
Abstract
Background
Fungal peroxidases are oxidoreductases that utilize hydrogen peroxide to catalyze lignin biodegradation.
Results
PER-K (peroxidase synthesis codon gene) was transformed from Aspergillus niger strain AN512 deposited in the National Center for Biotechnology Information with the accession number OK323140 to Escherichia coli strain (BL21-T7 with YEp356R recombinant plasmid) via calcium chloride heat-shock method. The impact of four parameters (CaCl2 concentrations, centrifugation time, shaking speed, growth intensity) on the efficacy of the transformation process was evaluated. Furthermore, peroxidase production after optimization was assessed both qualitatively and quantitatively, as well as SDS-PAGE analysis. The optimum conditions for a successful transformation process were as follows: CaCl2 concentrations (50 mM), centrifugation time (20 min), shaking speed (200 rpm), and growth optical density (0.45). PCR and gel electrophoresis detect DNA bands with lengths 175, 179, and 211 bps corresponding to UA3, AmpR, and PER-K genes respectively besides partially sequencing the PER-K gene. Pyrogallol/hydrogen peroxide assay confirmed peroxidase production, and the activity of the enzyme was determined to be 3924 U/L. SDS-PAGE analysis also confirms peroxidase production illustrated by the appearance of a single peroxidase protein band after staining with Coomassie blue R-250.
Conclusion
A successful peroxidase-gene (PER-K) transformation from fungi to bacteria was performed correctly. The enzyme activity was screened, and partial sequencing of PER-K gene was analyzed successively. The protein 3D structure was generated via in silico homology modeling, and determination of binding sites and biological annotations of the constructed protein were carried out via COACH and COFACTOR based on the I-TASSER structure prediction.
Similar content being viewed by others
Key points
-
The optimization of PER-K fungal gene transformation in Escherichia coli was performed.
-
The isoelectric point value of peroxidase is 6.98 with higher thermal stability.
-
The 3D structures of the recombinant gene revealed high-quality model.
Background
Peroxidases are a group of oxidative enzymes produced by living microbes either intra- or extracellular in a response to pollutant existence such as dyes and aromatic hydrocarbons in the same surrounding environments [1]. These compounds are oxidized through a free radical mechanism to render the products less toxic to the living cell either through loss of biological activity or reduction in the bioavailability or adsorption. Peroxidase enzymes catalyze oxidative reactions via hydrogen peroxide utilization and protect the microorganisms from oxidation by free radicals [2]. These enzymes have been involved in different biotechnological purposes such as cosmetics, pulping, paper manufacturing, and wastewater treatment [3]. Fungal peroxidase plays an important role in lignin biodegradation and other environmental applications, such as biobleaching, biopulping, and bioremediation of soil. Therefore, ligninolytic enzymes of fungal origin have been extensively studied [4, 5]. White-rot fungi Phanerochaete chrysosporium is the most resplendent producing strain for lignin-degrading enzymes. Both lignin peroxidase lipA and manganese peroxidase mnp genes in Aspergillus niger have been extensively isolated in several studies [6, 7]. The wide application of peroxidase production for industrial potentials is limited due to low yield and expensive production cost. The increased demands for peroxidase production shed the light to discover new microorganisms capable of efficient peroxidase production. For feasible and efficient industrial applications, large-scale production for these enzymes is needed. Peroxidases availability in the market including Bjerkandera adusta peroxidase, horseradish peroxidase [HRP], and streptavidin peroxidase from Streptomyces avidinii probably could not meet the increasing industrial demand for peroxidase [1]. Recombinant peroxidase enzyme technique is one of the best ways to achieve this purpose [8]. This technique is the calcium chloride heat-shock transformation process which enhances bacterial cells to uptake DNA from the surrounding environment. The calcium ions role is predicted to be a cation bridge in the cell suspension between the phosphate backbone of DNA and negative charges on phosphorylated lipid A in lipopolysaccharide (LPS) [9, 10]. The ice-cold CaCl2 solution activates the binding of DNA to the surface of the cell, which then enters the cell by a short period of heat shock [11]. Selection markers such as drug resistance are usually used to identify the successful transformation of the interested gene [12]. This technique is usually used to transform cells with plasmids for cloning, recombinant protein expression, and long-term storage of the plasmids [13]. Therefore, the present study aimed to transform the competent E. coli BL21-T7 with recombinant plasmid carrying A. niger peroxidase enzyme coding gene, to confirm the peroxidase enzyme production by the new recombinant E. coli, and to determine the culture conditions that support optimum peroxidase production recombinant strain. In addition, peroxidase production from transformed E. coli was estimated qualitatively and quantitatively as a new potential applicable source for extracellular peroxidase production.
