since impure products were nearly absent in UNG null cells [23]. UNG cleaves the N-glycosidic bond of uracil and leaves an abasic site (also known as apurinic/apyrimidinic site, AP site) [164,165,166,167,168,169,170]. The resulting abasic site can be recognized by a base excision repair enzyme, AP lyase, which converts abasic site to ssDNA break [167, 171,172,173,174,175,176,177]. Together with the nick within the target strand that is cleaved by Cas9 nickase, they were likely recognized as DSB by DNA repair machines, leading to the formation of indels. Alternatively, the abasic site is not transformed into nick, enabling NTS to serve as template to guide the repair of the TS. The repair of the TS might involve a trans-lesion synthesis pathway that uses a specialized DNA polymerase to bypass the lesion by adding a special nucleotide opposite to the abasic site [164, 177,178,179]. It is likely that the repaired TS may in turn serve as a template to guide the repair of NTS to fix the abasic site. Thereby, the trans-lesion synthesis pathway introduces an R base to substitute the deaminated C. The level and the type of C to R conversion are likely dependent on the species and sequence context where the deaminated C locates [180]. In mammalian cells, U s tends to be converted to Gs while in bacteria they tends to be converted to As [181,182,183,184,185]. Supplementing additional copies to CBE may further inhibit the endogenous UNG activity, thereby increasing the purity of the editing products by reducing the frequencies of C to R conversion or indel formation [23, 186, 187]. Unlike CBEs, ABEs rarely create impure editing outcomes, which is possibly due to that the activity of endogenous pathways dealing with inosine lesions is rather low [188]. Although ABEs also produce ssDNA breaks in the TS and ssDNA breaks themselves induce the formation of indel, the level of indels in ABE products are generally low (in most cases, < 1%) [189,190,191].
Prime editor
Base editing can efficiently install base transition mutations, i.e. the substitutions in-between purines or pyrimidines (from A to G, G to A, C to T or T to C), making it possible to correct human pathogenic diseases caused by such mutations [22, 30]. However, there were a large portion of genetic lesions caused by other types of mutation, such as base transversion, small fragment insertion and deletion etc., all of which were beyond the scope of base editing tools [192].
To fill above gaps and achieve more efficient, precise and flexible DNA changes, David Liu and co-workers proposed a new “search-and-replace” genome editing technology, called prime editing system [27]. In this system, Cas9 harboring H840A mutation was coupled with an engineered reverse transcriptase, M-MLV, to form prime editor (PE) that can achieve all 12 types point mutations, small fragment insertion and deletion (Fig. 2). Upon the guidance of prime editing guide RNA (pegRNA), PE fusion protein bind to the targeted DNA, nicked the NTS and then extend the 3’ end of NTS according to the information encoded by pegRNA [27]. Through these stepwise actions, PE incorporated new DNA sequences into the nicked NTS, which were then passed to its complementary strand, possibly through DNA repair or replication mechanisms. Thereby, PE accomplished the rewrite of the genetic information within the target site. The most beautiful part of PE system is the design of pegRNA that was composed of a sgRNA and a 3’end extended tail [27]. The 3’ tail of pegRNA contained two parts. One part was designed to complement to the 3’end of nick NTS, called primer binding site (PBS) and the other one encoding the desired edit served as reverse transcriptional template (RT-template). Once the NTS was nicked by nCas9 H840A, the 5’ portion of NTS was recognized by PBS to form a DNA-RNA duplex, which served as a good substrate for reverse transcriptase. The 5’ portion of NTS acting as a primer was extended by reverse transcriptase according the guidance of RT-template (Fig. 2).
Editing efficiency improvement of prime editor
Although PE system can install almost all types of DNA changes, it is overall less efficient than BE or Cas9 nuclease, which may in part due to that PE system required stepwise and concerted actions of multiple modulators [27]. Optimizing the parameters of nearly all steps is necessary to maximize the efficiency of PE. As mentioned above, the starting process of PE is to nick the NTS of the target site and bind its end with PBS. Hence, parameters enhancing this process may include the activity of Cas9 nickase and the thermodynamics of the PBS. As a matter of fact, David Liu’s lab has observed that mutations enhancing the activity of Cas9 nuclease also enhance the efficiency of PE. Caixia Gao’s lab observed that the length and sequence of PBS has a significant effect on the efficiency of PE, which is possibly related to the annealing temperature of PBS [193]. Moreover, the 3’ end of pegRNA, mainly the PBS, was thought to tend to be degraded in cells, resulting in the failure of pegRNA to prime the editing [194]. Interestingly, another line of evidence showed that the presence of PBS disrupted the structure of pegRNA by binding to its spacer sequence, resulting in the loss-of-function of pegRNA [195]. To sum up, both studies found that coupling an RNA motif harboring stem-loop structure improved the efficiency of PE. This finding was soon confirmed by other studies showing that coupling complicated structure to the 3’ end of pegRNA helped increasing the efficiency of PE [196, 197]. Enhancing the activity of RT provide another way to improve PE [198, 199]. Wen Xue’s lab showed that the removal of RNase H domain from RT improved PE [199]. Besides strengthening the elements of PE system, optimizing the design of PE with paired pegRNAs also improve the efficiency of PE, especially in cases of multiplex base conversion, large fragment deletion and insertion [28, 200,201,202,203]. Interestingly, paired pegRNA strategy also significantly decreased the frequency of undesired indels [28, 203]. In addition, modulating the endogenous mechanisms that are involved in each process of PE may affect its efficiency. The first evidence of such mechanism came from David Liu’s and Joanna Loizou’s labs [37, 38]. They independently demonstrated that the inhibition of mismatch repair pathway enhanced the efficiency and accuracy of PE. Importantly, David Liu’s lab discovered a set of enhanced prime editors by coupling dominant negative MLH1 to PE, which is a key player in the recognition of small insertion/deletion loops during the process of MMR [37]. In the evolved version of prime editors, called PE4 or PE5, dominant negative MLH1 was co-expressed with but not physically attached to PE2, resulting in the global inhibition of MLH1 activity and improved PE efficiency and decreased indel frequency [37]. Hopefully, in the near future we will see further improvements in the efficiency and precise of PE system.
CRISPR-related transposon and recombination systems
CRISPR-related transposon system
Targeted insertion of large DNA fragments into desired genomic loci holds great potential for the treatment of genetic diseases. Although such kind of insertions can be achieved by wild-type Cas9 mediated HDR or micro homologous recombination (MMEJ), the efficiencies are generally low and are limited by the cell type to be edited [204, 205]. Prime editor is also capable of inserting large DNA fragments into desired loci, but its efficiency is even lower [28, 206, 207]. Therefore, efficient targeted insertion of large DNA fragments into genome remains challenging.
Transposition is a special type of genetic recombination, which transfers specific genetic factors from one location to another by using the interaction of transposon elements and transposase enzymes [208]. In 2017, Peters et al. found a novel kind of CRISPR Cas system encoded by a Tn7 like transposon, which consists of several key proteins homologous to the core elements of Tn7 transposons, including two isomerase (TnsA and TnsB) and one regulatory protein (TnsC), and a set of CRSIPR system [209]. This system can process and bind crRNA but cannot cut target DNA. Instead, it can achieve gene transposition at specific sites [209]. In 2019, Klompe et al. used a three-plasmid system to express CRISPR transposase derived from Vibrio cholerae in Escherichia coli for the directional integration of large pieces of DNA [210]. In 2020, to simplify the process and improve the integration efficiency, the same researchers effectively combined key elements to construct INTEGRATE single particle system pSPIN, which uses a single promoter to simultaneously drive guide RNA and multi cis–trans mRNA to express key proteins and guide RNA [211]. It is worth noting that pSpin-mediated DNA transposition has no directionality, and the rotator can be inserted in both positive and negative directions. At the same time in 2019, Strecker, et al. have developed a similar CRISPR transposase system. Researchers have developed and used the CRISPR-related transposase (CAST) of the cyanobacteria Scytonema hofmanni, which is composed of Tn7-like transposase subunit and V-K CRISPR (Cas12k). CAST includes two variants: Scytonema hofmanni (ShCAST) and Anabaena cylindrica (AcCAST). The insertion of ShCAST mainly occurs between 60 and 66 bp 3 ' of PAM, while the insertion of AcCAST cargo into 49–56 bp 3 ′ of PAM. The gene insertion mediated by ShCAST is directional [212]. In the study, after the author replaced Cas12k with Cas9, no displacement was observed, indicating that Cas12k may have a specific role with other CAST components to facilitate the occurrence of transposition [212]. Since then, Chen et al. have developed a new synthesis system, Cas Transposon (CasTn), which combines the DNA integration capability of Himar1 transposase and the programmable genome targeting capability of dCas9 to achieve targeted gene transposition. The author has proved this in cell-free in vitro reactions and E. coli plasmid detection. With the further improvement of the system, CasTn may play a role in various organisms, because the Himar1-dCas9 protein does not need host factors to play a role [213]. Noteworthy, although all the above transposon systems have good gene integration ability in E. coli, there is lacking evidence demonstrating that these systems can also function efficiently in eukaryotic cells (Fig. 3a).

CRISPR-associated transposon and recombination systems. a Cas transposases include both Cas proteins and transposase-associated components. Cargo DNA is identified by its left end (LE) and right end (RE) sequences and bound by transposase proteins (Tns). Guide RNA binding to Cas nuclease brings transposase to the specific site, and transposase integrates the DNA cargo into the target site. The target site is duplicated and flanks the integrated LE–cargo–RE sequence. Each Cas-transposase complex has a specific requirement for the spacer length of guide RNA and a unique position preference of integration site. b reassembly of guide RNA-programmed recCas9 at the target sites
CRISPR-related recombination systems
Site-specific recombinase directly catalyzes the cleavage, chain exchange, and coordination of two double-stranded DNA sequences, to cause gene recombination phenomena such as insertion, deletion, or inversion of the target sequence [214,215,216,217]. Unlike DNA nuclease, the direct catalysis of recombinase usually does not trigger the error-prone DNA repair process, leading to the formation of indel. It does not rely on the endogenous cell DNA repair mechanism, and the recombinant product is relatively simple [214, 215]. Therefore, recombinant enzyme-mediated genome modification can produce more accurate and predictable genome changes than ribozyme-based genome editing and may be more efficient in non-dividing cells [216, 217]. Tyrosine and serine recombinases such as Cre, Flp, and Φ C31 integrase has been widely used to catalyze the recombination of exogenous DNA into model organisms [217, 218]. However, the use of these enzymes is limited by their inherent, nonprogrammable DNA sequence specificity. Chaikind et al. fused dCas9 into the catalytic domain of Gin recombinase β and the resulting recCas9 system. This “recCas9” system shows moderate efficiency on the plasmid matrix and can mediate a large number of genome deletions in mammalian cells with low efficiency [219] (Fig. 3b).
Therapeutic applications of Cas9 and its derivates
The versatility of Cas9 nuclease and its derivates has found its applications ranging from basic science to the clinic (Table 2). As discussed above, the versatility of Cas9 system now enabled virtually all types of genome editing, including targeted undesired indels formations, base substitutions, designed fragment deletion or insertions. Such editing strategies can be harnessed to disrupt or restore gene function, regulate gene expression and insert therapeutic DNA fragment, which can be used to treat a diverse set of disorders, including genetic diseases, metabolic diseases, cancer and infectious diseases etc. (Fig. 4).

Therapeutic Strategies of Cas9 nuclease, base editor and prime editor. Different Cas9-related editing tools (above) can produce different types of edits to the genome (middle) and thus can cause different changes to the corresponding genes (below)
Cas9 nucleases mediated gene editing
Cas9 nucleases mediated gene disruption
Since DSBs induced by Cas9s or other nucleases are mainly repaired by the mammalian cells to undesired indels, it is straightforward to harness the nuclease activity of Cas9s for the purpose of gene disruption (Table 3). Targeting gene coding regions, especially those containing conserve functional protein domains, to introduce frame-shifting indels or mis-sense mutation is a frequently used strategy in designing Cas9 based therapy. For example, 1. vascular endothelial growth factor receptor 2 (VEGFR2) is an important therapeutic target for angiogenesis-related disorders such as proliferative diabetic retinopathy and neovascular age-related macular degeneration [220]. Disruption of this receptor by AAV mediated retina expression of Cas9 system efficient abrogates angiogenesis in the mouse models of oxygen-induced retinopathy and laser-induced choroid neovascularization [221]. Another example came from transthyretin amyloidosis, a life-threatening disease stemmed from progressive accumulation of misfolded transthyretin (TTR) protein that mainly produced by the liver. LNP mediated expression of Cas9 system against liver TTR gene efficiently reduced the concentration of TTR protein in serum in both animal model and human patient [222]. In addition, targeting liver pcsk9 by AAV or LLN mediated expression of Cas9 systems significantly reduced plasma cholesterol levels in mice, preventing the genesis or the development of cardiovascular disease [39, 223].
Indel formation can also be harnessed to disrupt the regulator elements of genes, such as splicing donors or acceptors, transcription enhancers or suppressors and so on. BCL11a is an attractive target since its loss-of-function releases the repression of fetal γ-globin, which in turn can rescue the phenotype of beta-globin mutation related anemia [252]. Targeting the enhancer element of BCL11a or its binding sites within γ-globin locus are frequently used strategy to reactivate the expression of the latter [224, 252,253,254]. Delivery of CRISPR/Cas9 system targeting bcl11a enhancer into CD34 + hematopoietic stem cells via in vitro electroporation is an effective way to reactivate the expression of γ-globin in red blood cells [50, 224]. Recently, such strategy has achieved positive outcomes in human patients [50, 224]. The infusion of edited HSC increased the level of fetal globin, reduced the requirement of blood transfusion in in β-thalassemia and the incidence of vaso-occlusive episodes in sickle cell disease [50, 224].
