
Abstract
There is general agreement that auto-antibodies against ion channels and synaptic machinery proteins can induce limbic encephalitis. In immune-mediated cerebellar ataxias (IMCAs), various synaptic proteins, such as GAD65, voltage-gated Ca channel (VGCC), metabotropic glutamate receptor type 1 (mGluR1), and glutamate receptor delta (GluR delta) are auto-immune targets. Among them, the pathophysiological mechanisms underlying anti-VGCC, anti-mGluR1, and anti-GluR delta antibodies remain unclear. Despite divergent auto-immune and clinical profiles, these subtypes show common clinical features of good prognosis with no or mild cerebellar atrophy in non-paraneoplastic syndrome. The favorable prognosis reflects functional cerebellar disorders without neuronal death. Interestingly, these autoantigens are all involved in molecular cascades for induction of long-term depression (LTD) of synaptic transmissions between parallel fibers (PFs) and Purkinje cells (PCs), a crucial mechanism of synaptic plasticity in the cerebellum. We suggest that anti-VGCC, anti-mGluR1, and anti-GluR delta Abs-associated cerebellar ataxias share one common pathophysiological mechanism: a deregulation in PF-PC LTD, which results in impairment of restoration or maintenance of the internal model and triggers cerebellar ataxias. The novel concept of LTDpathies could lead to improvements in clinical management and treatment of cerebellar patients who show these antibodies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
During the last two decades, experimental and clinical studies have established the pathological roles of auto-antibodies against ion channels and synaptic receptors in limbic encephalitis [1,2,3,4,5]. Although auto-antibodies that target ion channels and synaptic machineries have been documented also in immune-mediated cerebellar ataxias (IMCAs), the types of auto-antibodies involved in IMCAs are different from those observed in limbic auto-immune encephalitis [6]. Anti-glutamate receptors, anti-GABA receptors and anti- leucine-rich glioma-inactivated 1(LGI1) antibodies (Abs) are rarely observed in IMCAs, whereas the association of CAs with anti-GAD65, anti-voltage-gated Ca channel (VGCC), anti-metabotropic glutamate receptor type 1 (mGluR1), and anti-glutamic receptor delta (GluR delta) Abs has been documented [7,8,9,10,11,12]. Especially, auto-antibodies against VGCC, mGluR and GluR delta are characteristically found in IMCAs, but not in auto-immune limbic encephalitis [6, 13]. These target molecules are involved in molecular cascades that induce long-term synaptic depression (LTD) of synaptic transmissions between parallel fibers (PFs) and Purkinje cells (PCs), a crucial form of synaptic plasticity in the cerebellum [6, 13].
In this review, we dissect the pathophysiological mechanisms underlying anti-VGCC, anti-mGluR1, and anti-GluR delta Abs-associated cerebellar ataxias (CAs), and address pathophysiological roles of impaired PF-PC LTD. Thus, we discuss (1) the roles of these auto-immune target molecules (VGCC, mGluR1 and GluR delta) in the induction of LTD, and (2) the clinical profiles of the IMCAs subtypes associated with these auto-antibodies. We argue that PF-PC LTD dysfunction is one of the final common pathophysiological mechanisms in these three subtypes of IMCAs. Auto-immune response might impair the restoration or maintenance of the internal model, resulting in the development of CAs.
Physiology of long-term depression between parallel fibers and Purkinje cells
Neuronal circuit of cerebellar cortex and PF-PC LTD
The cerebellar cortex receives two excitatory glutamatergic input fibers, namely, mossy fibers (MFs) and climbing fibers (CFs) (Fig. 1). MFs make synaptic connections with granule cells (GrCs) and cerebellar nucleus (CN) neurons. PFs, axons of GrCs, meet with multiple PCs, and form a glutamatergic synapse with a single PC [14, 15]. At the PF-boutons, action potentials activate VGCC (mainly P/Q-type VGCC) [16]. Glutamate, released from the PF terminals in a Ca2+-dependent manner, activates AMPA-type Glu-receptors at the dendritic spine of PC. The PF-PC synaptic input generates and modulates simple spikes, whose firing pattern is affected by voltage-gated K channels and Ca2+-dependent K channels [17,18,19]. The PF-PC synapse is bound by a set of synaptic adhesion molecules, i.e., neurexin-cerebellin1-GluR delta [20, 21]. By contrast, a single CF that originates from the inferior olive nucleus establishes strong multisite synapses on the dendrites of a single PC [14, 15].

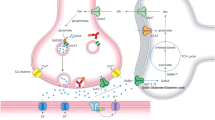
Schematic diagram of the pathophysiological mechanisms underlying anti-VGCC, anti-mGluR1, and anti-GluR delta-associated cerebellar ataxias. The antibody-mediated mechanisms include dysfunction of basal synaptic transmissions and long-term depression (LTD), leading to impairment of the internal model. These functional disorders are followed by cell-mediated cell death, depending on the auto-immune stimulus. PC; Purkinje cell, GC; granule cell, IN; inhibitory interneurons, CF; climbing fiber, PF; parallel fiber, MF; mossy fiber
Simultaneous and repetitive activation of PF and CF depresses PF-excitatory postsynaptic currents with a long-term time course, which is termed the LTD [15, 22]. Based on the finding that CF fire at high probability in case of motor failures [23], it has been proposed that CF carry an error signal of motor performance and PF-PC LTD provides the mechanism for motor learning [15], though this is still debated.
Molecular mechanisms underlying PF-PC LTD
Conjunctive stimulation of CF and PF causes LTD of PF-PC synaptic transmissions both in vivo [22] and in vitro [24,25,26]. CF input elicits an increase in [Ca2+]i through the VGCC (P/Q-type) (Fig. 2) [27]. PF inputs in dendritic spines activate the mGluR-PLCβ-IP3 signaling pathways, which elicits Ca2+ release from the Ca2+-stores in the endoplasmic reticulum (ER) through IP3 receptors and, consequently, increases [Ca2+]i (Fig. 2) [28].