Material and methods
Microbial strains and plasmid isolation
Two strains, E. coli BL21 — T7 and Aspergillus niger AN512, were cultivated from glycerol stock in nutrient agar (NA) (Oxoid, USA) and potato dextrose agar (PDA) (Oxoid, USA) for 24 h and 4 days for bacterial and fungal strains, respectively. Shuttle plasmid YEp356R (1.0μg/μl) ATCC 37737 was previously isolated using EasyPure Plasmid MiniPrep Kit, from E. coli Dh5α-k12. Selective step is carried out on MacConkey agar (Oxoid, USA) supplemented with 50 μg/ml ampicillin (Fig. S1) [14, 15]. All bacterial, fungal, and plasmid samples were obtained from Applied Microbial Genetics Department, Genetic Engineering and Biotechnology Division, NRC, Giza, Egypt.
Preparation of competent cells
Different concentrations of CaCl2, 0, 25, 50, 75, 100, 125, 150, and 200 mM were prepared, and improved buffer was prepared as follows: add 100 ml of glycerin into 50 mM CaCl2 solution, and then, complete the final volume to 1000 ml with ddH2O. E. coli BL21-T7 cells were picked from NA and grown on Luria-Bertani (LB) broth medium (Oxoid, USA) under vigorous incubation shaking condition (200 rpm) for 10 to 16 h until it reaches optical density (OD600) of 0.2 to 0.45. The resulted cell pellet was harvested by centrifugation undercooling (−4 °C) at 10,000 rpm for 5 min and washed three times by CaCl2 (50 mmol) supplemented with 17% glycerin. Finally, the cells were suspended in CaCl2 buffer and distributed in 1.5 ml Eppendorf [14, 15].
Construction of recombinant plasmid
After plasmid isolation, it digested with endonuclease buffers. The desired gene PER-K was also isolated from fungal DNA after its extraction with the same restriction enzymes which give complementary ends in plasmid and gene for easily recombination step. Digestion process was composed of 500 μl of DNA with 55 μl from both endonuclease buffers (1 μg DNA restriction 0.5 μg), and this ratio is also used for plasmid digestion. Restriction endonucleases and digestion buffers were illustrated in Tables S1 and S2.
Optimization of transformation conditions
Several steps were performed according to [16] for the determination of the efficiency of the transformation process. The different culture conditions, such as shaking speed (50, 70, 90, 120 150, and 200 rpm), growth turbidity using OD600 values (0–0.5), and centrifugation time (5, 10, 15, and 20 min), were tested to prepare E. coli competent cells and assessed by calculating the transformation efficiency. All optimization items were established in triplicates.
Genomic detection of successful transformant
The recombinant plasmid was isolated and extracted using QuickClean II Plasmid Purification Kit 50 rxns. Then, the plasmids were screened for their purity and concentration through NanoDrop and agarose gel electrophoresis, the appropriate samples were prepared for PCR reaction as a template, and specific primers were used for amplification plasmid genes (Per-K, UA3, and ampR). The primers used were illustrated in Table 1.
PCR detection and amplification of desired gene Per-K as well as two plasmid gene markers were performed under different specific conditions according to each gene. For Per-K, the PCR conditions were denaturation step for the first cycle adjusted at 95 °C for 1 min, extension step for the final cycle was at 4 °C overnight, while the rest of other cycles of the reaction was denaturation cycles (2–34) at 95 °C for 30 s, annealing cycles (2-34) at 56 °C for 30 s, and extension (cycles 2–34) at 72 °C for 40 s (Table 1).
In case of URA3, PCR conditions of denaturation step for the first cycle are adjusted at 93 °C for 1 min, and extension step for the final cycle was at 4 °C overnight, and the rest of other cycles of the reaction was as follows: denaturation cycles (2–37) at 93 °C for 30 s, annealing cycles (2–37) at 59 °C for 30 s, and extension (cycles 2–37) at 73 °C for 40 s (Table 1).
Regarding AmpR, PCR conditions comprised of denaturation step for the first cycle adjusted at 93 °C for 1 min, and extension step for the final cycle was at 4 °C overnight, and the rest of other cycles of the reaction was as the follows: denaturation cycles (2–33) at 93 °C for 30 s, annealing cycles (2–33) at 55 °C for 30 s, and extension (cycles 2–33) at 74 °C for 40 s (Table 1).