Besides producing undesired indels, Cas9 nuclease activity can also be rewired to produce targeted fragment deletions by coupling with dual sgRNAs against same loci [225]. This feature can be used to delete genomic fragment that contains mutations locating in the redundant sequences but impaired the expression of that gene. For example, DMD gene that encodes a muscle nutrition protein, dystrophin, contains 24 functional redundant spectrin-like repeats, within which mutations may impair RNA splicing or translation (pre-mature codon) thereby disrupting the expression of DMD and leading to muscle mass loss [255]. Dual sgRNA strategy has been demonstrated to be efficient in deleting the mutant exon and restore DMD gene expression and function [225]. Targeted fragment deletion can also be used to delete mis-spliced exons that contained stop codons and were resulted from mutations to splicing elements, such as cis -regulatory elements, core spliceosomal components and trans -acting regulatory factors. For example, Lu et al. used dual sgRNA strategy to deleted mis-spliced exons that contained stop codon in humanized ß-globin IVS-2 mouse model, resulting in the correction of abnormal ß-globin RNA splicing and improved β-thalassemia related syndrome [256]. A type of Leber congenital amaurosis 10 (LCA10), a rare inherited retinal dystrophy, is also caused by mis-spliced exon, which is stemmed from CEP290 IVS26 mutation [257]. Ruan et al.used AAV to deliver Cas9 and dual sgRNAs into the retinas of CEP290 IVS26 mutant mice and observed efficient targeted deletion of the mutant fragment. The deletion restored the expression of CEP290 protein and significantly rescued LCA related phenotypes [258].
Cas9 nucleases mediated gene konck-in
As mentioned above, DSBs can also be fixed via HDR pathway when sufficient repair template donors were provided. Therefore, Cas9 nuclease can be used to correct gene mutations or insert therapeutic fragment into the target region (Table 3). For example, Yang et al. developed a dual AAV system enabling the HDR-mediated in vivo correction of point mutation of OTC gene in mouse hepatocytes [44]. In this report, they infused two AAVs into neonatal or adult mice, one expressing Cas9 and the other expressing a guide RNA and the donor DNA. The infusion resulted in correction of the mutant OTC gene in about 10% hepatocytes and increased survival rate in neonatal mice but not in adult mice, which is possibly due to that HDR activity is limited to S/G2 phases of proliferating cells (Regulation of homologous recombination in eukaryotes. Enrichment of G2/M cell cycle phase in human pluripotent stem cells enhances HDR-mediated gene repair with customizable endonucleases.) [44]. Richards et al. also used AAV to deliver Cas9-mediated HDR editing system in to PKU mouse model and corrected the Pah point mutation in liver cells, partially restoring PAH activity and significantly reducing the level of blood phenylalanine [238]. Zhao et al. used similar strategy to correct a non-sense mutation of Ldlr gene in mouse model and observed partial rescue of LDLR protein expression and function, which significantly reduced the serum levels of total cholesterol, total triglycerides and LDL-cholesterol [239]. Besides correcting point mutation, Cas9-mediated HDR strategy can also be used to targeted insert a therapeutic DNA fragment into desired locus. In most cases, the inserted fragments were designed to therapeutic genes. For example, Lisjak et al. used this strategy to insert the coding region of human coagulation factor IX (HFIX) into albumin locus in hepatocytes in vivo, enabling the co-expression of HFIX with albumin. The ectopically expressed HFIX successfully rescued hemophilia related phenotypes [240]. Eyquem et al. used similar strategy to insert CD19-specific CAR into the T cell receptor α constant (TRAC) locus of T lymphocytes to allow the expression of CD-19 CAR under the control of TRAC promoter [241]. Besides HDR, another DNA repair mechanism, MMEJ, can also be used to insert therapeutic fragment. Compared with HDR that usually requires homologous arms of more than 500 bp, MMEJ strategy only requires 20–30-bp homologous arms, which significant simplifies the construction of donors and reduces size of donor DNA [205]. By using MMEJ strategy, Yao et al. inserted a full length fumalacetacetic acid hydrolase 1 (HT1) gene into the hepatocytes of HT1 deficient mice to rescue the liver damage phenotype [259] (Table 3).
Therapeutic applications of base editors
However, because NHEJ activity is prominent in mammalian cells, HDR or MMEJ strategy produced more unwanted indels than desired edits in most cases, making it not so efficient and safe for clinical applications. By contrast, base editors are more precise and efficient in introducing single base conversions. Such a feature is particularly useful for correcting point mutations or create desired functional base conversions (Table 3). For example, introducing missense or non-sense mutations within gene coding region can lead to either gain- or lose-of-function of target genes. Chadwick et al. delivered cytosine base editor to the liver of adult mice to generate stop codon within pcsk9 gene and observed significant reduction in plasma PCSK9 protein and cholesterol levels, demonstrating the therapeutic potential of base editing in silencing gene function [260]. More straightforwardly, base editors can also be used to correct diseases related point mutations. Rossidis et al. delivered cytosine base editor into the uterus of pregnant tyrosinemia type 1 mutant mice to correct Fah mutation [261]. They showed that in-utero delivery of base editor successfully rescued the lethal phenotype of neonatal Fah mutant mice, demonstrating the potential of in utero base editing in the treatment of genetic diseases causing in prenatal or neonatal death [261]. Zhou et al. and Villiger et al. delivered cytosine base editors derived from SaCas9 into the liver of neonatal and adult PKU mice respectively [243, 244]. They showed that AAV-8 delivered SaCas9 base editor efficiently corrected PAH point mutation and reduced the plasma phenylalanine to physiological level [243, 244]. In addition, base editing can also be used to manipulate RNA splicing via converting key bases within splicing regulatory elements. Li et al. used a non-popular cytosine base editor developed by themselves, eTAM, to destroy DMD splicing site, leading the skip of 4th exon that contains frame-shifting mutation, which genetically restored the open reading frame of DMD gene [262]. They showed that a single-dose of AAV9-eTAM achieved > 50% targeted exon skipping in the Dmd mRNAs and restored up to 90% dystrophin protein in the heart, leading to an increased life span of the Dmd mutant mice [262].
Delivery of Cas9 and its derivates
CRISPR-Cas9-mediated gene editing tools are very powerful, revolutionizing the field of gene therapy and showing encouraging results in a variety of applications. However, the in vivo delivery of editors remains a critical limitation for these editing tools to treat specific diseases. In vivo delivery methods of gene editing tools are classified into two main categories: viral and non-viral systems.
Viral delivery systems
Viruses have evolved naturally to deliver nucleic acids in vivo to different cell types, which makes them interest targets for delivering exogenous genes. Currently, several types of viral vectors have been developed and modified for the delivery of gene editing tools [263]. Among them, adeno-associated virus (AAV), as well as adenovirus have been widely used in preclinical studies of Cas9-mediated gene editing strategies [263] (Table 3).
AAV delivery
AAV is a single-stranded DNA virus, the smallest and simplest animal virus in the family Microviridae. AAV particles are envelope-free and consist of an icosahedral protein capsid of approximately 25 nm in diameter and a single-stranded DNA genome of ~ 4.7 kb [264]. Compared with other viral delivery systems, AAV has the advantages of low immunogenicity, high efficiency and high biocompatibility [265]. And importantly, unlike lentivirus and retrovirus, recombinant AAVs very rarely integrate into host genomes [266,267,268]. In addition, there are plenty of different types of capsid serotypes, directing AAVs to different target tissues in vivo [269]. These features nominate AAVs as promising gene therapy vehicles [270].
However, the small packaging capacity of AAVs is a major limitation for their application. AAVs have a packaging capacity of about 5 kb, including the transgene cascade and two flanking inverted terminal repeats (ITR) [271, 272]. Therefore, the room for exogenous transgene expression cascade is limited to ~ 4.7 kb, which limits them as delivery tools for Cas9-related gene editing, since Cas9 proteins are generally large. The most widely used Cas9, SpCas9, is about 4.3 kb, and its derivates, such as BE and PE, are even larger [22, 27, 30].
To overcome size limitations, researchers have developed several methods that allow Cas9-mediated gene editing tools to be packaged into AAV vectors for in vivo delivery. Several groups have developed strategies to split Cas9 and its derivates into two halves. Intein mediated protein splicing is the most frequently used splitting system. By optimizing the splitting sites, the efficiencies of split Cas9 systems are similar to or only slightly lower than those of their full-length equivalents, enabling practicable AAV mediated in vivo genome editing. For example, David Liu's group have designed a double AAV base editing strategy to treat the Hutchinson-Gilford premature aging syndrome (HGPS) mouse model and corrected the C·G-to-T·A mutation in the LMNA gene, achieving up to 30% gene correction efficiency in heart tissue [247]. Schwank and colleagues used a dual AAV9 strategy to deliver ABE targeting Pcsk9 to mice, achieving 60% base editing in the liver and significant reductions in serum pcsk9 protein levels as well as serum cholesterol levels [248]. Such strategy has also been used in central nervus tissue to knock out mutated Huntington (HTT) genes to correct pathogenic mutations in a mouse model of Niemann-Pick disease [273].
Alternatively, using smaller Cas9 can achieve single AAV delivery. For example, Cas9 from Staphylococcus aureus (SaCas9) is commonly used for single AAV transduction because its size is only 3.2 kb [112]. Such a compact size endows it to be co-packaged with the sgRNA expression cascade in a single AAV. Zhang and colleagues used a single AAV system to in vivo deliver SaCas9 to efficiently knock out Pcsk9 and lower serum cholesterol in mice [39]. In addition, an ongoing clinical trial uses a single AAV for subretinal delivery of SaCas9 double sgRNA has been lunched to delete a pathogenic mutation in the CEP290 gene in patients with Leber congenital amaurosis 10 [258]. In addition to SaCas9, many other compact Cas9 variants were identified and characterized, such as Nme2Cas9 (3.24 kb, PAM = N4CC) [96, 102, 274,275,276], CjCas9 (2.95 kb, PAM = N4RYAC) [70, 277, 278] and SauriCas9 (3.18 kb, PAM = N2GG) [82], all of which are capable of single AAV delivery. These Cas9 variants also broaden PAM recognition and expand the targeting range of the single AAV gene editor.
Adenoviral delivery
Adenovirus (Ad) is an icosahedral envelope-free virus, 90–100 nm in size, with a large (36 kb) genome [279]. Ad is the most commonly used viral vector (> 20%) in gene therapy clinical trials, possibly due to its large packaging capacity, genetic stability, high transduction efficiency, and ease of production [279]. Currently, there are 57 known Ad serotypes that can infect human and 100 serotypes that can infect primates, allowing researchers to modulate Ad tissue targeting by using different capsids [249]. In 2017, Musunuru and colleagues used Ad to deliver CBE into mice and observed a 28% editing efficiency of Pcsk9, successfully lowering plasma cholesterol levels in treated mice [260]. Lieber and colleagues used Ads to introduce ABE into HSC in vivo to disrupt the blocker binding site in the fetal hemoglobin promoter, significantly upregulating fetal hemoglobin [280]. Ad has also recently been used for in vivo introduction of Prime editor. Schwank and colleagues used Ad to deliver PE2 without the RNaseH structural domain into neonatal or adult mice and observed 58% and 36% editing efficiency, respectively [281]. Although adenoviruses can introduce gene editing tools in vivo and produce effective editing, their dosages are limited during application, possibly due to their high immunogenicity and cytotoxicity.
Non-viral delivery
In addition to the modification of existing viral vectors, other types of delivery systems can be used for transient in vivo delivery of exogenous genes, such as non-viral vectors (Table 3). Among them, lipid nanoparticles (LNP) are the most frequently used non-viral gene delivery systems, holding great potential for in vivo applications of gene editing tools. Besides, virus-like particles (VLP) are also developed to deliver these tools.
LNP delivery
Lipid nanoparticles (LNPs) have been used for decades to deliver nucleic acids, including siRNA and therapeutic mRNA, and in recent years LNPs have been widely used for in vivo delivery of gene editing tools [222, 226]. In most cases, these tools were delivered in the form of RNA, which is a negatively charged hydrophilic macromolecule and is electrostatically repelled by the same electrical properties as the cell membrane, making it difficult to enter the cell and susceptible to rapid degradation by the ubiquitous ribonuclease (RNase) [282]. Therefore, RNA needs a protective layer to "transport" it into the cell. Since cell membranes are mainly composed of lipids, liposome coating can make it easier for RNA to pass through the membrane and be released into the cytoplasm. To achieve this, liposomes require a positively charged lipid molecule that can attach to negatively charged RNA, in addition to structural lipids (which mimic the cell membrane and shield the positive charge) and poly (ethylene glycol)-anchored lipids (which prevent aggregation of LNPs and side reactions with the biological environment). Therefore, LNPs are typically composed of four molecules: cationic or ionizable lipids, co-lipids, poly (ethylene glycol) -lipids, and cholesterol [283].