Schematic diagram of long-term depression (LTD) at excitatory synapses between parallel fibers and Purkinje cells. The climbing fiber input elicits complex spikes through the activation of dendritic P/Q type Ca2+ channels, leading to an increase in intracellular calcium concentration ([Ca2+]i). On the other hand, the parallel fiber input activates metabotropic glutamate receptor-PLCβ-IP3 signaling pathways, resulting in an increase in [Ca2+]i. The conjunctive activation of these two pathways increases [Ca2+]i more than the additive level. The high [Ca2+]i activates PKCα, and PKCα phosphorylates GluA2 of the AMPA (α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptor, which results in detachment of the AMPA receptor from scaffold proteins and its internalization with PICK1 in an AP2 and clathrin-dependent manner. CF; climbing fibers, PF; parallel fibers, Glu; glutamate; AMAPA-R; AMPA receptor, mGluR1; metabotropic glutamate receptor, Cav2.1 (P/Q); P/Q type Ca2+ voltage-gated channel, PLC; phospholipase C, PKC; protein kinase C, IP3; Inositol triphosphate, GRIP; Glutamate receptor interactive protein, TARP; transmembrane AMPA receptor regulatory proteins, PICK1; protein interacting with C kinase, δ; GluR delta 2, PTPMEG; megakaryocyte protein phosphatase
Simultaneous activation of VGCC (P/Q-type) and mGluR1 elicits a series of events, including an increase in [Ca2+]i to levels higher than the additive level [29], which leads to the activation of PKCα, which in turn phosphorylates GluA2-C terminus, ultimately leading to the detachment of AMPA receptors, including phosphorylated GluA2, from the scaffold protein and its internalization with PICK1 in AP2- and clathrin-dependent manners (Fig. 2) [30]. Also CaMKII [31] and NMDA-receptor [32, 33] are involved in PF-PC LTD induction.
GluR delta, a synaptic adhesion molecule specific to the PF-PC synapse [21], is also involved in LTD. Antibodies against the N-terminus region (H2 ligand binding site) decrease the amplitude of evoked excitatory postsynaptic currents [34], possibly by suppressing the PF-PC synaptic interaction. In addition, the antibodies suppressed induction of LTD in culture preparations. Suppression of GluR delta expression in cultured PCs by antisense oligonucleotides also resulted in severe impairment of LTD [35]. These findings highlight the importance of GluR delta in LTD, for reasons other than maintenance of synaptic interaction. The cytoplasmic terminus of GluR delta binds to megakaryocyte protein phosphatase (PTPMEG), which dephosphorylates tyrosine 876 in GluA2. Dephosphorylation of this site is necessary for PKCα-induced phosphorylation of serine 880, an essential step in the internalization of AMPA receptors [36]. Thus, GluR delta is assumed to gate PF-PC LTD by coordinating the interaction between the two phosphorylation sites in GluA2 [36]. However, whether this dephosphorylation through GluR delta-PTPMEG interaction is impaired by antibodies against GluR delta extracellular region is unknown.
In conclusion, an increase in [Ca2+]i is triggered by conjunctive activation of VGCC and mGluR1, and the subsequent AP2-and clathrin-dependent endocytosis of AMPA receptors is an essential process in PF-PC LTD. Furthermore, endocytosis is gated by dephosphorylation of T876 of the GluA2-C terminus through GluR delta-PTPMEG interaction.
Internal model and PF-PC LTD
Internal model and cerebellar ataxias
Evidence suggest the presence of internal models embedded in the cerebellum to elaborate coordination of motor and cognitive commands [37, 38]. The internal model, that emulates the dynamics of body and environments, are essential for integrating the movements of the body parts without the need for sensory feedback [39]. An internal inverse model transforms the desired trajectory into motor commands [40, 41] and is involved mainly in cerebellar ocular motor controls [42]. On the other hand, an internal forward model computes the future state of the body based on the current estimate of the body and efferent signals of motor control [43,44,45]. Evidence suggests that online predictive computations that employ the internal forward model coordinate limb voluntary movements [46,47,48,49,50]. The predictive computation of the forward model affords coordination of multiple degrees of freedom and appropriate timing of muscle activities [39]. Since the deficits in predictive activation of the triceps muscles results in dysmetria, it is assumed that dysmetria occurs as a result of impaired predictive computation of the internal forward model in the cerebellum [39]. Other ataxic symptoms including adiadochokinesis and ataxic gaits could be attributed to impairments of the internal forward model [39].
Internal model and synaptic plasticity
Through cerebellar leaning, the internal model is assumed to be acquired and continuously updated following changes in the external factors. To execute the task of reaching, for example, the cerebellum organizes compound movements based on the physical properties of various body parts, including muscle strength, segment inertia, joint viscosity, and segmental interaction, which are stored in the cerebellum [51].
In the cerebellar cortex, multiple forms of synaptic plasticity at different sites are induced during procedural memory formation (Fig. 2) [52]. At the cerebellar input synapses (MF-GrC), high-frequency bursts (> 250 msec) of MF can induce MF–GrC LTP, whereas low-frequency bursts induce LTD [53, 54]. Repetitive stimulation of PF alone causes potentiation of PF-PC EPSP [25, 55]. This postsynaptic type of LTP can reset PF-PC LTD, and vice versa; PF-PC LTD can reset PF-PC LTP, thus PF-PC LTP and PF-PC LTD mutually counterbalance each other [56,57,58,59]. As for the inhibitory input to PC, GABA-mediated inhibitory synaptic transmission undergoes a long-lasting rebound potentiation (RP) after activation of excitatory CF inputs [60]. Thus, the multiple forms of synaptic plasticity in the cerebellar cortex challenges the concept of the critical role of LTD in cerebellar motor learning. Furthermore, in certain mutant mice that lack PF-PC LTD, show normal motor learning. Based on such conclusion, some researchers have argued that LTD is not essential for motor learning [61, 62].
Recent experiments have provided substantial evidence for the essential role of PF-PC LTD in motor learning. First, interpretation of the results of gene-manipulation experiments should be assessed carefully. In general, compensatory mechanisms of synaptic plasticity are probably expressed in gene-manipulated animals, and the conditions necessary to induce compensated synaptic plasticity could be different from the ordinal experimental conditions that induce PF-PC LTP or LTD in the wild type animal. Actually, several types of PF-PC LTD-inducing stimulation protocols have been used to induce LTD in the same mutant mouse [63], indicating that LTD-hypothesis could not be ruled out. Second, the use of a new optogenetic blocker of endocytosis allows reversible PF-PC LTD blockade. This tool (PhotonSABER) enables the temporal, spatial, and cell-type specific control of AMPA receptor endocytosis at active synapses. Blockade of PF-PC LTD by photostimulation in vivo resulted in impairment of vestibulo-ocular reflex (VOR)-adaptation [64]. This photoactivation of PhotonSABER neither affected RP nor PF-PC LTP. Although RP and PF-PC LTP were intact, VOR-adaptation was impaired following blockade of PF-PC LTD, thus suggesting the importance of LTD in cerebellar motor learning.