Isolation and cloning per-K gene
per-K gene is isolated from fungus through total genomic DNA extraction. Its purity was determined via NanoDrop and estimated at about 50 μg/ml. The extracted DNA was then digested with restriction digestion solution SalI digestion and HindIII (Table S2). On the other hand, plasmid was isolated from the E. coli host and its purity was determined by NanoDrop and estimated 1.00, and it also digested with the same restriction solutions that are mentioned before (Table S2). The recombination step is done in a 1.5 mL Eppendorf tube that is kept on the ice at −20 °C with different ratios of plasmid and digested DNA for the DNA library. The successful step was composed of 300 μl of digested DNA with 100 μl plasmid solution, and then, this mixture was mixed at very slow rotation speed with a thin, alcohol-sterilized glass rod in a clockwise direction for 15 s. After that, the mixture was kept in water bath 32 °C for 15 min; finally, it was mixed with sterile M9 medium with glucose and incubated without shaking for 4 h before transferring to LB broth with ampicillin for selecting our recombinant.
Determination of peroxidase molecular weight by SDS-PAGE
The molecular weight of peroxidase protein was determined through SDS–PAGE in a Mini Protean III Electrophoresis Cell (Bio-Rad), with 12% resolving and 4% stacking gel. Proteins were stained using the Coomassie blue staining technique, and the molecular weight was estimated by comparison to molecular weight ladder (10 to 180 kDa).
Detection of peroxidase activity
Peroxidase activity was evaluated using the method of Rayner and Boddy (1988). Briefly, a 2-day incubated culture at 30 °C was grown in nutrient agar plates, and the resulting colonies were immersed with 100 μl of 0.4% (v/v) hydrogen peroxide and 1% pyrogallol in water. Colonies that developed yellow-brown color was considered as positive for peroxidase production [17].
Total protein concentration in culture filtrate
Culture filtrate total protein was determined and calculated using the Bradford method for the quantitative determination of peroxidase activity. The isolate was incubated in tryptone soy broth (TSB) medium for 5 days, and the culture supernatant was separated by centrifugation at 6000 rpm for 25 min. A total of 50 μl of the supernatant was transferred to a clean test tube, and the volume was brought to 1 ml with phosphate buffer (0.1 M, pH 6.5). Five milliliters of protein reagent solution “Coomassie brilliant blue G-250 in ethanol” was added. The final reading was detected spectrophotometrically at 595 nm after 2 min and before 1 h of the reagent addition. The amount of total protein detected was calculated in μg/mL according to the standard equation: Y = 9011.1x–157.98 where x = absorbance at 595 nm [18].
Quantitative determination of peroxidase activity
Peroxidase activity was evaluated according to the rate of hydrogen peroxide oxidation of pyrogallol to purpurogallin as exhibited by modified method of Chance and Maehly. Reaction mixture of 350 μl was prepared, composed of 5% w/v pyrogallol dissolved in 100 mM potassium phosphate buffer (pH 6) and 25 μl of 5 days’ culture supernatant. The reaction mixture without culture supernatant was prepared as blank. The reaction was begun by the addition of 0.5% hydrogen peroxide (30%), and the readings were measured spectrophotometrically from the beginning with an interval of 30 s for 3 min at 420 nm [19, 20]. Peroxidase activity was calculated according to the equation U/ml: abs. (final)—abs. (beginning)/0.001.
Analysis of physicochemical parameters of peroxidase enzyme
To compute the physicochemical parameters of the peroxidase protein, ExPASy’s ProtParam program was used. These properties can be derived from a protein sequence which includes parameters such as molecular weight (M.Wt), instability index (II), aliphatic index (AI), theoretical pI, and grand average of hydropathicity (GRAVY). The instability index provides an approximation of our protein’s stability, an instability index less than 40 is projected to be stable, and a score greater than 40 indicates that the protein may be unstable [21].
Construction of the 3D enzymes structure by homology modeling
The amino acid sequences of the Aspergillus niger peroxidase enzymes were submitted to the SWISS-MODEL, and the 3D structure of the peroxidase enzymes was automatically generated by first transferring conserved atom coordinates provided by the desired template alignment [22]. Endo β-1,4-glucanase from Bacillus licheniformis was utilized as a template to perform the homology modeling of the fungal cellulase structure. The enzyme models were obtained as a PDB file, and the model was energy minimized via Gromos96 tools in the Swiss-PDB viewer [23].