Compared with cationic lipids, ionizable lipids (ILs) are less toxic and more effective in in vivo delivery [284]. When formulated as LNPs, ILs are designed to display electroneutrality at physiological pH, but behave positively charged in vivo in acidic LNP nuclei. This pH-adapted ionization results in enhanced efficacy with reduced toxicity, making them more suitable for nucleic acid delivery [284]. These lipids typically make up 30–50% of the total lipids in the formulation [285]. Cholesterol, a naturally abundant component of cell membranes, is one of the so-called structural lipids found mainly in the outer shell of LNPs and typically accounts for 20–50% of the total lipids in LNPs [285]. Phospholipids contribute to the encapsulation of nucleic acids and the stability of LNPs and usually represent only 10—20% of the total lipids in the formulation [285]. Phospholipids are used as structural lipids because they can spontaneously organize into lipid bilayers and have a high phase transition temperature thus ensuring the membrane stability of LNPs. Phospholipids are located at the periphery of LNPs, just like cell membranes, and PEG lipids are an important component in controlling the half-life and cellular uptake of LNPs [286]. During LNP assembly, PEG chains are located in the outer shell of the nanoparticles due to their hydrophilicity and large volume. PEG provides an external polymer layer for LNPs to hinder the adsorption of serum proteins and mononuclear phagocyte systems and prolong the in vivo circulation. PEG also prevents aggregation of nanoparticles during storage as well as in the blood [286]. Changing the properties of these components can produce LNPs with different properties, including different pharmacokinetic profiles and the ability to target different cell types.
The key advancement of the LNP formulation is that it enables efficient packaging and delivery of mRNA, allowing LNP to be widely used for in vivo delivery of exogenous mRNA, including in vivo introduction of Cas9-mediated gene editors. Anderson and colleagues co-packaged SpCas9 mRNA and chemically modified sgRNA via LNP and injected intravenously into mice, targeting Pcsk9 produced 80% editing efficiency and reduced and serum Pcsk9 to undetectable levels [287]. This result demonstrates for the first time the promise of therapeutic level gene editing in mice by LNP encapsulating both Cas9 nuclease mRNA and chemically modified sgRNA. Since then, researchers have shown that LNP can successfully introduce Cas9 in complex with sgRNA into crab-eating monkeys, which can achieve 73% TTR destruction in the liver and a corresponding reduction of serum TTR protein by more than 94% [288]. Next, clinical data showed that LNP introduced Cas9-sgRNA complex into patients could also reduce their serum TTR levels by up to 87% with low off-target editing. These results confirm the possibility of in vivo genome editing by LNP introduction of Cas9 and sgRNA complexes. In addition to delivering Cas9 nuclease mRNA, LNP has also been used to deliver base editor mRNA into the liver of mice and non-human primates. Xue and colleagues observed that delivery of ABE mRNA by LNP into the liver of a mouse model of tyrosinemia yielded a 12.5% base editing efficiency [289]. Schwank and colleagues observed a 10% base editing rate of phenylalanine hydroxylase mutations in the liver of mice with phenylketonuria model by delivery of LNP encapsulating SaCas9-BE3 mRNA [243]. Recently, it was demonstrated by Kathiresan, Schwank et al. that the splice site of PCSK9 can be effectively interrupted by LNP delivered ABE mRNA into the liver of mice as well as crab monkeys, thus disrupting the expression of functional PCSK9 protein. Accompanied with the delivery, substantial (90%) and sustained (> 8 months) inhibition of serum PCSK9 protein and 60% reduction in blood cholesterol were achieved in crab monkeys [248]. The above studies suggest that the LNP delivered base editor has high clinical therapeutic potential for liver related genetic or metabolic diseases.
Since most LNPs injected intravenously naturally accumulate in the liver, editing of other organs outside the liver is challenging. Dahlman and colleagues developed a strategy to screen hundreds of different LNPs simultaneously in vivo [290]. The strategy uses unique DNA barcodes to label different LNP formulations, allowing the LNPs to be traced by sequencing the barcodes [290]. Using this strategy, Dahlman and colleagues found an LNP that could target spleen endothelial cells. They also showed that this LNP could efficiently deliver Cas9 mRNA and sgRNA to mouse spleen endothelial cells, yielding editing efficiencies comparable to those of hepatocytes [291]. In addition, Siegwart et al. developed selective organ-targeted (SORT) LNPs by supplementing an additional charged lipid component to modulate the internal charge while without significantly disrupting the standard four-component nature of LNP [292, 293]. They found that altering the charge and concentration of this additional component was sufficient to redirect LNP to the lung or spleen. These SORT LNPs successfully delivered Cas9 mRNA and sgRNA specifically to the lungs of mice, achieving 15% lung tissue editing [292]. These studies have shown that the targeting of LNPs can be modulated by changing their composition.
In addition, besides systematic delivery, local injection has been demonstrated to be effective in several organs. A panel of pre-clinical studies have shown that local injection of lipid-coated RNP into the inner ear and retina of mice can be effective for nuclease editing and base editing [246].
Virus-like particle (VLP) delivery
In addition to LNP, the Virus-like Particle (VLP) are also capable of delivering gene editing tools. These particles are non-infectious viral protein assemblies that package the desired mRNA, protein or RNP for delivery to the appropriate tissues in vivo [294]. Because VLPs are derived from existing viral backbones or viral capsid like proteins, they have the similar delivery properties to their corresponding viruses, including cargo encapsulate, endosome escapement, and the ability to be reprogrammed to target different cell types [294]. However, unlike viruses, VLPs transiently deliver gene editors in the form of mRNA or RNP, which reduces the risk of off-target gene editing and viral genome integration [295].
Almost all reported VLP architectures for delivering mRNA or protein cargo are based on retroviruses because retroviruses have several characteristics that are well suited for VLP [296]. Immature retroviral particles are spherical and typically lack rigid structural symmetry, which allows higher loading flexibility as compared with non-enveloped icosahedral viruses. In addition, the large particle size of retroviruses (100–200 nm) provides more physical space for packaging large proteins such as Cas9 [296]. Finally, retroviruses are inherently modular in terms of cellular targeting and packaging [297]. Cell type specificity is determined by the envelope glycoprotein, while packaging is controlled by the capsid protein [297]. This modularity suggests that the VLP capsid structure, which effectively packages the desired "cargo", can easily bind to a variety of existing envelope glycoproteins to modulate targeting specificity.
David Liu's group has recently developed an engineered VLP (eVLP) based on Moloney MLV (MMLV) that greatly optimizes the protein packaging and delivery capabilities of the VLP [249]. eVLP can efficiently package Cas9 nuclease or base editors RNP and mediate effective therapeutic levels of gene editing in multiple organs of mice. Local injection of eVLP into mouse brain successfully delivered base editor RNP in vivo, resulting in robust base editing in VLP-rich transduced cells (60% editing) [249]. Subretinal injection of eVLP into a mouse model of genetic blindness effectively corrected the causative point mutation and improved visual function. Finally, a single intravenous injection of eVLP into mice achieved 63% editing of Pcsk9 in the liver and 78% knockdown of serum Pcsk9 levels, which is comparable to the efficiency that produced by AAV and LNP delivery [249]. VLP from lentivirus has also been developed to package Cas9 mRNA and sgRNA, which has been demonstrated to produce therapeutic level of genome editing when locally injected [298, 299]. VLP engineered from mammalian retrovirus-like protein PEG10 are also capable of delivering Cas9 mRNAs and sgRNAs, producing robust editing in vitro (~ 60%) [300]. Together, these investigations demonstrated the potential of VLPs for therapeutic delivery of gene editing tools.
Perspectives and challenges
The past ten years have witnessed the explosion of genome editing technology, with more and more Cas9 derived genome editing tools discovered and evolved. The ability of those tools to efficiently and precisely install a virtually wide range of changes to the genome has revolutionized the research of both basic life science and clinical medicine. In particular, these tools have provided fantastic new direction for the field of gene and cell therapy.
Noteworthy, many challenges need to be addressed before genome editing could be widely used in human patients. One of the major challenges would be safety issue. Nearly all editing tools have been reported to produce more or less levels of off-target or undesired editing, ranging from base substitutions or small scale indels (several to tens of base pairs) to large scale fragment deletions (hundreds to millions of base pairs) and even chromosomal scale variations in either sequence dependent or independent manner. Such unintended deleterious outcomes should be systematically evaluated when designing therapeutic strategies. Fortunately, several strategies have been developed to reduce such off-target effects, including high-specific and truncated sgRNAs, high-fidelity Cas9 variants, base editors with internally inlaid architecture or with engineered deaminases harboring hypoactive mutations. Alternatively, modulating endogenous pathways have also been4shown to reduce several types of undesired editing outcomes. Enhancing histone acetylation by HDAC inhibitors has been shown to improve product purity of cytosine base editors by strengthening the interaction between UGI and UNG. Inhibition of DNA mismatch repair pathway has been shown to reduce the frequency of prime editor produced unwanted indels. Another challenge came from the pre-existing adaptive immune responses to the Cas9 proteins. Since currently the most powerful and commonly used Cas9 proteins, SpCas9 and SaCas9, are derived from bacterial species that are prevalent in human, about 50% population harbor pre-existing immune response to them. Study from monkey revealed that such pre-existing immune response would eliminate the genome-edited cells. In addition, it was found that Cas9 delivered by AAV produces higher immune response than that delivered by LNP, suggesting that a transient expression of Cas9 is optimal to avoid the already existing Cas9 immune response. Moreover, since repeated administration of Cas9 in monkeys produces a stronger immune response than the initial administration, it is highly recommended to achieve the therapeutic goal with a single dose. Additionally, in vivo delivery of genome editors to target cells is another big challenge, especially considering that Cas9 derived editors are large in size. Both viral and non-viral delivery systems have been used for in vivo delivery of genome editors. However, viral systems, such as AAV, usually express the editors for a long term. Under such condition, sustained activity of the editor is supposed to amplify genotoxic effects. Moreover, long-term expression also increases the chances for the target cells to be cleared by immune system. Non-viral systems, especially lipid nano-particles, are efficient in delivering transgenes to several organs including liver, lung and spleen. Recently, LNP delivered Cas9 based editors has achieved significant success in manipulating liver related targets, such pcsk9, TTR and PAH. But as for organs other than the liver, LNP were not so efficient. Tremendous efforts have been made aiming to overcome these challenges and fill the gap between basic research and clinical translation by developing new editing tools, editing strategies and delivery systems. Hopefully, these efforts will speed up the therapeutic application of genome editing in the near future.
Availability of data and materials
Not applicable.
References
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–21. https://doi.org/10.1126/science.1225829.
Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A. 2012;109(39):E2579–86. https://doi.org/10.1073/pnas.1208507109.
Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol. 2013;31(3):233–9. https://doi.org/10.1038/nbt.2508.
Mali P, Yang L, Esvelt KM, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339(6121):823–6. https://doi.org/10.1126/science.1232033.
Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science (New York, NY). 2013;339(6121):819–23. https://doi.org/10.1126/science.1231143.
Long C, Amoasii L, Mireault AA, et al. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science (New York, NY). 2016;351(6271):400–3. https://doi.org/10.1126/science.aad5725.
Hwang WY, Fu Y, Reyon D, et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol. 2013;31(3):227–9. https://doi.org/10.1038/nbt.2501.
Jia Y, Xu RG, Ren X, et al. Next-generation CRISPR/Cas9 transcriptional activation in Drosophila using flySAM. Proc Natl Acad Sci U S A. 2018;115(18):4719–24. https://doi.org/10.1073/pnas.1800677115.
Shan Q, Wang Y, Li J, et al. Targeted genome modification of crop plants using a CRISPR-Cas system. Nat Biotechnol. 2013;31(8):686–8. https://doi.org/10.1038/nbt.2650.
Jeggo PA. DNA breakage and repair. Adv Genet. 1998;38:185–218. https://doi.org/10.1016/s0065-2660(08)60144-3.
Perez-Pinera P, Kocak DD, Vockley CM, et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods. 2013;10(10):973–6. https://doi.org/10.1038/nmeth.2600.
Thakore PI, D’Ippolito AM, Song L, et al. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat Methods. 2015;12(12):1143–9. https://doi.org/10.1038/nmeth.3630.
Kearns NA, Genga RM, Enuameh MS, Garber M, Wolfe SA, Maehr R. Cas9 effector-mediated regulation of transcription and differentiation in human pluripotent stem cells. Development. 2014;141(1):219–23. https://doi.org/10.1242/dev.103341.
Rojas-Fernandez A, Herhaus L, Macartney T, Lachaud C, Hay RT, Sapkota GP. Rapid generation of endogenously driven transcriptional reporters in cells through CRISPR/Cas9. Sci Rep. 2015;5:9811. https://doi.org/10.1038/srep09811.
Black JB, McCutcheon SR, Dube S, et al. Master Regulators and Cofactors of Human Neuronal Cell Fate Specification Identified by CRISPR Gene Activation Screens. Cell Rep. 2020;33(9):108460. https://doi.org/10.1016/j.celrep.2020.108460.
Morita S, Horii T, Kimura M, Hatada I. Synergistic Upregulation of Target Genes by TET1 and VP64 in the dCas9-SunTag Platform. Int J Mol Sci. 2020;21(5):1574. https://doi.org/10.3390/ijms21051574.
Liu J, Li B, Yang L, Ren N, Xu M, Huang Q. Increasing Genome Editing Efficiency of Cas9 Nucleases by the Simultaneous Use of Transcriptional Activators and Histone Acetyltransferase Activator. CRISPR J. 2022;5(6):854–67. https://doi.org/10.1089/crispr.2022.0001.