In conclusion, CAs can be attributed to impaired predictive computation of the internal forward model in the cerebellum. The internal model is formed and continuously updated by cerebellar learning processes. Motor learning depends on various forms of synaptic plasticity in the cerebral cortex through an increase in the contrast of input signals, efficiency of learning, and accuracy of learned behavior. However, PF-PC LTD seems to play a crucial role in motor learning by adjusting the final stage of the cerebellar cortical integration.
LTD-related synaptopathies in immune-mediated cerebellar ataxias
Anti-VGCC antibody-associated cerebellar ataxia
Clinical profiles
Anti-VGCC antibodies were initially described in association with Lambert-Eaton myasthenia syndrome (LESM) [3]. However, the association of CAs with anti-VGCC Abs has also been described in patients with paraneoplastic cerebellar degeneration (PCD) with or without LEMS [65]. The most frequent associated cancer is small cell lung cancer (SCLC) [66]. Auto-antibodies against the P/Q-type VGCC, the main antigen, was detected in 78% of patients with PCDs and LEMS, and in 20% of anti-Hu Ab-negative patients with PCDs without LEMS [66]. The patients showed subacute pancerebellar ataxias, and there were no differences in the clinical profiles of PCD patients with and without anti-VGCC Ab [67]. Anti-VGCC Abs were also identified in patients with non-paraneoplastic IMCAs [65] and a large-scale study showed that anti-VGCC Abs were positive in 8 of 67 patients who showed chronic cerebellar degeneration [68].
The therapeutic response depends on the background. A study on 16 anti-VGCC Ab-positive patients with PCD and SCLC found poor benefits to immunotherapies [67]. Of these patients, one showed complete recovery, five showed stabilization at a low Rankin score, and five stabilized or worsened at high Rankin scores. The median survival time of these patients was 12 months. On the other hand, good prognosis was reported in patients with non-paraneoplastic conditions [68]. Immunotherapies have been used in patients with paraneoplastic and non-paraneoplastic conditions, and these included intravenous immunoglobulins (IVIg), prednisone, and mycophenolate mofetil (Table 1).
Physiological actions of antibodies
A polyclonal peptide Ab against the major immunogenic region in P/Q-type VGCCs (the extracellular domain-III S5–6 loop) impaired the functions of neuronal and recombinant P/Q-type VGCC, and elicited a decrease in Ca2+ currents, leading to impaired synaptic transmission between PF and PC [73]. A reduction in P/Q-type VGCC was also observed in the autopsies of three patients with PCDs and LEMS [74]. In experimental studies, ataxic symptoms were induced in mice by intrathecal administration of serum IgGs obtained from anti-P/Q type VGCC Ab-positive patients with PCDs and LEMS [75]. However, the actions of anti-VGCC Ab on LTD have not been studied.
Anti-mGluR1 antibody-associated cerebellar ataxia
Clinical profiles
The association of anti-mGluR1 Ab with CAs has been reported initially in two patients with Hodgkin’s lymphoma [76] and one patient with prostate adenocarcinoma [69]. The response to immunotherapy varied among the three patients; the two patients with Hodgkin’s lymphoma responded well to the combination of plasma exchange, IVIg and oral prednisone, whereas the other patient with prostate cancer showed no objective improvement after plasma exchange.
On the other hand, the association of anti-mGluR Ab with CAs was also described in non-paraneoplastic conditions [70, 77]. The clinical course is now better known for portraying a series of 11 new patients and 19 previously reported patients (Table 1) [71]. The main neurological manifestations were subacute cerebellar gait and limb ataxias in 25 of these 30 patients (86%), sometimes associated with extra-cerebellar symptoms, such as behavioral changes (irritability, apathy, mood, personality change, psychosis with hallucinations, and catatonia), cognitive changes (memory problems, executive functions and spatial orientation deficits) or dysgeusia. Seizures were uncommon. Three of the 26 patients (11%) had paraneoplastic conditions (cutaneous T lymphoma and Hodgkin’s lymphoma). Serological tests identified anti-mGluR1 Ab in both the serum and CSF, together with evidence of pleocytosis in the CSF. MRI showed normal in 12 of the 19 patients (63%) at the onset. Abnormal findings included T2/FLAIR hyperintensities or leptomeningeal gadolinium enhancement. At follow-up, MRI showed cerebellar atrophy in 10 of the 12 (83%). Twenty-five of the 30 (83%) patients received immunotherapies, including IVIg, steroids, mycophenolate mofetil, cyclophosphamide, and rituximab alone or in combinations. Ten patients (40%) showed significant improvements or complete resolution of symptoms, and 13 patients (52%) showed stabilization or mild improvement. Although two patients not associated with tumors died, one patient initialized improved and died due to unknown etiologies. Thus, only one patient showed progressive worsening of CAs.
Physiological actions of antibodies
IgGs purified from the sera of patients with Hodgkin’s lymphoma blocked glutamate-stimulated formation of inositol phosphates in mGluR1α-expressing Chinese-hamster-ovary cells [76]. The IgGs blocked the induction of LTD in tissue slices [72]. Application of IgGs in the subarachnoid space elicited ataxic gaits in mice, which disappeared after absorption of anti-mGluR1 Ab [75, 76], while administration of the same Ab in the flocculus also evoked acute disturbances in compensatory eye movements [72]. A recent study confirmed that CSF from the patients decreased mGluR1 clusters in cultured neurons [71].
Anti-GluR delta antibody-associated cerebellar ataxia
Clinical profiles
The association of anti-GluR delta Ab with CAs has been described in a few patients with non-paraneoplastic conditions [78,79,80,81]. The conditions affected children aged 8 months to 13-years and medical history showed infection or vaccination preceding CAs. These patients exhibited prominent gait ataxia associated with variable degree of limb ataxia and dysarthria with an acute time-course. Serological tests were positive for anti-GluR delta Ab in the serum and CSF and laboratory tests showed pleocytosis without oligoclonal bands in the CSF, while MRI demonstrated no evidence of cerebellar atrophy. Most of these patients responded well to immunotherapy of IVIg or IVMP coupled with clinical improvement in CAs (Table 1). Another 25-month-old girl with chronic recurrent CA positive for anti-GluR delta Ab also showed a good response to corticosteroid therapy [82].