Identification of the enzymes catalytic residues
The active-site residues of the peroxidase enzyme were predicted using the I-TASSER web server (https://zhanggroup.org/I-TASSER/). I-TASSER web server detects catalytic residues in the primary structural alignment, which was then viewed in PyMOL. According to a previously reported approach, the probable active-site residues were superimposed on a template structure in this case [24]. COACH, a meta-server, was then used to predict the protein-ligand interaction site. To construct the final ligand binding site predictions, the predictions were merged with data from the COFACTOR, FINDSITE, and ConCavity analyses.
Results
Positive transformant selection
Out of 225 isolated colonies, about 14 single colonies were able to grow on M9 minimal medium supplemented with 1% glucose and incubated at 37 °C for 24 h with the lowest shaking (50 rpm), nutrient agar medium was also used to cultivate the promising 14 colonies as standard plates. The fourteen different transformant colonies were confirmed for successful cloning with the recombinant plasmid through cultivation on MacConkey agar supplemented with 30 μg/mL ampicillin. All colonies were grown well with pinky color. MacConkey agar is considered a good selective medium for our E. coli host strain as it was gram-negative lactose-fermenting bacteria besides the presence of ampicillin allows us to easily pick up grown cells with successful recombinant plasmid.
Optimization of plasmid transformation
Optimization for plasmid transformation was performed through CaCl2 adjustment, so different CaCl2 solution concentrations were used, and transformation efficiency increased at concentration 25 mM and gradually increased by increasing solution concentration and reached its maximum value at 50 mM of CaCl2 solution and then decreased gradually until 150 mM (Fig. 1a). In the present study, the highest transformation efficiency reached its maximum when the OD600 values were between 0.41 and 0.45 (Fig. 1b). The transformation efficiency of competent cells was also affected by centrifugation time where it was neglectable value at centrifugation less than 5 min and begun to increase after 5 min, and its efficiency reaches a maximum at 20 min and then decreased gradually until be neglectable again (Fig. 1c). Shaker’s speed during competent cells preparation is also considered a significant factor in the transformation efficiency. Transformation efficiency in our study was increased gradually from 50 rpm until it reached its maximum of 11 × 106/μg at 200 rpm (Fig. 1d).

Optimization for plasmid transformation. a CaCl2 concentrations. b Different absorbance values. c Different centrifugation time. d Different shaking speeds. TE, transformation efficiency
PCR detection of the recombinant plasmid
Three genes (our insert per-K, UA3, and AmpR genes) were used as target sequences for PCR amplification in order to detect our insert plasmid into host cells. Agarose gel electrophoresis and DNA sequences (Fig. S2) showed that Per-K, UA3, and AmpR amplicons were 211, 175, and 179 bps, respectively. Per-K sequence scored through online BLAST, 100% query cover, with a total and maximum score of 390 with Aspergillus officinalis peroxidase 72-like (LOC109840690), mRNA (Figs. S3 and S4 Table 2).
PER-K gene isolation and cloning
The complete gene sequence was analyzed. The open reading frame (ORF) started from 141 to 1145 bps, while the total nucleotide sequence was 1330 bps (Fig. S5). The amino acid polypeptide resulted from ORF was 334 which gives protein with an expected molecular weight near to 33.4 KDa as analyzed through SnapGene Viewer software (version 4.1.3).
Partial purification of peroxidase and molecular weight determination
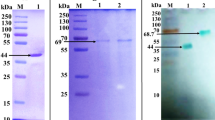
The results showed that 5.9% of ammonium sulfate was given complete precipitation of the enzyme with maximum enzyme activity. After the precipitation of peroxidase with ammonium sulfate, the sediment was redissolved in citrate-phosphate buffer (pH 7.2) and dialyzed against the same buffer. The obtained purified bacterial peroxidase showed a single protein band (55 KDa) on SDS-PAGE after staining with Coomassie blue R-250 (Fig. 2).

Molecular weight of recombinant peroxidase by electrophoretic analysis on 7.5% SDS-PAGE. PM, molecular weight marker proteins. PEP, purified peroxidase. CR, crude extract
Peroxidase activity
E. coli cloned strain was detected as a positive for peroxidase enzyme production, illustrated by the appearance of yellow-brown color after immersing growth with 1% pyrogallol and 0.4% hydrogen peroxide. The quantitative evaluation of peroxidase production and activity was calculated by subtracting the final absorbance of the reaction mixture (0.456) from the initial absorbance (0.064) at 420 nm divided by 0.001. The obtained peroxidase enzymatic activity detected was 392.4 U/ml after 5 days of the incubation period.