Nunez JK, Chen J, Pommier GC, et al. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell. 2021;184(9):2503-2519 e17. https://doi.org/10.1016/j.cell.2021.03.025.
Anton T, Bultmann S. Site-specific recruitment of epigenetic factors with a modular CRISPR/Cas system. Nucleus. 2017;8(3):279–86. https://doi.org/10.1080/19491034.2017.1292194.
Morita S, Noguchi H, Horii T, et al. Targeted DNA demethylation in vivo using dCas9-peptide repeat and scFv-TET1 catalytic domain fusions. Nat Biotechnol. 2016;34(10):1060–5. https://doi.org/10.1038/nbt.3658.
Hilton IB, D’Ippolito AM, Vockley CM, et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol. 2015;33(5):510–7. https://doi.org/10.1038/nbt.3199.
Gaudelli NM, Komor AC, Rees HA, et al. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature. 2017;551(7681):464–71. https://doi.org/10.1038/nature24644.
Komor AC, Zhao KT, Packer MS, et al. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T: A base editors with higher efficiency and product purity. Sci Adv. 2017;3(8):eaao4774. https://doi.org/10.1126/sciadv.aao4774.
Neugebauer ME, Hsu A, Arbab M, et al. Evolution of an adenine base editor into a small, efficient cytosine base editor with low off-target activity. Nat Biotechnol. 2022. https://doi.org/10.1038/s41587-022-01533-6
Velimirovic M, Zanetti LC, Shen MW, et al. Peptide fusion improves prime editing efficiency. Nat Commun. 2022;13(1):3512. https://doi.org/10.1038/s41467-022-31270-y.
Chen L, Zhu B, Ru G, et al. Re-engineering the adenine deaminase TadA-8e for efficient and specific CRISPR-based cytosine base editing. Nat Biotechnol. 2022. https://doi.org/10.1038/s41587-022-01532-7
Anzalone AV, Randolph PB, Davis JR, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576(7785):149–57. https://doi.org/10.1038/s41586-019-1711-4.
Tao R, Wang Y, Jiao Y, et al. Bi-PE: bi-directional priming improves CRISPR/Cas9 prime editing in mammalian cells. Nucleic Acids Res. 2022;50(11):6423–34. https://doi.org/10.1093/nar/gkac506.
Tao R, Wang Y, Hu Y, et al. WT-PE: Prime editing with nuclease wild-type Cas9 enables versatile large-scale genome editing. Signal Transduct Target Ther. 2022;7(1):108. https://doi.org/10.1038/s41392-022-00936-w.
Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533(7603):420–4. https://doi.org/10.1038/nature17946.
Kulcsar PI, Talas A, Ligeti Z, Krausz SL, Welker E. SuperFi-Cas9 exhibits remarkable fidelity but severely reduced activity yet works effectively with ABE8e. Nat Commun. 2022;13(1):6858. https://doi.org/10.1038/s41467-022-34527-8.
Wang X, Li J, Wang Y, et al. Efficient base editing in methylated regions with a human APOBEC3A-Cas9 fusion. Nat Biotechnol. 2018;36(10):946–9. https://doi.org/10.1038/nbt.4198.
Ma Y, Zhang J, Yin W, Zhang Z, Song Y, Chang X. Targeted AID-mediated mutagenesis (TAM) enables efficient genomic diversification in mammalian cells. Nat Methods. 2016;13(12):1029–35. https://doi.org/10.1038/nmeth.4027.
Shimatani Z, Kashojiya S, Takayama M, et al. Targeted base editing in rice and tomato using a CRISPR-Cas9 cytidine deaminase fusion. Nat Biotechnol. 2017;35(5):441–3. https://doi.org/10.1038/nbt.3833.
Richter MF, Zhao KT, Eton E, et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat Biotechnol. 2020;38(7):883–91. https://doi.org/10.1038/s41587-020-0453-z.
Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol. 2017;18(8):495–506. https://doi.org/10.1038/nrm.2017.48.
Chen PJ, Hussmann JA, Yan J, et al. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell. 2021;184(22):5635-5652 e29. https://doi.org/10.1016/j.cell.2021.09.018.
Ferreira da Silva J, Oliveira GP, Arasa-Verge EA, et al. Prime editing efficiency and fidelity are enhanced in the absence of mismatch repair. Nat Commun. 2022;13(1):10.1038/s41467-022-28442–1.
Ran FA, Cong L, Yan WX, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520(7546):186–91. https://doi.org/10.1038/nature14299.
Mianne J, Nasri A, Van CN, et al. CRISPR/Cas9-mediated gene knockout and interallelic gene conversion in human induced pluripotent stem cells using non-integrative bacteriophage-chimeric retrovirus-like particles. BMC Biol. 2022;20(1):8. https://doi.org/10.1186/s12915-021-01214-x.
Foy SP, Jacoby K, Bota DA, et al. Non-viral precision T cell receptor replacement for personalized cell therapy. Nature. 2022. https://doi.org/10.1038/s41586-022-05531-1
Hiatt J, Hultquist JF, McGregor MJ, et al. A functional map of HIV-host interactions in primary human T cells. Nat Commun. 2022;13(1):1752. https://doi.org/10.1038/s41467-022-29346-w.
Lin SC, Haga K, Zeng XL, Estes MK. Generation of CRISPR-Cas9-mediated genetic knockout human intestinal tissue-derived enteroid lines by lentivirus transduction and single-cell cloning. Nat Protoc. 2022;17(4):1004–27. https://doi.org/10.1038/s41596-021-00669-0.
Yang Y, Wang L, Bell P, et al. A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat Biotechnol. 2016;34(3):334–8. https://doi.org/10.1038/nbt.3469.
Sheriff A, Guri I, Zebrowska P, et al. ABE8e adenine base editor precisely and efficiently corrects a recurrent COL7A1 nonsense mutation. Sci Rep. 2022;12(1):19643. https://doi.org/10.1038/s41598-022-24184-8.
Kim Y, Hong SA, Yu J, et al. Adenine base editing and prime editing of chemically derived hepatic progenitors rescue genetic liver disease. Cell Stem Cell. 2021;28(9):1614-1624 e5. https://doi.org/10.1016/j.stem.2021.04.010.
Newby GA, Yen JS, Woodard KJ, et al. Base editing of haematopoietic stem cells rescues sickle cell disease in mice. Nature. 2021;595(7866):295–302. https://doi.org/10.1038/s41586-021-03609-w.
Osborn MJ, Newby GA, McElroy AN, et al. Base Editor Correction of COL7A1 in Recessive Dystrophic Epidermolysis Bullosa Patient-Derived Fibroblasts and iPSCs. J Invest Dermatol. 2020;140(2):338-347 e5. https://doi.org/10.1016/j.jid.2019.07.701.
Wu Y, Zeng J, Roscoe BP, et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat Med. 2019;25(5):776–83. https://doi.org/10.1038/s41591-019-0401-y.
Fu B, Liao J, Chen S, et al. CRISPR-Cas9-mediated gene editing of the BCL11A enhancer for pediatric beta(0)/beta(0) transfusion-dependent beta-thalassemia. Nat Med. 2022;28(8):1573–80. https://doi.org/10.1038/s41591-022-01906-z.
Han Y, Tan X, Jin T, et al. CRISPR/Cas9-based multiplex genome editing of BCL11A and HBG efficiently induces fetal hemoglobin expression. Eur J Pharmacol. 2022;918:174788. https://doi.org/10.1016/j.ejphar.2022.174788.
Khosravi MA, Abbasalipour M, Concordet JP, et al. Targeted deletion of BCL11A gene by CRISPR-Cas9 system for fetal hemoglobin reactivation: A promising approach for gene therapy of beta thalassemia disease. Eur J Pharmacol. 2019;854:398–405. https://doi.org/10.1016/j.ejphar.2019.04.042.
Rebar EJ, Greisman HA, Pabo CO. Phage display methods for selecting zinc finger proteins with novel DNA-binding specificities. Methods Enzymol. 1996;267:129–49. https://doi.org/10.1016/s0076-6879(96)67010-4.
Harmatz P, Prada CE, Burton BK, et al. First-in-human in vivo genome editing via AAV-zinc-finger nucleases for mucopolysaccharidosis I/II and hemophilia B. Mol Ther. 2022;30(12):3587–600. https://doi.org/10.1016/j.ymthe.2022.10.010.
Christian M, Cermak T, Doyle EL, et al. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics. 2010;186(2):757–61. https://doi.org/10.1534/genetics.110.120717.
Maliga P. Engineering the plastid and mitochondrial genomes of flowering plants. Nat Plants. 2022;8(9):996–1006. https://doi.org/10.1038/s41477-022-01227-6.
Chen Y, Yu J, Niu Y, et al. Modeling Rett Syndrome Using TALEN-Edited MECP2 Mutant Cynomolgus Monkeys. Cell. 2017;169(5):945-955 e10. https://doi.org/10.1016/j.cell.2017.04.035.
Grissa I, Vergnaud G, Pourcel C. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics. 2007;8:172. https://doi.org/10.1186/1471-2105-8-172.
Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol. 1987;169(12):5429–33. https://doi.org/10.1128/jb.169.12.5429-5433.1987.
Jansen R, Embden JD, Gaastra W, Schouls LM. Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol. 2002;43(6):1565–75. https://doi.org/10.1046/j.1365-2958.2002.02839.x.
Deltcheva E, Chylinski K, Sharma CM, et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471(7340):602–7. https://doi.org/10.1038/nature09886.
Brouns SJ, Jore MM, Lundgren M, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science (New York, NY). 2008;321(5891):960–4. https://doi.org/10.1126/science.1159689.
Bolotin A, Quinquis B, Sorokin A, Ehrlich SD. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology. 2005;151(Pt 8):2551–61. https://doi.org/10.1099/mic.0.28048-0.
Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, Soria E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol. 2005;60(2):174–82. https://doi.org/10.1007/s00239-004-0046-3.
Lander ES. The Heroes of CRISPR. Cell. 2016;164(1–2):18–28. https://doi.org/10.1016/j.cell.2015.12.041.
Cho SW, Kim S, Kim JM, Kim JS. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol. 2013;31(3):230–2. https://doi.org/10.1038/nbt.2507.
Yuen CTL, Thean DGL, Chan BKC, et al. High-fidelity KKH variant of Staphylococcus aureus Cas9 nucleases with improved base mismatch discrimination. Nucleic Acids Res. 2022;50(3):1650–60. https://doi.org/10.1093/nar/gkab1291.
Helfer-Hungerbuehler AK, Shah J, Meili T, Boenzli E, Li P, Hofmann-Lehmann R. Adeno-Associated Vector-Delivered CRISPR/SaCas9 System Reduces Feline Leukemia Virus Production In Vitro. Viruses. 2021;13(8):1636. https://doi.org/10.3390/v13081636.
Ronspies M, Schindele P, Wetzel R, Puchta H. CRISPR-Cas9-mediated chromosome engineering in Arabidopsis thaliana. Nat Protoc. 2022;17(5):1332–58. https://doi.org/10.1038/s41596-022-00686-7.
Kim E, Koo T, Park SW, et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat Commun. 2017;8:14500. https://doi.org/10.1038/ncomms14500.
Francis C, Amiji M. Expanding CRISPR repertoire using CjCas9 as a smaller editing tool. Mol Ther Nucleic Acids. 2022;30:64–5. https://doi.org/10.1016/j.omtn.2022.09.013.
Dugar G, Leenay RT, Eisenbart SK, et al. CRISPR RNA-Dependent Binding and Cleavage of Endogenous RNAs by the Campylobacter jejuni Cas9. Mol Cell. 2018;69(5):893–9057. https://doi.org/10.1016/j.molcel.2018.01.032.
Nakagawa R, Ishiguro S, Okazaki S, et al. Engineered Campylobacter jejuni Cas9 variant with enhanced activity and broader targeting range. Commun Biol. 2022;5(1):211. https://doi.org/10.1038/s42003-022-03149-7.
Hou Z, Zhang Y, Propson NE, et al. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc Natl Acad Sci U S A. 2013;110(39):15644–9. https://doi.org/10.1073/pnas.1313587110.
Hoffmann MD, Mathony J, Upmeier Zu Belzen J, et al. Optogenetic control of Neisseria meningitidis Cas9 genome editing using an engineered, light-switchable anti-CRISPR protein. Nucleic Acids Res. 2021;49(5):e29. https://doi.org/10.1093/nar/gkaa1198.
Amrani N, Gao XD, Liu P, et al. NmeCas9 is an intrinsically high-fidelity genome-editing platform. Genome Biol. 2018;19(1):214. https://doi.org/10.1186/s13059-018-1591-1.
Rousseau BA, Hou Z, Gramelspacher MJ, Zhang Y. Programmable RNA Cleavage and Recognition by a Natural CRISPR-Cas9 System from Neisseria meningitidis. Mol Cell. 2018;69(5):906-914 e4. https://doi.org/10.1016/j.molcel.2018.01.025.
Hirano H, Gootenberg JS, Horii T, et al. Structure and Engineering of Francisella novicida Cas9. Cell. 2016;164(5):950–61. https://doi.org/10.1016/j.cell.2016.01.039.