Physiological actions of antibodies
Although subarachnoidal injection of Ab against the H2 ligand binding site of GluR delta elicited the development of ataxic phenotype in mice and the Ab impaired simplified LTD in cultured PC [34], the actions of anti-GluR delta Ab on LTD in mature PC with intact physiological neuronal circuitry remain to be investigated in adult cerebellar slices.
Possible pathophysiological mechanisms
Diversity and composite in auto-immune mechanisms
Anti-VGCC, anti-mGluR1, and anti-GluR delta Abs-associated CAs show divergent clinical profiles, for example, different background (paraneoplastic or non-paraneoplastic) and age preponderance. In addition to the clinical diversity among these subtypes, compound and overlapped immune mechanisms appear to be involved even in each etiology, especially in anti-VGCC Ab-associated CA. Both in vitro and in vivo studies have shown that antibodies towards VGCC impaired the release of neurotransmitters, leading to the development of CAs. Despite this evidence of functional disorders, cerebellar cell loss was found in postmortem studies [74]. This discrepancy suggests that antibody-induced functional disorders are followed by additional cell-mediated mechanisms that lead to cell death. It is suggested that in paraneoplastic conditions, the auto-immune signal is augmented in a positive feed-back fashion [83]. For example, persistence of the auto-immune responses to cancer cells would continue to cause secretion of cytokines that elicit sustained vascular hyperexcitability and infiltration of immune cells. Death of cancer cells subsequently leads to lasting release of intracellular antigens. These persistent and amplified auto-immune conditions might recruit sequential responses, from antibody-mediated functional disorder to additional cytotoxic T-cell-mediated cell death [75].
In conclusion, various backgrounds (paraneoplastic, infectious or unknown conditions) can trigger auto-immune attacks against the cerebellar circuits in anti-VGCC, anti-mGluR1, and anti-GluR delta Abs-associated CAs, where multiple auto-immune reactions can be elicited depending on the quality, intensity or duration of the auto-immune stimulus. Sequential responses, from humoral functional disorders to cell-mediated irreversible cell death, might sometimes occur (Fig. 1).
Functional disorders and internal model impairments
In spite of the diversity and composite of auto-immune mechanisms, anti-VGCC, anti-mGluR1, and anti-GluR delta Abs-associated CAs also appear to have a single final common pathway. Patients with these three subtypes commonly show good prognosis without or only mild cerebellar atrophy when associated with non-paraneoplastic conditions, which is contrast to the poor prognosis in anti-GAD ataxia. The anti-GAD ataxia sometimes shows resistance to immunotherapy, resulting in severe CAs associated with marked cerebellar atrophy and cell death [84]. Thus, the good therapeutic response in these three etiologies can be an outstanding feature of IMCAs, and reflects staying of functional disorders, dysfunction of the internal model. Taken together, we propose that anti-VGCC, anti-mGluR1, and anti-GluR delta Abs commonly distort the internal model leading to the development of cerebellar functional disorders.
LTD as a common final pathophysiological pathway?
One possible mechanism for the functional disorders is deficit in the basal synaptic transmission in the cerebellar circuits. In addition to anti-VGCC Ab [73], impaired synaptic transmission on PC was documented in anti-mGluR1 Ab [85] and anti-GluR delta Ab [34]. Disturbance of the basal synaptic transmission subsequently lead to distortion of the internal model (Fig. 1).
The alternative mechanism is a deficit in PF-PC LTD, since VGCC and mGluR1 are molecules required for the increase in Ca2+ concentrations and subsequent induction of PF-PC LTD, while GluR delta plays an important role in AMPA receptor trafficking. Consistently, it has been demonstrated that IgGs from some of these patients impaired the induction of LTD [72]. The physiological relationship between LTD dysfunction and internal model impairment is as follows (Fig. 1).
(1) When these auto-antibodies interfere with the basal synaptic transmission, any impairment of the internal model should be compensated for by synaptic plasticity, including PF-PC LTD. Thus, PF-PC LTD dysfunction can distort internal model restoration.
(2) It has been a matter of debate whether PF-PC LTD is required to develop normal motor performance [86]. However, recent studies suggest that PF-PC might be involved in execution of the internal model. First, using the method of conditional knockout mice (a tetracycline-controlled gene expression system), acute blockade of mGluR impaired PF-PC LTD and simultaneously elicited motor incoordination without affecting basal synaptic transmission [85]. Second, a study on neural activities in monkeys indicates a facilitative role of PF-PC LTD in the online predictive controls [87]. The cerebellar output, before the movements, is generated by phasic suppression of PCs and concomitant activation of the dentate nucleus cells (disinhibition mechanism) [87]. Impairments in the disinhibition elicit a delay in movement initiation, which was termed asthenia by Holmes G [88]. Notably, the phasic suppression on PCs is assumed to be tuned by facilitation by PF-PC LTD [87].
It is uncertain whether dysfunction of LTD can directly alter the internal model. This hypothesis will be validated using in vitro and in vivo experiments in which PF-PC LTD is directly blocked by molecular manipulation of AMPA receptor endocytosis (e.g., PhotonSABER method [64] or manipulation of transmembrane AMPA receptor regulating proteins [89]). In these in vivo models, impairments in execution of the internal model will be examined by monitoring predictive muscle activations, an index for on line predictive controls [39]. The long-term alterations will be analyzed using in learning paradigms.
Conclusion
Anti-VGCC, anti-mGluR1, and anti-GluR delta Abs-associated CAs show divergent clinical profiles in auto-immune background (paraneoplastic or non-paraneoplastic) and susceptible age. However, these patients commonly show good prognosis without cerebellar atrophy in non-paraneoplastic conditions, suggesting functional disorders in the internal model. These autoantibodies would impair basal synaptic transmissions in the cerebellar cortex. Besides, the dysregulated synaptic plasticity-related dysfunction might be overlapped, since VGCC, mGluR1, and GluR delta are involved in the induction of PF-PC LTD. Deficits in restoration of the internal model might occur. Furthermore, dysregulated PF-PC LTD might impair the execution processes of the internal model. In order to highlight the synaptic plasticity-related dysfunction, we propose the concept of “LTDpathies” to gather cerebellar ataxias associated with antibodies targeting the critical mechanism of PF-PC LTD. Auto-antibodies impair fundamental paths of a key-plasticity in the cerebellar cortex. “LTDpathies” share a favorable prognosis, highlighting that this group of disorders does not target the cerebellar reserve in terms of neuronal destruction, but rather blocks adaptative mechanism in the cerebellum.