Physicochemical properties of peroxidase
The ProtParam tool was used to predict the physicochemical parameters of the peroxidase protein enzyme. The physical parameters showed that the enzyme’s molecular weight is 36,650.92 Da, and the peroxidase enzyme’s instability index is 48.26. Aspergillus niger AN512 peroxidase has a calculated isoelectric point pI value of6.98 and a higher aliphatic index (85.57), suggesting that it is a thermally stable protein. Meanwhile, the peroxidase enzyme’s negatively grand average of hydropathicity (GRAVY) values revealed its hydrophilicity (−0.156). The total number of negatively charged residues (Asp + Glu) is 37, while the total number of positively charged residues (Arg + Lys) is also 37 (Table 3).
Modeling the 3D structures of Aspergillus niger AN512 peroxidase enzymes
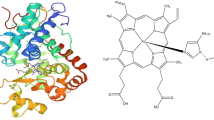
The amino acid sequences of Aspergillus niger AN512 peroxidase were subjected to homology modeling via SWISS-MODELweb server to generate the 3D structures of the peroxidase enzyme. The cellulase enzyme was constructed using an Arabidopsis thaliana peroxidase (52.49% sequence similarity). Figure 3a illustrates the generated 3D structures of Aspergillus niger AN512 peroxidase. Furthermore, the I-TASSER web server was also used to generate high-quality model predictions of 3D structure (Fig. 3b) and biological function of protein Aspergillus niger AN512 peroxidase protein.

a The SWISS-MODEL generated 3D structures of Aspergillus niger AN512 peroxidase enzymes. b The predicted 3D structures by I-TASSER
Validation of homology model
To evaluate the predicted 3D structure of the homology model, Ramachandran’s plot of the model was constructed to determine the stereochemical quality of the protein structure by analyzing residue-by-residue geometry. The backbone conformation and overall stereochemical quality of cellulase of Aspergillus niger AN512 were calculated by analyzing the phi (Φ) and psi (ψ) torsion angles, and the results were illustrated in the Ramachandran plots in Fig. 4.

Ramachandran’s plot calculations on the 3D models of peroxidase of Aspergillus niger AN512 computed by the SWISS-MODEL web-server to show the favored regions for backbone dihedral angles against of amino acid residues in protein structure a) General (No Proline or Glycine) b) Glycine Only c) Pre-Proline Only d) Proline only
Determination of binding site
Biological annotations of the target protein were measured by COACH and COFACTOR based on the I-TASSER structure prediction. While COFACTOR uses structure comparison and protein-protein networks to deduce protein functions (ligand-binding sites, EC, and GO), COACH is a meta-server technique that collects various function annotation results (on ligand-binding sites) from the COFACTOR, TM-SITE, and S-SITE programs. According to a prediction by I-TASSER algorithm for the protein 3D structure, 5 ligands, calcium (2+) at binding site residues (74, 77, 78, 79, 81, 83), b) 3-bromoquinolin-4-amine (2NW) at binding site residues (200, 202, 203, 204, 205, 266, 273, 274, 277), N,2-dihydroxybenzamide (SHA) at binding site residues (69, 72, 73, 100, 170) were detected Fig. 5.

Predicted binding sites in complex with ligands
Discussion
The concentration of CaCl2 solution is an important factor affecting the transformation efficiency of competent cells [25]. Li et al. reported that the transformation efficiency of competent cells increased by rising the concentration of CaCl2 solution an reached its maximum at 75 mM and then decreased rapidly when the concentration exceeded 100 mM [26]. This result is slightly similar to result in the present study as maximum transformation efficiency reached 50 mM and decreased as CaCl2 concentration increased until 150 mM. This inverse proportion between CaCl2 concentration and transformation efficiency may be due to lipid array on cell membrane is destroyed by 75 to 100 mM Ca2+, and then, a liquid crystal would be formed. The bacteria would be swollen when incubated in hypotonic calcium chloride solution, at 0 °C, and DNA in the mixture can be formed into hydroxyapatite (anti-DNase) and then stick to the surface of cells [27, 28]. The cellular absorption ability of exogenous DNA will be increased after heat shock at 42 °C. This process may be inhibited, and hence, the transformation efficiency will be decreased when the concentration of calcium chloride solution exceeds 100 mM.
The early logarithmic growth period of E. coli was the very important factor, the best growth condition, and easily be induced when the OD600 meets 0.35 to 0.45 Meanwhile, during this phase, the bacteria are in the best tolerance condition suffering the physical damage, and this could increase the transformation efficiency [29, 30]. Sambrook and Russell [16] found out that the absorptivity of E. coli obviously decreased because of the mutation of hofQ during the stationary phases.