Acharya S, Mishra A, Paul D, et al. Francisella novicida Cas9 interrogates genomic DNA with very high specificity and can be used for mammalian genome editing. Proc Natl Acad Sci U S A. 2019;116(42):20959–68. https://doi.org/10.1073/pnas.1818461116.
Mekler V, Kuznedelov K, Severinov K. Quantification of the affinities of CRISPR-Cas9 nucleases for cognate protospacer adjacent motif (PAM) sequences. J Biol Chem. 2020;295(19):6509–17. https://doi.org/10.1074/jbc.RA119.012239.
Chen F, Ding X, Feng Y, Seebeck T, Jiang Y, Davis GD. Targeted activation of diverse CRISPR-Cas systems for mammalian genome editing via proximal CRISPR targeting. Nat Commun. 2017;8:14958. https://doi.org/10.1038/ncomms14958.
Hu Z, Wang S, Zhang C, et al. A compact Cas9 ortholog from Staphylococcus Auricularis (SauriCas9) expands the DNA targeting scope. PLoS Biol. 2020;18(3):e3000686. https://doi.org/10.1371/journal.pbio.3000686.
Hu Z, Zhang C, Wang S, et al. Discovery and engineering of small SlugCas9 with broad targeting range and high specificity and activity. Nucleic Acids Res. 2021;49(7):4008–19. https://doi.org/10.1093/nar/gkab148.
Nishimasu H, Ran FA, Hsu PD, et al. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156(5):935–49. https://doi.org/10.1016/j.cell.2014.02.001.
Jiang F, Doudna JA. CRISPR-Cas9 Structures and Mechanisms. Annu Rev Biophys. 2017;46:505–29. https://doi.org/10.1146/annurev-biophys-062215-010822.
Jinek M, Jiang F, Taylor DW, et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science (New York, NY). 2014;343(6176):1247997. https://doi.org/10.1126/science.1247997.
Jiang F, Zhou K, Ma L, Gressel S, Doudna JA. STRUCTURAL BIOLOGY. A Cas9-guide RNA complex preorganized for target DNA recognition. Science (New York, NY). 2015;348(6242):1477–81. https://doi.org/10.1126/science.aab1452.
Jiang F, Taylor DW, Chen JS, et al. Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science (New York, NY). 2016;351(6275):867–71. https://doi.org/10.1126/science.aad8282.
Cofsky JC, Soczek KM, Knott GJ, Nogales E, Doudna JA. CRISPR-Cas9 bends and twists DNA to read its sequence. Nat Struct Mol Biol. 2022;29(4):395–402. https://doi.org/10.1038/s41594-022-00756-0.
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8(11):2281–308. https://doi.org/10.1038/nprot.2013.143.
Kleinstiver BP, Prew MS, Tsai SQ, et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015;523(7561):481–5. https://doi.org/10.1038/nature14592.
Hu JH, Miller SM, Geurts MH, et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. 2018;556(7699):57–63. https://doi.org/10.1038/nature26155.
Nishimasu H, Shi X, Ishiguro S, et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science (New York, NY). 2018;361(6408):1259–62. https://doi.org/10.1126/science.aas9129.
Walton RT, Christie KA, Whittaker MN, Kleinstiver BP. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science (New York, NY). 2020;368(6488):290–6. https://doi.org/10.1126/science.aba8853.
Chatterjee P, Jakimo N, Jacobson JM. Minimal PAM specificity of a highly similar SpCas9 ortholog. Sci Adv. 2018;4(10):eaau0766. https://doi.org/10.1126/sciadv.aau0766.
Agudelo D, Carter S, Velimirovic M, et al. Versatile and robust genome editing with Streptococcus thermophilus CRISPR1-Cas9. Genome Res. 2020;30(1):107–17. https://doi.org/10.1101/gr.255414.119.
Cui Z, Tian R, Huang Z, et al. FrCas9 is a CRISPR/Cas9 system with high editing efficiency and fidelity. Nat Commun. 2022;13(1):1425. https://doi.org/10.1038/s41467-022-29089-8.
Kleinstiver BP, Prew MS, Tsai SQ, et al. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat Biotechnol. 2015;33(12):1293–8. https://doi.org/10.1038/nbt.3404.
Wang S, Mao H, Hou L, et al. Compact SchCas9 Recognizes the Simple NNGR PAM. Adv Sci (Weinh). 2022;9(4):e2104789. https://doi.org/10.1002/advs.202104789.
Yamada M, Watanabe Y, Gootenberg JS, et al. Crystal Structure of the Minimal Cas9 from Campylobacter jejuni Reveals the Molecular Diversity in the CRISPR-Cas9 Systems. Mol Cell. 2017;65(6):1109-1121 e3. https://doi.org/10.1016/j.molcel.2017.02.007.
Esvelt KM, Mali P, Braff JL, Moosburner M, Yaung SJ, Church GM. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat Methods. 2013;10(11):1116–21. https://doi.org/10.1038/nmeth.2681.
Edraki A, Mir A, Ibraheim R, et al. A Compact, High-Accuracy Cas9 with a Dinucleotide PAM for In Vivo Genome Editing. Mol Cell. 2019;73(4):714-726 e4. https://doi.org/10.1016/j.molcel.2018.12.003.
Harrington LB, Paez-Espino D, Staahl BT, et al. A thermostable Cas9 with increased lifetime in human plasma. Nat Commun. 2017;8(1):1424. https://doi.org/10.1038/s41467-017-01408-4.
Fedorova I, Arseniev A, Selkova P, et al. DNA targeting by Clostridium cellulolyticum CRISPR-Cas9 Type II-C system. Nucleic Acids Res. 2020;48(4):2026–34. https://doi.org/10.1093/nar/gkz1225.
Hirano S, Abudayyeh OO, Gootenberg JS, et al. Structural basis for the promiscuous PAM recognition by Corynebacterium diphtheriae Cas9. Nat Commun. 2019;10(1):1968. https://doi.org/10.1038/s41467-019-09741-6.
Hille F, Richter H, Wong SP, Bratovic M, Ressel S, Charpentier E. The Biology of CRISPR-Cas: Backward and Forward. Cell. 2018;172(6):1239–59. https://doi.org/10.1016/j.cell.2017.11.032.
Huang J, Rowe D, Subedi P, et al. CRISPR-Cas12a induced DNA double-strand breaks are repaired by multiple pathways with different mutation profiles in Magnaporthe oryzae. Nat Commun. 2022;13(1):7168. https://doi.org/10.1038/s41467-022-34736-1.
Naqvi MM, Lee L, Montaguth OET, Diffin FM, Szczelkun MD. CRISPR-Cas12a-mediated DNA clamping triggers target-strand cleavage. Nat Chem Biol. 2022;18(9):1014–22. https://doi.org/10.1038/s41589-022-01082-8.
Shebanova R, Nikitchina N, Shebanov N, et al. Efficient target cleavage by Type V Cas12a effectors programmed with split CRISPR RNA. Nucleic Acids Res. 2022;50(2):1162–73. https://doi.org/10.1093/nar/gkab1227.
Gier RA, Budinich KA, Evitt NH, et al. High-performance CRISPR-Cas12a genome editing for combinatorial genetic screening. Nat Commun. 2020;11(1):3455. https://doi.org/10.1038/s41467-020-17209-1.
Zetsche B, Gootenberg JS, Abudayyeh OO, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163(3):759–71. https://doi.org/10.1016/j.cell.2015.09.038.
Nishimasu H, Cong L, Yan WX, et al. Crystal Structure of Staphylococcus aureus Cas9. Cell. 2015;162(5):1113–26. https://doi.org/10.1016/j.cell.2015.08.007.
Miller SM, Wang T, Randolph PB, et al. Continuous evolution of SpCas9 variants compatible with non-G PAMs. Nat Biotechnol. 2020;38(4):471–81. https://doi.org/10.1038/s41587-020-0412-8.
Novakova Z, Milosevic M, Kutil Z, et al. Generation and characterization of human U-2 OS cell lines with the CRISPR/Cas9-edited protoporphyrinogen oxidase IX gene. Sci Rep. 2022;12(1):17081. https://doi.org/10.1038/s41598-022-21147-x.
Shan W, Yang X, Ren Q, Wang Q. Generation of SCN1A Knock out induced pluripotent stem cell (iPSC) line. Stem Cell Res. 2021;55:102452. https://doi.org/10.1016/j.scr.2021.102452.
Chen H, Durinck S, Patel H, et al. Population-wide gene disruption in the murine lung epithelium via AAV-mediated delivery of CRISPR-Cas9 components. Mol Ther Methods Clin Dev. 2022;27:431–49. https://doi.org/10.1016/j.omtm.2022.10.016.
Tipanee J, Samara-Kuko E, Gevaert T, Chuah MK, VandenDriessche T. Universal allogeneic CAR T cells engineered with Sleeping Beauty transposons and CRISPR-CAS9 for cancer immunotherapy. Mol Ther. 2022;30(10):3155–75. https://doi.org/10.1016/j.ymthe.2022.06.006.
He X, Zhang Z, Xue J, et al. Low-dose AAV-CRISPR-mediated liver-specific knock-in restored hemostasis in neonatal hemophilia B mice with subtle antibody response. Nat Commun. 2022;13(1):7275. https://doi.org/10.1038/s41467-022-34898-y.
Luo C, Wang Q, Guo R, et al. A novel Pseudorabies virus vaccine developed using HDR-CRISPR/Cas9 induces strong humoral and cellular immune response in mice. Virus Res. 2022;322:198937. https://doi.org/10.1016/j.virusres.2022.198937.
Ishibashi R, Maki R, Kitano S, Miyachi H, Toyoshima F. Development of an in vivo cleavable donor plasmid for targeted transgene integration by CRISPR-Cas9 and CRISPR-Cas12a. Sci Rep. 2022;12(1):17775. https://doi.org/10.1038/s41598-022-22639-6.
Ko N, Shim J, Kim HJ, et al. A desirable transgenic strategy using GGTA1 endogenous promoter-mediated knock-in for xenotransplantation model. Sci Rep. 2022;12(1):9611. https://doi.org/10.1038/s41598-022-13536-z.
Tan J, Zhao Y, Wang B, et al. Efficient CRISPR/Cas9-based plant genomic fragment deletions by microhomology-mediated end joining. Plant Biotechnol J. 2020;18(11):2161–3. https://doi.org/10.1111/pbi.13390.
Nishiguchi KM, Fujita K, Miya F, Katayama S, Nakazawa T. Single AAV-mediated mutation replacement genome editing in limited number of photoreceptors restores vision in mice. Nat Commun. 2020;11(1):482. https://doi.org/10.1038/s41467-019-14181-3.
Iyer S, Suresh S, Guo D, et al. Precise therapeutic gene correction by a simple nuclease-induced double-stranded break. Nature. 2019;568(7753):561–5. https://doi.org/10.1038/s41586-019-1076-8.
Kim SI, Matsumoto T, Kagawa H, et al. Microhomology-assisted scarless genome editing in human iPSCs. Nat Commun. 2018;9(1):939. https://doi.org/10.1038/s41467-018-03044-y.
Xiao Q, Min T, Ma S, Hu L, Chen H, Lu D. Intracellular generation of single-strand template increases the knock-in efficiency by combining CRISPR/Cas9 with AAV. Mol Genet Genomics. 2018;293(4):1051–60. https://doi.org/10.1007/s00438-018-1437-2.
Wong EA, Capecchi MR. Homologous recombination between coinjected DNA sequences peaks in early to mid-S phase. Mol Cell Biol. 1987;7(6):2294–5. https://doi.org/10.1128/mcb.7.6.2294-2295.1987.
Rothkamm K, Kruger I, Thompson LH, Lobrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol. 2003;23(16):5706–15. https://doi.org/10.1128/MCB.23.16.5706-5715.2003.
Harris RS, Petersen-Mahrt SK, Neuberger MS. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol Cell. 2002;10(5):1247–53. https://doi.org/10.1016/s1097-2765(02)00742-6.
Caldecott KW. Single-strand break repair and genetic disease. Nat Rev Genet. 2008;9(8):619–31. https://doi.org/10.1038/nrg2380.
Kim YB, Komor AC, Levy JM, Packer MS, Zhao KT, Liu DR. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat Biotechnol. 2017;35(4):371–6. https://doi.org/10.1038/nbt.3803.
Wang Y, Zhou L, Tao R, et al. sgBE: a structure-guided design of sgRNA architecture specifies base editing window and enables simultaneous conversion of cytosine and adenosine. Genome Biol. 2020;21(1):222. https://doi.org/10.1186/s13059-020-02137-6.
Wang Y, Zhou L, Liu N, Yao S. BE-PIGS: a base-editing tool with deaminases inlaid into Cas9 PI domain significantly expanded the editing scope. Signal Transduct Target Ther. 2019;4:36. https://doi.org/10.1038/s41392-019-0072-7.
Cheng TL, Li S, Yuan B, Wang X, Zhou W, Qiu Z. Expanding C-T base editing toolkit with diversified cytidine deaminases. Nat Commun. 2019;10(1):3612. https://doi.org/10.1038/s41467-019-11562-6.
Huang TP, Zhao KT, Miller SM, et al. Circularly permuted and PAM-modified Cas9 variants broaden the targeting scope of base editors. Nat Biotechnol. 2019;37(6):626–31. https://doi.org/10.1038/s41587-019-0134-y.
Jiang L, Long J, Yang Y, et al. Internally inlaid SaCas9 base editors enable window specific base editing. Theranostics. 2022;12(10):4767–78. https://doi.org/10.7150/thno.70869.