Availability of data and materials
The concept reported in this manuscript is not associated with raw data.
Abbreviations
- Ab:
-
Antibody
- CFs:
-
Climbing fibers
- CN:
-
Cerebellar nucleus
- GluR delta:
-
Glutamic receptor delta
- GrCs:
-
Granule cells
- IMCAs:
-
Immune-mediated cerebellar ataxias
- IVIg:
-
Intravenous immunoglobulins
- LESM:
-
Lambert-Eaton myasthenia syndrome
- LGI1:
-
Leucine-rich glioma-inactivated 1
- LTD:
-
Long-term synaptic depression
- MFs:
-
Mossy fibers
- mGluR1:
-
Metabotropic glutamate receptor type 1
- PCD:
-
Paraneoplastic cerebellar degeneration
- PCs:
-
Purkinje cells
- PFs :
-
Parallel fibers
- PTPMEG:
-
Megakaryocyte protein phosphatase
- RP:
-
Rebound potentiation
- SCLC:
-
Small cell lung cancer
- VGCC:
-
Voltage-gated Ca channel
- VOR:
-
Vestibulo-ocular reflex
References
Graus F, Saiz A, Dalmau J. Antibodies and neuronal autoimmune disorders of the CNS. J Neurol. 2010;257(4):509–17. https://doi.org/10.1007/s00415-009-5431-9.
Lancaster E, Martinex-Hernandez E, Dalmau J. Encephalitis and antibodies to synaptic and neuronal cell surface proteins. Neurology. 2011;77(2):179–89. https://doi.org/10.1212/WNL.0b013e318224afde.
Lancaster E, Dalmau J. Neuronal autoantigens-pathogenesis, associated disorders and antibody testing. Nat Rev Neurol. 2012;8(7):380–92. https://doi.org/10.1038/nrneurol.2012.99.
Dalmau J, Rosenfeld MR. Autoimmune encephalitis update. Neuro-Oncology. 2014;16(6):771–8. https://doi.org/10.1093/neuonc/nou030.
Dalmau J, Geis C, Graus F. Autoantibodies to synaptic receptors and neuronal cell surface protein in autoimmune diseases of the central nervous system. Physiol Rev. 2017;97(2):839–87. https://doi.org/10.1152/physrev.00010.2016.
Mitoma H, Honnorat J, Yanaguchi K, Manto M. Fundamental mechanisms of autoantibody-induced impairments on ion channels and synapses in immune-mediated cerebellar ataxias. Int J Mol Sci. 2020;21(14):E4936. https://doi.org/10.3390/ijms21144936.
Hadjivassiliou M. Immune-mediated acquired ataxias. Handb Clin Neurol. 2012;103:189–99. https://doi.org/10.1016/B978-0-444-51892-7.00011-5.
Joubert B, Rotásky J, Honnorat J. Immune-mediated ataxias. Handb Clin Neurol. 2018;155:313–32. https://doi.org/10.1016/B978-0-444-64189-2.00021-4.
Joubert B, Honnorat J. Nonparaneoplastic autoimmune cerebellar ataxia. Curr Opin Neurol. 2019;32(3):484–92. https://doi.org/10.1097/WCO.0000000000000678.
Mitoma H, Adhikari K, Aeschlimann D, Chattopadhyay P, Hadjivassiliou M, Hampe CS, et al. Consensus paper: Neuroimmune mechanisms of cerebellar ataxias. Cerebellum. 2016;15(2):213–32. https://doi.org/10.1007/s12311-015-0664-x.
Mitoma H, Hadjivassiliou M, Honnorat J. Guidelines for treatment of immune-mediated cerebellar ataxias. Cerebellum Ataxias. 2015;2:14. https://doi.org/10.1186/s40673-015-0034-y.
Mitoma H, Manto M, Hampe CS. Immune-mediated cerebellar ataxias: practical guidelines and therapeutic challenges. Curr Neuropharmacol. 2019;17(1):33–58. https://doi.org/10.2174/1570159X16666180917105033.
Ito M. The cerebellum and neural control. New York: Raven press; 1984.
Eccles JC, Ito M, Szentagothai J. The cerebellum as a neuronal machine. New York: Springer; 1967.
Ito M. Cerebellar long-term depression: characterization, signal transduction, and functional roles. Physiol Rev. 2001;81(3):1143–95. https://doi.org/10.1152/physrev.2001.81.3.1143.
Lorenzon NM, Lutz CM, Frankel WN, Beam KG. Altered calcium channel currents in Purkinje cells of the neurological mutant mouse leaner. J Neurosci. 1998;18(12):4482–9. https://doi.org/10.1523/JNEUROSCI.18-12-04482.1998.
Haghdoosdt-Yazdi H, Janahmadi M, Behzadi G. Iberiotoxin-sensitive large conductance Ca2+ −dependent K+ (BK) channels regulate the spike configuration in the burst firing of cerebellar Purkinje neurons. Brain Res. 2008;1212:1–8. https://doi.org/10.1016/j.brainres.2008.03.030.
Benton MD, Lewis AH, Bant JS, Raman IM. Iberiotoxin-sensitive and -insensitive BK currents in Purkinje neuron somata. J Neurophysiol. 2013;109(10):2528–41. https://doi.org/10.1152/jn.00127.2012.
Tian J, Tep C, Benedick A, Saidi N, Ryu JC, Kim ML, Sadasivan S, et al. p75 regulates Purkinje cell firing by modulating SK channel activity through Rac1. J Biol Chem. 2014;289(45):31458–72. https://doi.org/10.1074/jbc.M114.589937.
Matsuda K, Miura E, Miyazaki T, Kakegawa K, Emi K, Narumi S, et al. Cbln1 is a ligand for an orphan glutamate receptor delta 2, a bidirectional synapse organizer. Science. 2010;328(5976):363–8. https://doi.org/10.1126/science.1185152.
Yuzaki M, Aricescu AR. A GluD coming-of-age story. Trends Neurosci. 2017;40(3):138–50. https://doi.org/10.1016/j.tins.2016.12.004.