Chan et al. [25] reported that centrifugation time had little effect on transformation efficiency, but when the time is exceeding 10 min, some dead bacteria would form sediment together with activity bacteria, which could degrade transformation efficiency to a certain extent. On the other hand, almost all of active bacteria could form sediment when centrifugation time was 5 min. So, using centrifugation time from 5 to 10 min for preparation of competent cells will give maximum efficiency.
Catalase-peroxidase gene from the pathogenic fungus Penicillium marneffei was isolated by Pongpom et al. [31], and DNA sequence analysis of this gene revealed an ORF encoding a 748 amino acid polypeptide with a predicted molecular mass of 82.4 KDa within the amino acid sequence was 45/69% identical to that of catalase-peroxidases from many bacteria and fungi [29, 31].
The E. coli cloned strain was similar to that reported by Falade et al. [1] They isolated freshwater bacterial strains capable of peroxidase production by applying the same identification technique. Also, Al-Senaidy and Ismael [32] obtained peroxides enzyme with 55 KDa. Earlier reports also performed on bacterial peroxidases purified from Bacillus sp. VUS Dawkar et al. [33] showed peroxides enzyme with molecular weight of 43 KDa. Isolation and cloning of fungus per-K gene in E. coli expression host have numerous advantages including easy extraction method and huge productivity in shorter time in comparison with fungus isolate. Also the used E. coli host is more safe than Aspergillus niger isolate on the human health in case of using it for commercial production of the enzyme.
Ammonium sulfate is a salt used in the precipitation of enzymes due to its high solubility; therefore, it was used in the precipitation of different enzymes [34]. Partial purification was performed by gradual saturation ratios ranged from 40 to 90% of ammonium sulfate to precipitate crude enzyme, and then, the peroxidase activity was checked in each fraction to check the most suitable saturation according to precipitation of peroxidase enzyme. A total of 90% of ammonium sulfate was given completely precipitation of the enzyme with maximum enzyme activity which is similar to these reported by Kalyani et al. [35] and Mustafa [36]. The result of peroxidase activity was in approval with the studies obtained by Falade et al. [1] who isolated two bacterial strains capable for producing a significantly high peroxidase activity with 5250 and 5833 U/L for both strains. Other studies reported a lower [24] peroxidase activity for different Streptomyces strains with 270 and 535 U/L. [37, 38] The reason for different peroxidase production between different isolates is not yet clearly understood; however, the excessive production of peroxidase in the cloned E. coli strain confirmed the successful recombinant process with the desired results obtained. Peroxidase-producing microbes possess high potential for industrial applications. One of the main significantly important application is dye treatment process in wastewater streams. Peroxidase-producing microorganisms also serve as important cause in the pretreatment of lignocellulose material which eventually leads to the conversion of lignocellulose, complex molecules to ethanol. Fungi contain diverse important genes encoded several proteins and enzymes used in medical, pharmaceutical, and industrial uses [2, 39,40,41]. The physical parameters of the obtained protein were predicted, and results showed that Aspergillus niger AN512 peroxidase has a calculated isoelectric point pI value of 6.98 and a higher thermal stability. The amino acid sequences of Aspergillus niger AN512 peroxidase were subjected to homology modeling via SWISS-MODEL web server to generate the 3D structures of the peroxidase enzyme. The Ramachandran’s plots of the model were constructed to determine the stereochemical quality of the protein structure by analyzing residue-by-residue geometry, and results revealed that the obtained model quality is high according to constructed Ramachandran’s plots.
Conclusion
This study showed that the optimum conditions for a successful transformation of the PER-K gene of Aspergillus niger AN 512 inside E. coli BL21-T7 were include 50 mM CaCl2 concentration, 20-min centrifugation time, 200 rpm shaking speed, and growth optical density of 0.45. PCR detection of plasmid DNA was through amplification of UA3, AmpR, and PER-K genes. Partial sequencing of the PER-K gene (under submission in NCBI GeneBank) has an activity of 3924 U/L and molecular weight of 55 KDa. Recombinant peroxidase enzyme technique can be used to achieve large-scale production for this enzyme for feasible and efficient industrial applications.
Availability of data and materials
All authors declare that the data supporting the findings of this study are available within the article.