Jiang W, Feng S, Huang S, et al. BE-PLUS: a new base editing tool with broadened editing window and enhanced fidelity. Cell Res. 2018;28(8):855–61. https://doi.org/10.1038/s41422-018-0052-4.
Jia K, Cui YR, Huang S, et al. Phage peptides mediate precision base editing with focused targeting window. Nat Commun. 2022;13(1):1662. https://doi.org/10.1038/s41467-022-29365-7.
Xiong X, Li Z, Liang J, Liu K, Li C, Li JF. A cytosine base editor toolkit with varying activity windows and target scopes for versatile gene manipulation in plants. Nucleic Acids Res. 2022;50(6):3565–80. https://doi.org/10.1093/nar/gkac166.
Tan J, Zhang F, Karcher D, Bock R. Engineering of high-precision base editors for site-specific single nucleotide replacement. Nat Commun. 2019;10(1):439. https://doi.org/10.1038/s41467-018-08034-8.
Tan J, Zeng D, Zhao Y, et al. PhieABEs: a PAM-less/free high-efficiency adenine base editor toolbox with wide target scope in plants. Plant Biotechnol J. 2022;20(5):934–43. https://doi.org/10.1111/pbi.13774.
Rees HA, Komor AC, Yeh WH, et al. Improving the DNA specificity and applicability of base editing through protein engineering and protein delivery. Nat Commun. 2017;8:15790. https://doi.org/10.1038/ncomms15790.
Kim D, Kim DE, Lee G, Cho SI, Kim JS. Genome-wide target specificity of CRISPR RNA-guided adenine base editors. Nat Biotechnol. 2019;37(4):430–5. https://doi.org/10.1038/s41587-019-0050-1.
Kim D, Lim K, Kim ST, et al. Genome-wide target specificities of CRISPR RNA-guided programmable deaminases. Nat Biotechnol. 2017;35(5):475–80. https://doi.org/10.1038/nbt.3852.
Grunewald J, Zhou R, Lareau CA, et al. A dual-deaminase CRISPR base editor enables concurrent adenine and cytosine editing. Nat Biotechnol. 2020;38(7):861–4. https://doi.org/10.1038/s41587-020-0535-y.
Zhang X, Zhu B, Chen L, et al. Dual base editor catalyzes both cytosine and adenine base conversions in human cells. Nat Biotechnol. 2020;38(7):856–60. https://doi.org/10.1038/s41587-020-0527-y.
Li C, Zhang R, Meng X, et al. Targeted, random mutagenesis of plant genes with dual cytosine and adenine base editors. Nat Biotechnol. 2020;38(7):875–82. https://doi.org/10.1038/s41587-019-0393-7.
Gehrke JM, Cervantes O, Clement MK, et al. An APOBEC3A-Cas9 base editor with minimized bystander and off-target activities. Nat Biotechnol. 2018;36(10):977–82. https://doi.org/10.1038/nbt.4199.
Grunewald J, Zhou R, Garcia SP, et al. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature. 2019;569(7756):433–7. https://doi.org/10.1038/s41586-019-1161-z.
Jin S, Zong Y, Gao Q, et al. Cytosine, but not adenine, base editors induce genome-wide off-target mutations in rice. Science (New York, NY). 2019;364(6437):292–5. https://doi.org/10.1126/science.aaw7166.
Zhou C, Sun Y, Yan R, et al. Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature. 2019;571(7764):275–8. https://doi.org/10.1038/s41586-019-1314-0.
Zuo E, Sun Y, Wei W, et al. Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science (New York, NY). 2019;364(6437):289–92. https://doi.org/10.1126/science.aav9973.
Pacesa M, Lin CH, Clery A, et al. Structural basis for Cas9 off-target activity. Cell. 2022;185(22):4067–408121. https://doi.org/10.1016/j.cell.2022.09.026.
Bravo JPK, Liu MS, Hibshman GN, et al. Structural basis for mismatch surveillance by CRISPR-Cas9. Nature. 2022;603(7900):343–7. https://doi.org/10.1038/s41586-022-04470-1.
Cromer MK, Barsan VV, Jaeger E, et al. Ultra-deep sequencing validates safety of CRISPR/Cas9 genome editing in human hematopoietic stem and progenitor cells. Nat Commun. 2022;13(1):4724. https://doi.org/10.1038/s41467-022-32233-z.
Fu R, He W, Dou J, et al. Systematic decomposition of sequence determinants governing CRISPR/Cas9 specificity. Nat Commun. 2022;13(1):474. https://doi.org/10.1038/s41467-022-28028-x.
Pacesa M, Loeff L, Querques I, Muckenfuss LM, Sawicka M, Jinek M. R-loop formation and conformational activation mechanisms of Cas9. Nature. 2022;609(7925):191–6. https://doi.org/10.1038/s41586-022-05114-0.
Kosicki M, Tomberg K, Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol. 2018;36(8):765–71. https://doi.org/10.1038/nbt.4192.
Jin S, Fei H, Zhu Z, et al. Rationally Designed APOBEC3B Cytosine Base Editors with Improved Specificity. Mol Cell. 2020;79(5):728-740 e6. https://doi.org/10.1016/j.molcel.2020.07.005.
Zuo E, Sun Y, Yuan T, et al. A rationally engineered cytosine base editor retains high on-target activity while reducing both DNA and RNA off-target effects. Nat Methods. 2020;17(6):600–4. https://doi.org/10.1038/s41592-020-0832-x.
Nguyen Tran MT, Mohd Khalid MKN, Wang Q, et al. Engineering domain-inlaid SaCas9 adenine base editors with reduced RNA off-targets and increased on-target DNA editing. Nat Commun. 2020;11(1):4871. https://doi.org/10.1038/s41467-020-18715-y.
Schmelas C, Grimm D. Split Cas9, Not Hairs - Advancing the Therapeutic Index of CRISPR Technology. Biotechnol J. 2018;13(9):e1700432. https://doi.org/10.1002/biot.201700432.
Long J, Liu N, Tang W, et al. A split cytosine deaminase architecture enables robust inducible base editing. FASEB J. 2021;35(12):e22045. https://doi.org/10.1096/fj.202100123R.
Boiteux S, Guillet M. Abasic sites in DNA: repair and biological consequences in Saccharomyces cerevisiae. DNA Repair (Amst). 2004;3(1):1–12. https://doi.org/10.1016/j.dnarep.2003.10.002.
Hagen L, Pena-Diaz J, Kavli B, Otterlei M, Slupphaug G, Krokan HE. Genomic uracil and human disease. Exp Cell Res. 2006;312(14):2666–72. https://doi.org/10.1016/j.yexcr.2006.06.015.
Krokan HE, Drablos F, Slupphaug G. Uracil in DNA–occurrence, consequences and repair. Oncogene. 2002;21(58):8935–48. https://doi.org/10.1038/sj.onc.1205996.
Krokan HE, Nilsen H, Skorpen F, Otterlei M, Slupphaug G. Base excision repair of DNA in mammalian cells. FEBS Lett. 2000;476(1–2):73–7. https://doi.org/10.1016/s0014-5793(00)01674-4.
Krokan HE, Otterlei M, Nilsen H, et al. Properties and functions of human uracil-DNA glycosylase from the UNG gene. Prog Nucleic Acid Res Mol Biol. 2001;68:365–86. https://doi.org/10.1016/s0079-6603(01)68112-1.
Lindahl T. An N-glycosidase from Escherichia coli that releases free uracil from DNA containing deaminated cytosine residues. Proc Natl Acad Sci U S A. 1974;71(9):3649–53. https://doi.org/10.1073/pnas.71.9.3649.
Trasvina-Arenas CH, Demir M, Lin WJ, David SS. Structure, function and evolution of the Helix-hairpin-Helix DNA glycosylase superfamily: Piecing together the evolutionary puzzle of DNA base damage repair mechanisms. DNA Repair (Amst). 2021;108:103231. https://doi.org/10.1016/j.dnarep.2021.103231.
Kouzminova EA, Kuzminov A. Patterns of chromosomal fragmentation due to uracil-DNA incorporation reveal a novel mechanism of replication-dependent double-stranded breaks. Mol Microbiol. 2008;68(1):202–15. https://doi.org/10.1111/j.1365-2958.2008.06149.x.
Bhakat KK, Sengupta S, Mitra S. Fine-tuning of DNA base excision/strand break repair via acetylation. DNA Repair (Amst). 2020;93:102931. https://doi.org/10.1016/j.dnarep.2020.102931.
Davletgildeeva AT, Kuznetsova AA, Novopashina DS, et al. Comparative Analysis of Exo- and Endonuclease Activities of APE1-like Enzymes. Int J Mol Sci. 2022;23(5):2869. https://doi.org/10.3390/ijms23052869.
Nicholl ID, Nealon K, Kenny MK. Reconstitution of human base excision repair with purified proteins. Biochemistry. 1997;36(24):7557–66. https://doi.org/10.1021/bi962950w.
Schrader CE, Guikema JE, Wu X, Stavnezer J. The roles of APE1, APE2, DNA polymerase beta and mismatch repair in creating S region DNA breaks during antibody class switch. Philos Trans R Soc Lond B Biol Sci. 2009;364(1517):645–52. https://doi.org/10.1098/rstb.2008.0200.
Stavnezer J, Linehan EK, Thompson MR, et al. Differential expression of APE1 and APE2 in germinal centers promotes error-prone repair and A: T mutations during somatic hypermutation. Proc Natl Acad Sci U S A. 2014;111(25):9217–22. https://doi.org/10.1073/pnas.1405590111.
Swanson RL, Morey NJ, Doetsch PW, Jinks-Robertson S. Overlapping specificities of base excision repair, nucleotide excision repair, recombination, and translesion synthesis pathways for DNA base damage in Saccharomyces cerevisiae. Mol Cell Biol. 1999;19(4):2929–35. https://doi.org/10.1128/MCB.19.4.2929.
Beard WA, Wilson SH. DNA polymerase beta and other gap-filling enzymes in mammalian base excision repair. Enzymes. 2019;45:1–26. https://doi.org/10.1016/bs.enz.2019.08.002.
Krijger PH, Tsaalbi-Shtylik A, Wit N, van den Berk PC, de Wind N, Jacobs H. Rev1 is essential in generating G to C transversions downstream of the Ung2 pathway but not the Msh2+Ung2 hybrid pathway. Eur J Immunol. 2013;43(10):2765–70. https://doi.org/10.1002/eji.201243191.
Arbab M, Shen MW, Mok B, et al. Determinants of Base Editing Outcomes from Target Library Analysis and Machine Learning. Cell. 2020;182(2):463-480 e30. https://doi.org/10.1016/j.cell.2020.05.037.
Zhao D, Li J, Li S, et al. Glycosylase base editors enable C-to-A and C-to-G base changes. Nat Biotechnol. 2021;39(1):35–40. https://doi.org/10.1038/s41587-020-0592-2.
Kurt IC, Zhou R, Iyer S, et al. CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nat Biotechnol 2020;39(1):41–6. https://doi.org/10.1038/s41587-020-0609-x.
Sretenovic S, Liu S, Li G, et al. Exploring C-To-G Base Editing in Rice, Tomato, and Poplar. Front Genome Ed. 2021;3:756766. https://doi.org/10.3389/fgeed.2021.756766.
Chen S, Liu Z, Lai L, Li Z. Efficient C-to-G Base Editing with Improved Target Compatibility Using Engineered Deaminase-nCas9 Fusions. CRISPR J. 2022;5(3):389–96. https://doi.org/10.1089/crispr.2021.0124.
Chen L, Park JE, Paa P, et al. Programmable C: G to G: C genome editing with CRISPR-Cas9-directed base excision repair proteins. Nat Commun. 2021;12(1):1384. https://doi.org/10.1038/s41467-021-21559-9.
Rees HA, Liu DR. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet. 2018;19(12):770–88. https://doi.org/10.1038/s41576-018-0059-1.
Wang L, Xue W, Yan L, et al. Enhanced base editing by co-expression of free uracil DNA glycosylase inhibitor. Cell Res. 2017;27(10):1289–92. https://doi.org/10.1038/cr.2017.111.
Lau AY, Wyatt MD, Glassner BJ, Samson LD, Ellenberger T. Molecular basis for discriminating between normal and damaged bases by the human alkyladenine glycosylase, AAG. Proc Natl Acad Sci U S A. 2000;97(25):13573–8. https://doi.org/10.1073/pnas.97.25.13573.
Yeh WH, Chiang H, Rees HA, Edge ASB, Liu DR. In vivo base editing of post-mitotic sensory cells. Nat Commun. 2018;9(1):2184. https://doi.org/10.1038/s41467-018-04580-3.
Ryu SM, Koo T, Kim K, et al. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat Biotechnol. 2018;36(6):536–9. https://doi.org/10.1038/nbt.4148.
Kang BC, Yun JY, Kim ST, et al. Precision genome engineering through adenine base editing in plants. Nat Plants. 2018;4(7):427–31. https://doi.org/10.1038/s41477-018-0178-x.
Landrum MJ, Lee JM, Benson M, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44(D1):D862–8. https://doi.org/10.1093/nar/gkv1222.
Lin Q, Jin S, Zong Y, et al. High-efficiency prime editing with optimized, paired pegRNAs in plants. Nat Biotechnol. 2021;39(8):923–7. https://doi.org/10.1038/s41587-021-00868-w.