Ito M, Sakurai M, Tongroach P. Climbing fibre induced depression of both mossy fibre responsiveness and glutamate sensitivity of cerebellar Purkinje cells. J Physiol. 1982;324:113–34. https://doi.org/10.1113/jphysiol.1982.sp014103.
Kitazawa S, Kimura T, Yin PB. Cerebellar complex spikes encode both destinations and errors in arm movements. Nature. 1998;392(6675):494–7. https://doi.org/10.1038/33141.
Hirano T, Ohmori H. Voltage-gated and synaptic currents in rat Purkinje cells in dissociated cell cultures. Proc Natl Acad Sci U S A. 1986;83(6):1945–9. https://doi.org/10.1073/pnas.83.6.1945.
Sakurai M. Synaptic modification of parallel fibre-Purkinje cell transmission in in vitro Guinea-pig cerebellar slices. J Physiol. 1987;394:463–80. https://doi.org/10.1113/jphysiol.1987.sp016881.
Linden DJ, Connor JA. Participation of postsynaptic PKC in cerebellar long-term depression in culture. Science. 1991;254(5038):1656–9. https://doi.org/10.1126/science.1721243.
Linás R, Sugimori M. Electrophysiological properties of in vitro Purkinje cell dendrites in mammalian cerebellar slices. J Physiol. 1980;305:197–213. https://doi.org/10.1113/jphysiol.1980.sp013358.
Ito M. The molecular organization of cerebellar long-term depression. Nat Rev Neurosci. 2002;3(11):896–902. https://doi.org/10.1038/nrn962.
Wang SS, Denk W, Hausser M. Coincidence detection in single dendritic spines mediated by calcium release. Nat Neurosci. 2000;3(12):1266–73. https://doi.org/10.1038/81792.
Wang YT, Linden DJ. Expression of cerebellar long-term depression requires postsynaptic clathrin-mediated endocytosis. Neuron. 2000;25(3):635–47. https://doi.org/10.1016/s0896-6273(00)81066-1.
Hansel C, de Jeu M, Belmeguenai A, Houtman SH, Buitendijk GHS, Andreev D, et al. alphaCaMKII is essential for cerebellar LTD and motor learning. Neuron. 2006;51(6):835–43. https://doi.org/10.1016/j.neuron.2006.08.013.
Piochon C, Levenes C, Ohtsuki G, Hansel C. Purkinje cell NMDA receptors assume a key role in synaptic gain control in the mature cerebellum. J Neurosci. 2010;30(45):15330–5. https://doi.org/10.1523/JNEUROSCI.4344-10.2010.
Kono M, Kakegawa W, Yoshida K, Yuzaki M. Interneuronal NMDA receptors regulate long-term depression and motor learning in the cerebellum. J Physiol. 2019;597(3):903–20. https://doi.org/10.1113/JP276794.
Hirai H, Launey T, Mikawa S, Torashima T, Yanagihara D, Kasaura T, et al. New role of delta2-glutamate receptors in AMPA receptor trafficking and cerebellar function. Nat Neurosci. 2003;6(8):869–76. https://doi.org/10.1038/nn1086.
Jeromin A, Huganir RL, Linden DJ. Suppression of the glutamate receptor delta 2 subunit produces a specific impairment in cerebellar long-term depression. J Neurophysiol. 1996;76(5):3578–83. https://doi.org/10.1152/jn.1996.76.5.3578.
Kohda K, Kakegawa W, Matsuda S, Yamamoto T, Hirano H, Yuzaki M. The δ glutamate receptor gates long-term depression by coordinating interactions between two AMPA receptor phosphorylation sites. Proc Natl Acad Sci U S A. 2013;110(10):E948–57. https://doi.org/10.1073/pnas.1218380110.
Ito M. Bases and implications of learning in the cerebellum - adaptive control and internal model mechanism. Prog Brain Res. 2005;148:95–109. https://doi.org/10.1016/S0079-6123(04)48009-1.
Ito M. Control of mental activities by internal models in the cerebellum. Nat Rev Neurosci. 2008;9(4):304–13. https://doi.org/10.1038/nrn2332.
Cabaraux P, Gandini J, Kakei S, Manto M, Mitoma H, Tanaka H. Dysmetria and errors in predictions: the role of internal forward model. Int J Mol Sci. 2020;21(18):6900. https://doi.org/10.3390/ijms21186900.
Atkeson CG. Learning arm kinematics and dynamics. Annu Rev Neurosci. 1989;12:157–83. https://doi.org/10.1146/annurev.ne.12.030189.001105.
Kawato M, Furukawa K, Suzuki R. A hierarchical neural-network model for control and learning of voluntary movement. Biol Cybern. 1987;57(3):169–85. https://doi.org/10.1007/BF00364149.
Kawato M, Gomi N. The cerebellum and VOR/OKR learning models. Trends Neurosci. 1992;15(11):445–53. https://doi.org/10.1016/0166-2236(92)90008-v.
Wolpert DM, Ghahramani Z, Jordan MI. An internal model for sensorimotor integration. Science. 1995;269(5232):1880–2. https://doi.org/10.1126/science.7569931.
Wolpert DM, Miall RC. Forward models for physiological motor control. Neural Netw. 1996;9(8):1265–79. https://doi.org/10.1016/s0893-6080(96)00035-4.
Todorov E. Optimality principles in sensorimotor control. Nat Neurosci. 2004;7(9):907–15. https://doi.org/10.1038/nn1309.
Popa LS, Hewitt AL, Ebner TJ. Predictive and feedback performance errors are signaled in the simple spike discharge of individual Purkinje cells. J Neurosci. 2012;32(44):15345–58. https://doi.org/10.1523/JNEUROSCI.2151-12.2012.
Popa LS, Hewitt AL, Ebner TJ. The cerebellum for jocks and nerds alike. Front Syst Neurosci. 2014;8:113. https://doi.org/10.3389/fnsys.2014.00113.
Popa LS, Streng ML, Ebner TJ. Long-term predictive and feedback encoding of motor signals in the simple spike discharge of Purkinje cells. eNeuro. 2017;4(2):ENEURO.0036–17.2017. https://doi.org/10.1523/ENEURO.0036-17.2017.