References
Falade A, Jaouani A, Mabinya L, Okoh A, Nwodo U (2019) Exoproduction and molecular characterization of peroxidase from Ensifer adhaerens. Appl Sci 9:3121
Mohamed AF, et al (2021) Genetic and Histopathological Alterations in Caco-2 and HuH-7 Cells Treated with Secondary Metabolites of Marine fungi. J Gastrointest Canc 53:480–95.https://doi.org/10.1007/s12029-021-00640-y
Atala ML, Ghafil JA, Zgair AK (2019) Production of peroxidase from Providencia spp. bacteria. J Pharm Sci Res 11:2322–2326
Conesa A, van den Hondel CA, Punt PJ (2000) Studies on the production of fungal peroxidases in Aspergillus niger. Appl Environ Microbiol 66:3016–3023
Hamed A, et al (2020) Meleagrin from marine fungus Emericella dentata Nq45: crystal structure and diverse biological activity studies. Nat Prod Res 35:21, 3830-3838. https://doi.org/10.1080/14786419.2020.1741583
Padma N, Sudha H (2013) Optimization of lignin peroxidase, manganese peroxidase, and lac production from Ganoderma lucidum under solid state fermentation of pineapple leaf
Lin M-I, Nagata T, Katahira M (2018) High yield production of fungal manganese peroxidases by E. coli through soluble expression, and examination of the activities. Protein Expr Purif 145:45–52
Khan S et al (2016) Role of recombinant DNA technology to improve life. Int J Genomics 2016:2405954
DeLucia AM et al (2011) Lipopolysaccharide (LPS) inner-core phosphates are required for complete LPS synthesis and transport to the outer membrane in Pseudomonas aeruginosa PAO1. MBio 2:e00142–e00111
Yoon K-W, Doo E-H, Kim S-W, Park J-B (2008) In situ recovery of lycopene during biosynthesis with recombinant Escherichia coli. J Biotechnol 135:291–294
Bergmans H, Van Die I, Hoekstra W (1981) Transformation in Escherichia coli: stages in the process. J Bacteriol 146:564–570
Jang C-W, Magnuson T (2013) A novel selection marker for efficient DNA cloning and recombineering in E. coli. PLoS One 8:e57075
Goldstein D et al (2005) A REVIEW Human safety and genetically modified plants: a review of antibiotic resistance markers and future transformation selection technologies. J Appl Microbiol 99:7–23
Elbing KL, Brent R (2019) Recipes and tools for culture of Escherichia coli. Curr Protoc Mol Biol 125:e83
Aryal S (2018) Potato dextrose agar (PDA)-principle. In: Uses, composition, procedure and colony characteristics. https://microbenotes.com/potato-dextrose-agar-pda/
Sambrook J (2001) Preparation and analysis of Eukaryotic Genome DNA. Molecular cloning: a laboratory manual 1, 6-1
Rayner ADM, Boddy L (1988) Fungal Decomposition of Wood: Its Biology and Ecology. Wiley, New York, pp 587
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Ghadermarzi M, Moosavi-Movahedi A (1996) Determination of the kinetic parameters for the “suicide substrate” inactivation of bovine liver catalase by hydrogen peroxide. J Enzyme Inhib Med Chem 10:167–175
Maehly AC, Chance B (1954) The assay of catalases and peroxidases. Methods Biochem Anal 1:357–424. https://doi.org/10.1002/9780470110171.ch14
Guruprasad K, Reddy BB, Pandit MW (1990) Correlation between stability of a protein and its dipeptide composition: a novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng Des Sel 4:155–161
Waterhouse A et al (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46:W296–W303
Bienert S et al (2017) The SWISS-MODEL repository—new features and functionality. Nucleic Acids Res 45:D313–D319
Batumalaie K, Edbeib MF, Mahat NA, Huyop F, Wahab RA (2018) In silico and empirical approaches toward understanding the structural adaptation of the alkaline-stable lipase KV1 from Acinetobacter haemolyticus. J Biomol Struct Dyn 36:3077–3093
Chan W-T, Verma CS, Lane DP, Gan SK-E (2013) A comparison and optimization of methods and factors affecting the transformation of Escherichia coli. Biosci Rep 33:e00086
Green MR, Sambrook J (2018) The Hanahan method for preparation and transformation of competent Escherichia coli: high-efficiency transformation. Cold Spring Harb Protoc 2018. https://doi.org/10.1101/pdb.prot101188
Panja S, Saha S, Jana B, Basu T (2006) Role of membrane potential on artificial transformation of E. coli with plasmid DNA. J Biotechnol 127:14–20
Ahmad I, Rubbab T, Deeba F, Naqvi SMS (2014) Optimization of E. coli culture conditions for efficient DNA uptake by electroporation. Turk J Biol 38:568–573
Tsen S-D et al (2002) Natural plasmid transformation in Escherichia coli. J Biomed Sci 9:246–252