Nelson JW, Randolph PB, Shen SP, et al. Engineered pegRNAs improve prime editing efficiency. Nat Biotechnol. 2022;40(3):402–10. https://doi.org/10.1038/s41587-021-01039-7.
Liu Y, Yang G, Huang S, et al. Enhancing prime editing by Csy4-mediated processing of pegRNA. Cell Res. 2021;31(10):1134–6. https://doi.org/10.1038/s41422-021-00520-x.
Li X, Wang X, Sun W, et al. Enhancing prime editing efficiency by modified pegRNA with RNA G-quadruplexes. J Mol Cell Biol. 2022;14(4):mjac022. https://doi.org/10.1093/jmcb/mjac022.
Zhang G, Liu Y, Huang S, et al. Enhancement of prime editing via xrRNA motif-joined pegRNA. Nat Commun. 2022;13(1):1856. https://doi.org/10.1038/s41467-022-29507-x.
Zong Y, Liu Y, Xue C, et al. Author Correction: An engineered prime editor with enhanced editing efficiency in plants. Nat Biotechnol. 2022;40(9):1394–402. https://doi.org/10.1038/s41587-022-01308-z.
Zheng C, Liang SQ, Liu B, et al. A flexible split prime editor using truncated reverse transcriptase improves dual-AAV delivery in mouse liver. Mol Ther. 2022;30(3):1343–51. https://doi.org/10.1016/j.ymthe.2022.01.005.
Jiang T, Zhang XO, Weng Z, Xue W. Deletion and replacement of long genomic sequences using prime editing. Nat Biotechnol. 2022;40(2):227–34. https://doi.org/10.1038/s41587-021-01026-y.
Choi J, Chen W, Suiter CC, et al. Precise genomic deletions using paired prime editing. Nat Biotechnol. 2022;40(2):218–26. https://doi.org/10.1038/s41587-021-01025-z.
Anzalone AV, Gao XD, Podracky CJ, et al. Programmable deletion, replacement, integration and inversion of large DNA sequences with twin prime editing. Nat Biotechnol. 2021;45(5):731–40. https://doi.org/10.1038/s41587-021-01133-w.
Zhuang Y, Liu J, Wu H, et al. Increasing the efficiency and precision of prime editing with guide RNA pairs. Nat Chem Biol. 2022;18(1):29–37. https://doi.org/10.1038/s41589-021-00889-1.
Heyer WD, Ehmsen KT, Liu J. Regulation of homologous recombination in eukaryotes. Annu Rev Genet. 2010;44:113–39. https://doi.org/10.1146/annurev-genet-051710-150955.
Nakade S, Tsubota T, Sakane Y, et al. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nat Commun. 2014;5:5560. https://doi.org/10.1038/ncomms6560.
Anzalone AV, Gao XD, Podracky CJ, et al. Programmable deletion, replacement, integration and inversion of large DNA sequences with twin prime editing. Nat Biotechnol. 2022;40(5):731–40. https://doi.org/10.1038/s41587-021-01133-w.
Wang J, He Z, Wang G, et al. Efficient targeted insertion of large DNA fragments without DNA donors. Nat Methods. 2022;19(3):331–40. https://doi.org/10.1038/s41592-022-01399-1.
Modzelewski AJ, Gan Chong J, Wang T, He L. Mammalian genome innovation through transposon domestication. Nat Cell Biol. 2022;24(9):1332–40. https://doi.org/10.1038/s41556-022-00970-4.
Peters JE, Makarova KS, Shmakov S, Koonin EV. Recruitment of CRISPR-Cas systems by Tn7-like transposons. Proc Natl Acad Sci U S A. 2017;114(35):E7358–66. https://doi.org/10.1073/pnas.1709035114.
Klompe SE, Vo PLH, Halpin-Healy TS, Sternberg SH. Transposon-encoded CRISPR-Cas systems direct RNA-guided DNA integration. Nature. 2019;571(7764):219–25. https://doi.org/10.1038/s41586-019-1323-z.
Vo PLH, Ronda C, Klompe SE, et al. CRISPR RNA-guided integrases for high-efficiency, multiplexed bacterial genome engineering. Nat Biotechnol. 2021;39(4):480–9. https://doi.org/10.1038/s41587-020-00745-y.
Strecker J, Ladha A, Gardner Z, et al. RNA-guided DNA insertion with CRISPR-associated transposases. Science (New York, NY). 2019;365(6448):48–53. https://doi.org/10.1126/science.aax9181.
Bhatt S, Chalmers R. Targeted DNA transposition in vitro using a dCas9-transposase fusion protein. Nucleic Acids Res. 2019;47(15):8126–35. https://doi.org/10.1093/nar/gkz552.
Capecchi MR. Altering the genome by homologous recombination. Science (New York, NY). 1989;244(4910):1288–92. https://doi.org/10.1126/science.2660260.
Thomas KR, Folger KR, Capecchi MR. High frequency targeting of genes to specific sites in the mammalian genome. Cell. 1986;44(3):419–28. https://doi.org/10.1016/0092-8674(86)90463-0.
Turan S, Zehe C, Kuehle J, Qiao J, Bode J. Recombinase-mediated cassette exchange (RMCE) - a rapidly-expanding toolbox for targeted genomic modifications. Gene. 2013;515(1):1–27. https://doi.org/10.1016/j.gene.2012.11.016.
Gaj T, Sirk SJ, Barbas CF 3rd. Expanding the scope of site-specific recombinases for genetic and metabolic engineering. Biotechnol Bioeng. 2014;111(1):1–15. https://doi.org/10.1002/bit.25096.
Jakobsen JE, Johansen MG, Schmidt M, et al. Generation of minipigs with targeted transgene insertion by recombinase-mediated cassette exchange (RMCE) and somatic cell nuclear transfer (SCNT). Transgenic Res. 2013;22(4):709–23. https://doi.org/10.1007/s11248-012-9671-6.
Chaikind B, Bessen JL, Thompson DB, Hu JH, Liu DR. A programmable Cas9-serine recombinase fusion protein that operates on DNA sequences in mammalian cells. Nucleic Acids Res. 2016;44(20):9758–70. https://doi.org/10.1093/nar/gkw707.
Holmes K, Roberts OL, Thomas AM, Cross MJ. Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition. Cell Signal. 2007;19(10):2003–12. https://doi.org/10.1016/j.cellsig.2007.05.013.
Huang X, Zhou G, Wu W, et al. Genome editing abrogates angiogenesis in vivo. Nat Commun. 2017;8(1):112. https://doi.org/10.1038/s41467-017-00140-3.
Finn JD, Smith AR, Patel MC, et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep. 2018;22(9):2227–35. https://doi.org/10.1016/j.celrep.2018.02.014.
Jiang C, Mei M, Li B, et al. A non-viral CRISPR/Cas9 delivery system for therapeutically targeting HBV DNA and pcsk9 in vivo. Cell Res. 2017;27(3):440–3. https://doi.org/10.1038/cr.2017.16.
Frangoul H, Ho TW, Corbacioglu S. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and beta-Thalassemia. Reply. N Engl J Med. 2021;384(23):e91. https://doi.org/10.1056/NEJMc2103481.
Wang JZ, Wu P, Shi ZM, Xu YL, Liu ZJ. The AAV-mediated and RNA-guided CRISPR/Cas9 system for gene therapy of DMD and BMD. Brain Dev. 2017;39(7):547–56. https://doi.org/10.1016/j.braindev.2017.03.024.
Kenjo E, Hozumi H, Makita Y, et al. Low immunogenicity of LNP allows repeated administrations of CRISPR-Cas9 mRNA into skeletal muscle in mice. Nat Commun. 2021;12(1):7101. https://doi.org/10.1038/s41467-021-26714-w.
Gee P, Lung MSY, Okuzaki Y, et al. Extracellular nanovesicles for packaging of CRISPR-Cas9 protein and sgRNA to induce therapeutic exon skipping. Nat Commun. 2020;11(1):1334. https://doi.org/10.1038/s41467-020-14957-y.
Zheng R, Li Y, Wang L, et al. CRISPR/Cas9-mediated metabolic pathway reprogramming in a novel humanized rat model ameliorates primary hyperoxaluria type 1. Kidney Int. 2020;98(4):947–57. https://doi.org/10.1016/j.kint.2020.04.049.
Martinez-Turrillas R, Martin-Mallo A, Rodriguez-Diaz S, et al. In vivo CRISPR-Cas9 inhibition of hepatic LDH as treatment of primary hyperoxaluria. Mol Ther Methods Clin Dev. 2022;25:137–46. https://doi.org/10.1016/j.omtm.2022.03.006.
Gu X, Wang D, Xu Z, et al. Prevention of acquired sensorineural hearing loss in mice by in vivo Htra2 gene editing. Genome Biol. 2021;22(1):86. https://doi.org/10.1186/s13059-021-02311-4.
Santiago-Fernandez O, Osorio FG, Quesada V, et al. Development of a CRISPR/Cas9-based therapy for Hutchinson-Gilford progeria syndrome. Nat Med. 2019;25(3):423–6. https://doi.org/10.1038/s41591-018-0338-6.
Lim CKW, Gapinske M, Brooks AK, et al. Treatment of a Mouse Model of ALS by In Vivo Base Editing. Mol Ther. 2020;28(4):1177–89. https://doi.org/10.1016/j.ymthe.2020.01.005.
McCullough KT, Boye SL, Fajardo D, et al. Somatic Gene Editing of GUCY2D by AAV-CRISPR/Cas9 Alters Retinal Structure and Function in Mouse and Macaque. Hum Gene Ther. 2019;30(5):571–89. https://doi.org/10.1089/hum.2018.193.
Martinez-Lage M, Torres-Ruiz R, Puig-Serra P, et al. In vivo CRISPR/Cas9 targeting of fusion oncogenes for selective elimination of cancer cells. Nat Commun. 2020;11(1):5060. https://doi.org/10.1038/s41467-020-18875-x.
Tang N, Cheng C, Zhang X, et al. TGF-beta inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight. 2020;5(4):e133977. https://doi.org/10.1172/jci.insight.133977.
Yin C, Zhang T, Qu X, et al. In Vivo Excision of HIV-1 Provirus by saCas9 and Multiplex Single-Guide RNAs in Animal Models. Mol Ther. 2017;25(5):1168–86. https://doi.org/10.1016/j.ymthe.2017.03.012.
Fine EJ, Appleton CM, White DE, et al. Trans-spliced Cas9 allows cleavage of HBB and CCR5 genes in human cells using compact expression cassettes. Sci Rep. 2015;5:10777. https://doi.org/10.1038/srep10777.
Richards DY, Winn SR, Dudley S, et al. AAV-Mediated CRISPR/Cas9 Gene Editing in Murine Phenylketonuria. Mol Ther Methods Clin Dev. 2020;17:234–45. https://doi.org/10.1016/j.omtm.2019.12.004.
Zhao H, Li Y, He L, et al. In Vivo AAV-CRISPR/Cas9-Mediated Gene Editing Ameliorates Atherosclerosis in Familial Hypercholesterolemia. Circulation. 2020;141(1):67–79. https://doi.org/10.1161/CIRCULATIONAHA.119.042476.
Lisjak M, De Caneva A, Marais T, et al. Promoterless Gene Targeting Approach Combined to CRISPR/Cas9 Efficiently Corrects Hemophilia B Phenotype in Neonatal Mice. Front Genome Ed. 2022;4:785698. https://doi.org/10.3389/fgeed.2022.785698.
Eyquem J, Mansilla-Soto J, Giavridis T, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543(7643):113–7. https://doi.org/10.1038/nature21405.
Chu SH, Packer M, Rees H, et al. Rationally Designed Base Editors for Precise Editing of the Sickle Cell Disease Mutation. CRISPR J. 2021;4(2):169–77. https://doi.org/10.1089/crispr.2020.0144.
Villiger L, Grisch-Chan HM, Lindsay H, et al. Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat Med. 2018;24(10):1519–25. https://doi.org/10.1038/s41591-018-0209-1.
Zhou L, Su J, Long J, et al. A universal strategy for AAV delivery of base editors to correct genetic point mutations in neonatal PKU mice. Mol Ther Methods Clin Dev. 2022;24:230–40. https://doi.org/10.1016/j.omtm.2022.01.001.
Villiger L, Rothgangl T, Witzigmann D, et al. In vivo cytidine base editing of hepatocytes without detectable off-target mutations in RNA and DNA. Nat Biomed Eng. 2021;5(2):179–89. https://doi.org/10.1038/s41551-020-00671-z.
Gao X, Tao Y, Lamas V, et al. Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature. 2018;553(7687):217–21. https://doi.org/10.1038/nature25164.
Koblan LW, Erdos MR, Wilson C, et al. In vivo base editing rescues Hutchinson-Gilford progeria syndrome in mice. Nature. 2021;589(7843):608–14. https://doi.org/10.1038/s41586-020-03086-7.
Rothgangl T, Dennis MK, Lin PJC, et al. In vivo adenine base editing of PCSK9 in macaques reduces LDL cholesterol levels. Nat Biotechnol. 2021;39(8):949–57. https://doi.org/10.1038/s41587-021-00933-4.
Banskota S, Raguram A, Suh S, et al. Engineered virus-like particles for efficient in vivo delivery of therapeutic proteins. Cell. 2022;185(2):250-265 e16. https://doi.org/10.1016/j.cell.2021.12.021.