Kakei S, Lee J, Mitoma H, Tanaka H, Manto M, Hampe CS. Contribution of the cerebellum to predictive motor control and its evaluation in ataxic patients. Front Hum Neurosci. 2019;13:216. https://doi.org/10.3389/fnhum.2019.00216.
Tanaka H, Ishikawa T, Kakei S. Neural evidence of the cerebellum as a state predictor. Cerebellum. 2019;18(3):349–71. https://doi.org/10.1007/s12311-018-0996-4.
Thach WT. Does the cerebellum initiate movement? Cerebellum. 2014;13(1):139–50. https://doi.org/10.1007/s12311-013-0506-7.
Gao Z, van Beugen BJ, De Zeeuw CI. Distributed synergistic plasticity and cerebellar learning. Nat Rev Neurosci. 2012;13(9):619–35. https://doi.org/10.1038/nrn3312.
Gall D, Prestori F, Sola E, D’Errico A, Roussel C, Forti L, et al. Intracellular calcium regulation by burst discharge determines bidirectional long-term synaptic plasticity at the cerebellum input stage. J Neurosci. 2005;25(19):4813–22. https://doi.org/10.1523/JNEUROSCI.0410-05.2005.
Maffei A, Prestori F, Shibuki K, Rossi P, Taglietti V, D’Angelo E. NO enhances presynaptic currents during cerebellar mossy fiber-granule cell LTP. J Neurophysiol. 2003;90(4):2478–83. https://doi.org/10.1152/jn.00399.2003.
Piochona C, Titleya HK, Simmonsa DH, Grassellia G, Elgersmab Y, Hansela C. Calcium threshold shift enables frequency-independentcontrol of plasticity by an instructive signal. Proc Natl Acad Sci U S A. 2016;113(46):13221–6. https://doi.org/10.1073/pnas.1613897113.
Lev-Ram V, Mehta SB, Kleinfeld D, Tsien RY. Reversing cerebellar long-term depression. Proc Natl Acad Sci U S A. 2003;100(26):15989–93. https://doi.org/10.1073/pnas.2636935100.
Lev-Ram V, Wong ST, Storm DR, Tsien RY. A new form of cerebellar long-term potentiation is postsynaptic and depends on nitric oxide but not cAMP. Proc Natl Acad Sci U S A. 2002;99(12):8389–93. https://doi.org/10.1073/pnas.122206399.
Kakegawa W, Yuzaki M. A mechanism underlying AMPA receptor trafficking during cerebellar long-term potentiation. Proc Natl Acad Sci U S A. 2005;102(49):17846–51. https://doi.org/10.1073/pnas.0508910102.
Schonewille M, Belmeguenai A, Koekkoek SK, Houtman SH, Boele HJ, van Beugen BJ, et al. Purkinje cell-specific knockout of the protein phosphatase PP2B impairs potentiation and cerebellar motor learning. Neuron. 2010;67(4):618–28. https://doi.org/10.1016/j.neuron.2010.07.009.
Kano M, Kano M, Fukunaga K, Konnerth A. Ca2+-induced rebound potentiation of gamma-aminobutyric acid-mediated currents requires activation of Ca2+/calmodulin-dependent kinase II. Proc Natl Acad Sci U S A. 1996;93(23):13351–6. https://doi.org/10.1073/pnas.93.23.13351.
Schonewille M, Gao Z, Boele HJ, Veloz MFV, Amerika WE, Simek AAM, et al. Reevaluating the role of LTD in cerebellar motor learning. Neuron. 2011;70(1):43–50. https://doi.org/10.1016/j.neuron.2011.02.044.
Boele HJ, Peter S, Ten Brinke MM, Verdonschot L, IJpelaar ACH, Rizopoulos D, et al. Impact of parallel fiber to Purkinje cell long-term depression is unmasked in absence of inhibitory input. Sci Adv. 2018;4(10):eaas9426. https://doi.org/10.1126/sciadv.aas9426.
Yamaguchi K, Itohara S, Ito M. Reassessment of long-term depression in cerebellar Purkinje cells in mice carrying mutated GluA2 C terminus. Proc Natl Aca Sci USA. 2016;113(36):10192–7. https://doi.org/10.1073/pnas.1609957113.
Kakegawa W, Katoh A, Narumi S, Miura E, Motohashi J, Takahashi A, et al. Optogenetic control of synaptic AMPA receptor endocytosis reveals roles of LTD in motor learning. Neuron. 2018;99(5):985–98.e6. https://doi.org/10.1016/j.neuron.2018.07.034.
Clouston PD, Saper CB, Arbizu T, Johnston I, Lang B, Newsom-Davis J, et al. Paraneoplastic cerebellar degeneration. III. Cerebellar degeneration, cancer, and the Lambert-Eaton myasthenic syndrome. Neurology. 1992;42(10):1944–50. https://doi.org/10.1212/wnl.42.10.1944.
Mason WP, Graus F, Lang B, Honnorat J, Delattre JY, Valldeoriola F, et al. Small-cell lung cancer, paraneoplastic cerebellar degeneration and the Lambert-Eaton myasthenic syndrome. Brain. 1997;120(Pt8):1279–300. https://doi.org/10.1093/brain/120.8.1279.
Graus F, Lang B, Pozo-Rosich P, Saiz A, Casamitjana R, Vincent A. P/Q type calcium-channel antibodies in paraneoplastic cerebellar degeneration with lung cancer. Neurology. 2002;59(5):764–6. https://doi.org/10.1212/wnl.59.5.764.
Bürk K, Wick M, Roth G, Decker P, Voltz R. Antineuronal antibodies in sporadic late-onset cerebellar ataxia. J Neurol. 2010;257(1):59–62. https://doi.org/10.1007/s00415-009-5262-8.
Iorio R, Damato V, Mirabella M, Vita MG, Hulsenboom E, Plantone D, et al. Cerebellar degeneration associated with mGluR1 autoantibodies as a paraneoplastic manifestation of prostate adenocarcinoma. J Neuroimmunol. 2013;263(1–2):155–8. https://doi.org/10.1016/j.jneuroim.2013.07.015.
Lopez-Chiriboga AS, Komorowski L, Kümpfel T, Probst C, Hinson SR, Pittock SJ, et al. Metabotropic glutamate receptor type 1 autoimmunity: clinical features and treatment outcomes. Neurology. 2016;86(11):1009–13. https://doi.org/10.1212/WNL.0000000000002476.