Singh M, Yadav A, Ma X, Amoah E (2010) Plasmid DNA transformation in Escherichia coli: effect of heat shock temperature, duration, and cold incubation of CaCl2 treated cells. IJBB 6:561–568
Pongpom P, Cooper CR Jr, Vanittanakom N (2005) Isolation and characterization of a catalase-peroxidase gene from the pathogenic fungus, Penicillium marneffei. Med Mycol 43:403–411
Al-Senaidy AM, Ismael MA (2011) Purification and characterization of membrane-bound peroxidase from date palm leaves (Phoenix dactylifera L.). Saudi. J Biol Sci 18:293–298
Dawkar VV, Jadhav UU, Telke AA, Govindwar SP (2009) Peroxidase from Bacillus sp. VUS and its role in the decolorization of textile dyes. Biotechnol Bioprocess Eng 14:361–368
Yin L-J, Lin H-H, Xiao Z-R (2010) Purification and characterization of a cellulase from Bacillus subtilis YJ1. J Mar Sci Technol 18:466–471
Kalyani DC, Phugare SS, Shedbalkar UU, Jadhav JP (2011) Purification and characterization of a bacterial peroxidase from the isolated strain Pseudomonas sp. SUK1 and its application for textile dye decolorization. Ann Microbiol 61:483–491
Mustafa HSI (2014) Staphylococcus aureus can produce catalase enzyme when adding to human WBCs as a source of H 2 O 2 productions in human plasma or serum in the laboratory. Open J Med Microbiol 4:249
Mercer DK, Iqbal M, Miller P, McCarthy A (1996) Screening actinomycetes for extracellular peroxidase activity. Appl Environ Microbiol 62:2186–2190
Tuncer M, Kuru A, Sahin N, Isikli M, Isik K (2009) Production and partial characterization of extracellular peroxidase produced by Streptomyces sp. F6616 isolated in Turkey. Ann Microbiol 59:323–334
Mohamed AA, Abu-Elghait M, Ahmed NE, Salem SS (2021) Eco-friendly mycogenic synthesis of ZnO and CuO nanoparticles for in vitro antibacterial, antibiofilm, and antifungal applications. Biol Trace Elem Res 199:2788–2799
Abu-Elghait M, Hasanin M, Hashem AH, Salem SS (2021) Ecofriendly novel synthesis of tertiary composite based on cellulose and myco-synthesized selenium nanoparticles: characterization, antibiofilm and biocompatibility. Int J Biol Macromol 175:294–303
Shebl RI et al (2021) Staphylococcus aureus derived hyaluronic acid and bacillus Calmette-Guérin purified proteins as immune enhancers to rabies vaccine and related immuno-histopathological alterations. CEVR 10:229
Funding
Not applicable
Author information
Authors and Affiliations
Contributions
MK: conceptualization, methodology, resources, investigation, software, writing—original draft, and cloning steps. KMAK: conceptualization, methodology, and resources. HAK: resources, investigation of peroxidase production, and writing—review. MSEMB: methodology, resources, investigation, software, writing—original draft, and docking study. AAH: methodology, software, writing—review, and partial purification of peroxidase by SDS. MAE: conceptualization, methodology, investigation, software, writing—review, editing, and submitting the paper to the journal. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval consent to participate
This article does not contain any studies with human participants performed by any of the authors.
Consent for publication
The authors permit the publisher to publish all data in this work.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
Yeast episomal vector with a UA3 marker for construction of lacZ fusions. The MCS is reversed in YEp356. Fig. S2. DNA sequences and Agarose gel electrophoresis analysis of PCR product of Per-K, UA3 and AmpR amplicons. Fig. S3. Phylogenetic tree of Per-K gene sequence of this study and the most genetically related taxa on NCBI, this tree was designed through MEGA 7 software. Fig. S4. DNA nucleotides alignment between our Per-K query and the most related two DNA sequences, alignment run through online Clustal Omega and finished through Jalview software. Fig. S5. Complete gene sequence open reading frame (ORF) started from 141 to 1145 bps. Table S1. Restriction endonucleases used in fungus genomic DNA and vector digestion. Table S2. Digestion buffers.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khedr, M., Khalil, K.M.A., Kabary, H.A. et al. Molecular docking and nucleotide sequencing of successive expressed recombinant fungal peroxidase gene in E.coli. J Genet Eng Biotechnol 20, 94 (2022). https://doi.org/10.1186/s43141-022-00377-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43141-022-00377-6