Choi EH, Suh S, Foik AT, et al. In vivo base editing rescues cone photoreceptors in a mouse model of early-onset inherited retinal degeneration. Nat Commun. 2022;13(1):1830. https://doi.org/10.1038/s41467-022-29490-3.
Georgiadis C, Rasaiyaah J, Gkazi SA, et al. Base-edited CAR T cells for combinational therapy against T cell malignancies. Leukemia. 2021;35(12):3466–81. https://doi.org/10.1038/s41375-021-01282-6.
Brendel C, Guda S, Renella R, et al. Lineage-specific BCL11A knockdown circumvents toxicities and reverses sickle phenotype. J Clin Invest. 2016;126(10):3868–78. https://doi.org/10.1172/JCI87885.
Brendel C, Negre O, Rothe M, et al. Preclinical Evaluation of a Novel Lentiviral Vector Driving Lineage-Specific BCL11A Knockdown for Sickle Cell Gene Therapy. Mol Ther Methods Clin Dev. 2020;17:589–600. https://doi.org/10.1016/j.omtm.2020.03.015.
Esrick EB, Lehmann LE, Biffi A, et al. Post-Transcriptional Genetic Silencing of BCL11A to Treat Sickle Cell Disease. N Engl J Med. 2021;384(3):205–15. https://doi.org/10.1056/NEJMoa2029392.
Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50(3):509–17. https://doi.org/10.1016/0092-8674(87)90504-6.
Lu D, Gong X, Fang Y, et al. Correction of RNA splicing defect in beta(654)-thalassemia mice using CRISPR/Cas9 gene-editing technology. Haematologica. 2022;107(6):1427–37. https://doi.org/10.3324/haematol.2020.278238.
den Hollander AI, Koenekoop RK, Yzer S, et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am J Hum Genet. 2006;79(3):556–61. https://doi.org/10.1086/507318.
Ruan GX, Barry E, Yu D, Lukason M, Cheng SH, Scaria A. CRISPR/Cas9-Mediated Genome Editing as a Therapeutic Approach for Leber Congenital Amaurosis 10. Mol Ther. 2017;25(2):331–41. https://doi.org/10.1016/j.ymthe.2016.12.006.
Yao X, Wang X, Liu J, et al. CRISPR/Cas9 - Mediated Precise Targeted Integration In Vivo Using a Double Cut Donor with Short Homology Arms. EBioMedicine. 2017;20:19–26. https://doi.org/10.1016/j.ebiom.2017.05.015.
Chadwick AC, Wang X, Musunuru K. In Vivo Base Editing of PCSK9 (Proprotein Convertase Subtilisin/Kexin Type 9) as a Therapeutic Alternative to Genome Editing. Arterioscler Thromb Vasc Biol. 2017;37(9):1741–7. https://doi.org/10.1161/ATVBAHA.117.309881.
Rossidis AC, Stratigis JD, Chadwick AC, et al. In utero CRISPR-mediated therapeutic editing of metabolic genes. Nat Med. 2018;24(10):1513–8. https://doi.org/10.1038/s41591-018-0184-6.
Li J, Wang K, Zhang Y, et al. Therapeutic Exon Skipping Through a CRISPR-Guided Cytidine Deaminase Rescues Dystrophic Cardiomyopathy in Vivo. Circulation. 2021;144(22):1760–76. https://doi.org/10.1161/CIRCULATIONAHA.121.054628.
Raguram A, Banskota S, Liu DR. Therapeutic in vivo delivery of gene editing agents. Cell. 2022;185(15):2806–27. https://doi.org/10.1016/j.cell.2022.03.045.
Atchison RW, Casto BC, Hammon WM. Adenovirus-Associated Defective Virus Particles. Science (New York, NY). 1965;149(3685):754–6. https://doi.org/10.1126/science.149.3685.754.
Zhou X, Zeng X, Fan Z, et al. Adeno-associated virus of a single-polarity DNA genome is capable of transduction in vivo. Mol Ther. 2008;16(3):494–9. https://doi.org/10.1038/sj.mt.6300397.
Nakai H, Yant SR, Storm TA, Fuess S, Meuse L, Kay MA. Extrachromosomal recombinant adeno-associated virus vector genomes are primarily responsible for stable liver transduction in vivo. J Virol. 2001;75(15):6969–76. https://doi.org/10.1128/JVI.75.15.6969-6976.2001.
Clark KR, Sferra TJ, Lo W, Qu G, Chen R, Johnson PR. Gene transfer into the CNS using recombinant adeno-associated virus: analysis of vector DNA forms resulting in sustained expression. J Drug Target. 1999;7(4):269–83. https://doi.org/10.3109/10611869909085510.
Schnepp BC, Clark KR, Klemanski DL, Pacak CA, Johnson PR. Genetic fate of recombinant adeno-associated virus vector genomes in muscle. J Virol. 2003;77(6):3495–504. https://doi.org/10.1128/jvi.77.6.3495-3504.2003.
Sonntag F, Kother K, Schmidt K, et al. The assembly-activating protein promotes capsid assembly of different adeno-associated virus serotypes. J Virol. 2011;85(23):12686–97. https://doi.org/10.1128/JVI.05359-11.
Rutledge EA, Halbert CL, Russell DW. Infectious clones and vectors derived from adeno-associated virus (AAV) serotypes other than AAV type 2. J Virol. 1998;72(1):309–19. https://doi.org/10.1128/JVI.72.1.309-319.1998.
Srivastava A, Lusby EW, Berns KI. Nucleotide sequence and organization of the adeno-associated virus 2 genome. J Virol. 1983;45(2):555–64. https://doi.org/10.1128/JVI.45.2.555-564.1983.
Samulski RJ, Chang LS, Shenk T. Helper-free stocks of recombinant adeno-associated viruses: normal integration does not require viral gene expression. J Virol. 1989;63(9):3822–8. https://doi.org/10.1128/JVI.63.9.3822-3828.1989.
Yang S, Chang R, Yang H, et al. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J Clin Invest. 2017;127(7):2719–24. https://doi.org/10.1172/JCI92087.
Huang TP, Heins ZJ, Miller SM, et al. High-throughput continuous evolution of compact Cas9 variants targeting single-nucleotide-pyrimidine PAMs. Nat Biotechnol 2022;114(8):2060–5. https://doi.org/10.1038/s41587-022-01410-2.
Wen J, Cao T, Wu J, et al. Single AAV-mediated CRISPR-Nme2Cas9 efficiently reduces mutant hTTR expression in a transgenic mouse model of transthyretin amyloidosis. Mol Ther. 2022;30(1):164–74. https://doi.org/10.1016/j.ymthe.2021.05.010.
Ibraheim R, Tai PWL, Mir A, et al. Self-inactivating, all-in-one AAV vectors for precision Cas9 genome editing via homology-directed repair in vivo. Nat Commun. 2021;12(1):6267. https://doi.org/10.1038/s41467-021-26518-y.
Chung SH, Sin TN, Dang B, et al. CRISPR-based VEGF suppression using paired guide RNAs for treatment of choroidal neovascularization. Mol Ther Nucleic Acids. 2022;28:613–22. https://doi.org/10.1016/j.omtn.2022.04.015.
Chen F, Ding X, Feng Y, Seebeck T, Jiang Y, Davis GD. Improving CRISPR Gene Editing Efficiency by Proximal dCas9 Targeting. Bio Protoc. 2017;7(15):e2432. https://doi.org/10.21769/BioProtoc.2432.
Lee CS, Bishop ES, Zhang R, et al. Adenovirus-Mediated Gene Delivery: Potential Applications for Gene and Cell-Based Therapies in the New Era of Personalized Medicine. Genes Dis. 2017;4(2):43–63. https://doi.org/10.1016/j.gendis.2017.04.001.
Li C, Georgakopoulou A, Newby GA, et al. In vivo base editing by a single i.v. vector injection for treatment of hemoglobinopathies. JCI Insight. 2022;7(19):e162939. https://doi.org/10.1172/jci.insight.162939.
Bock D, Rothgangl T, Villiger L, et al. In vivo prime editing of a metabolic liver disease in mice. Sci Transl Med. 2022;14(636):eabl9238. https://doi.org/10.1126/scitranslmed.abl9238.
Witzigmann D, Kulkarni JA, Leung J, Chen S, Cullis PR, van der Meel R. Lipid nanoparticle technology for therapeutic gene regulation in the liver. Adv Drug Deliv Rev. 2020;159:344–63. https://doi.org/10.1016/j.addr.2020.06.026.
Paunovska K, Loughrey D, Dahlman JE. Drug delivery systems for RNA therapeutics. Nat Rev Genet. 2022;23(5):265–80. https://doi.org/10.1038/s41576-021-00439-4.
Buschmann MD, Carrasco MJ, Alishetty S, Paige M, Alameh MG, Weissman D. Nanomaterial Delivery Systems for mRNA Vaccines. Vaccines (Basel). 2021;9(1):65. https://doi.org/10.3390/vaccines9010065.
Reichmuth AM, Oberli MA, Jaklenec A, Langer R, Blankschtein D. mRNA vaccine delivery using lipid nanoparticles. Ther Deliv. 2016;7(5):319–34. https://doi.org/10.4155/tde-2016-0006.
Horejs C. From lipids to lipid nanoparticles to mRNA vaccines. Nat Rev Mater. 2021;6(12):1075–6. https://doi.org/10.1038/s41578-021-00379-9.
Yin H, Song CQ, Suresh S, et al. Structure-guided chemical modification of guide RNA enables potent non-viral in vivo genome editing. Nat Biotechnol. 2017;35(12):1179–87. https://doi.org/10.1038/nbt.4005.
Gilleron J, Querbes W, Zeigerer A, et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat Biotechnol. 2013;31(7):638–46. https://doi.org/10.1038/nbt.2612.
Jiang T, Henderson JM, Coote K, et al. Chemical modifications of adenine base editor mRNA and guide RNA expand its application scope. Nat Commun. 2020;11(1):1979. https://doi.org/10.1038/s41467-020-15892-8.
Dahlman JE, Kauffman KJ, Xing Y, et al. Barcoded nanoparticles for high throughput in vivo discovery of targeted therapeutics. Proc Natl Acad Sci U S A. 2017;114(8):2060–5. https://doi.org/10.1073/pnas.1620874114.
Sago CD, Lokugamage MP, Paunovska K, et al. High-throughput in vivo screen of functional mRNA delivery identifies nanoparticles for endothelial cell gene editing. Proc Natl Acad Sci U S A. 2018;115(42):E9944–52. https://doi.org/10.1073/pnas.1811276115.
Cheng Q, Wei T, Farbiak L, Johnson LT, Dilliard SA, Siegwart DJ. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat Nanotechnol. 2020;15(4):313–20. https://doi.org/10.1038/s41565-020-0669-6.
Wang X, Liu S, Sun Y, et al. Preparation of selective organ-targeting (SORT) lipid nanoparticles (LNPs) using multiple technical methods for tissue-specific mRNA delivery. Nat Protoc. 2022;11(1):5060. https://doi.org/10.1038/s41596-022-00755-x.
Lyu P, Wang L, Lu B. Virus-Like Particle Mediated CRISPR/Cas9 Delivery for Efficient and Safe Genome Editing. Life (Basel). 2020;10(12):366. https://doi.org/10.3390/life10120366.
Chandler RJ, Sands MS, Venditti CP. Recombinant Adeno-Associated Viral Integration and Genotoxicity: Insights from Animal Models. Hum Gene Ther. 2017;28(4):314–22. https://doi.org/10.1089/hum.2017.009.
Zhang W, Cao S, Martin JL, Mueller JD, Mansky LM. Morphology and ultrastructure of retrovirus particles. AIMS Biophys. 2015;2(3):343–69. https://doi.org/10.3934/biophy.2015.3.343.
Cronin J, Zhang XY, Reiser J. Altering the tropism of lentiviral vectors through pseudotyping. Curr Gene Ther. 2005;5(4):387–98. https://doi.org/10.2174/1566523054546224.
Yin D, Ling S, Wang D, et al. Targeting herpes simplex virus with CRISPR-Cas9 cures herpetic stromal keratitis in mice. Nat Biotechnol. 2021;39(5):567–77. https://doi.org/10.1038/s41587-020-00781-8.
Ling S, Yang S, Hu X, et al. Lentiviral delivery of co-packaged Cas9 mRNA and a Vegfa-targeting guide RNA prevents wet age-related macular degeneration in mice. Nat Biomed Eng. 2021;5(2):144–56. https://doi.org/10.1038/s41551-020-00656-y.
Segel M, Lash B, Song J, et al. Mammalian retrovirus-like protein PEG10 packages its own mRNA and can be pseudotyped for mRNA delivery. Science (New York, NY). 2021;373(6557):882–9. https://doi.org/10.1126/science.abg6155.
Acknowledgements
None.
Funding
This work was supported by National Natural Science Foundation of China (No. 81974238) and 1·3·5 project for disciplines of excellence, West China Hospital, Sichuan University (ZYJC21018).
Author information
Authors and Affiliations
Contributions
Lifang Zhou and Shaohua Yao wrote the manuscript. Lifang Zhou created all the the figures and tables. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Yes.
Competing interests
The authors declare no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhou, L., Yao, S. Recent advances in therapeutic CRISPR-Cas9 genome editing: mechanisms and applications. Mol Biomed 4, 10 (2023). https://doi.org/10.1186/s43556-023-00115-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43556-023-00115-5