Spatola M, Pedrol MP, Maudes E, Simabukuro M, Muñiz-Castrillo S, Pinto AL, et al. Clinical features prognostic factors, and antibody effects in anti-mGlur1 encephalitis. Neurology. 2020;95(22):e3012–25. https://doi.org/10.1212/WNL.0000000000010854.
Coesmans M, Smitt PA, Linden DJ, Shigemoto R, Hirano T, Yamakawa Y, et al. Mechanisms underlying cerebellar motor deficits due to mGluR1-autoantibodies. Ann Neurol. 2003;53(3):325–36. https://doi.org/10.1002/ana.10451.
Liao YJ, Safa P, Chen YR, Sobel RA, Boyden ES, Tsien RW. Anti-Ca2+ channel antibody attenuates Ca2+ currents and mimics cerebellar ataxia in vivo. Proc Natl Acad Sci U S A. 2008;105(7):2705–10. https://doi.org/10.1073/pnas.0710771105.
Fukuda T, Motomura M, Nakao Y, Shiraishi H, Yoshimura T, Iwanaga K, et al. Reduction of P/Q-type calcium channels in the postmortem cerebellum of paraneoplastic cerebellar degeneration with Lambert-Eaton myasthenic syndrome. Ann Neurol. 2003;53(1):21–8. https://doi.org/10.1002/ana.10392.
Martin-Garcia E, Mannara F, Gutierrez-Cuesta J, Sabater L, Dalmau J, Maldonado R, et al. Intrathecal injection of P/Q type voltage-gated calcium channel antibodies from paraneoplastic cerebellar degeneration cause ataxia in mice. J Neuroimmunol. 2013;261(1–2):53–9. https://doi.org/10.1016/j.jneuroim.2013.05.003.
Sillevis Smitt P, Kinoshita A, De Leeuw B, Moll W, Coesmans M, Jaarsma D, et al. Paraneoplastic cerebellar ataxia due to antibodies toward against a glutamate receptor. N Engl J Med. 2000;342(1):21–7. https://doi.org/10.1056/NEJM200001063420104.
Marignier R, Chenevier F, Rogemond V, Sillevis Smitt P, Renoux C, Cavillon G, et al. Metabotropic glutamate receptor type 1 autoantibody-associated cerebellitis: a primary autoimmune disease? Arch Neurol. 2010;67(5):627–30. https://doi.org/10.1001/archneurol.2010.51.
Sugiyama N, Hamano S, Mochizuki M, Tanaka M, Takahashi Y. A case of chronic cerebellitis with anti-glutamate receptor delta 2 antibody. No To Hattatsu. 2004;36(1):60–3 PMID: 14737866.
Shimokaze T, Kato M, Yoshimura Y, Takahashi Y, Hayasaka K. A case of acute cerebellitis accompanied by autoantibodies against glutamate receptor delta2. Brain and Development. 2007;29(4):224–6. https://doi.org/10.1016/j.braindev.2006.08.011.
Shiihara T, Kato M, Konno A, Takahashi Y, Hayasaka K. Acute cerebellar ataxia and consecutive cerebellitis produced by glutamate receptor delta2 autoantibody. Brain and Development. 2007;29(4):254–6. https://doi.org/10.1016/j.braindev.2006.09.004.
Usui D, Mitsuda N, Hosokawa T, Fujieda M, Takahashi Y, Wakiguchi H. A case of persistent cerebellar ataxia complicated by conversion disorder-confirmed by positive cerebrospinal fluid glutamate receptor delta2 and epsilon2 antibodies. No To Hattatsu. 2011;43(1):41–5 PMID: 21400931.
Ichikawa K, Kikuchi M, Takeshita S, Nezu A. A case of chronic recurrent cerebellar ataxia responding to steroid therapy. Brain and Development. 2009;31(1):83–5. https://doi.org/10.1016/j.braindev.2008.04.010.
Zaenker P, Gray ES, Ziman MR. Autoantibody production in cancer-the humoral immune response toward autologous antigens in cancer patients. Autoimmun Rev. 2016;15(5):477–83. https://doi.org/10.1016/j.autrev.2016.01.017.
Mitoma H, Manto M, Hampe CS. Pathogenic roles of glutamate decarboxylase 65 auto-antibodies in cerebellar ataxias. J Immunol Res. 2017;2017:2913297. https://doi.org/10.1155/2017/2913297.
Nakao H, Nakao K, Kano M, Aiba A. Metabotropic glutamate receptor subtype-1 is essential for motor coordination in the adult cerebellum. Neurosci Res. 2007;57(4):538–43. https://doi.org/10.1016/j.neures.2006.12.014.
Rinaldo L, Hansel C. Ataxias and cerebellar dysfunction: involvement of synaptic plasticity deficits? Funct Neurol. 2010;25(3):135–9 PMID: 21232209.
Ishikawa T, Tomatsu S, Lee J, Hoffman DS, Kakei S. Releasing dentate nucleus cells from Purkinje cell inhibition generates output from the cerebrocerebellum. PLoS One. 2014;9(10):e108774. https://doi.org/10.1371/journal.pone.0108774.
Ishikawa T, Kakei S, Mitoma H. Overlooked Holmes’ clinical signs: reevaluation by recent physiological findings. Cerebellum Ataxias. 2015;2:13. https://doi.org/10.1186/s40673-015-0033-z.
Matsuda S, Kakegawa W, Budisantoso T, Nomura T, Kohda K, Yuzaki M. Sargazin regulates AMPA receptor trafficking through adaptor protein complexes during long-term depression. Nat Commun. 2013;4:2759. https://doi.org/10.1038/ncomms3759.
Ethical committee request
Not applicable.
Consent to participate
All authors are consent to participate.
Code availability
There is no software application or custom code associated with the paper.
Declaration
This work is not associated with a specific funding.
Funding
There is no specific funding related to this study.
Author information
Authors and Affiliations
Contributions
Project administration, M.M., H.M.; Writing, H.M., K.Y.; Editing J.H., M.M. All authors have read and agreed to the publication.
Corresponding author
Ethics declarations
Consent for publication
All authors are consent for publication.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mitoma, H., Honnorat, J., Yamaguchi, K. et al. Cerebellar long-term depression and auto-immune target of auto-antibodies: the concept of LTDpathies. Mol Biomed 2, 2 (2021). https://doi.org/10.1186/s43556-020-00024-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43556-020-00024-x