
Abstract
Background
Spathodea campanulata P. Beauv. known as the African tulip tree has potential medicinal properties that have been shown traditionally for the treatment of various ailments. The aim of the present study was isolation, identification, and evaluation of the cytotoxic activity of phytochemicals from the chloroform extract of S. campanulata.
Result
Three compounds were isolated by using column chromatography and preparative TLC from chloroform extract of leaves of S. campanulata. The structures of the isolated compounds were elucidated by using spectroscopic methods, including, FTIR, ESI-TOF MS, 1H NMR, and 13C NMR spectroscopy. In vitro cytotoxic activity of compounds was evaluated by using SRB assay against human leukemia cancer cell lines (HL-60). Results were expressed in IC50 values. Stigmasta-5,22-dien-3-ol, octadecenamide, and umbelliferone were isolated and identified from chloroform extract. The isolated compounds showed cytotoxicity with decreasing cell viability in a dose-dependent manner, but it was found low as compared to positive control, i.e., Adriamycin against HL-60 cell lines.
Conclusion
The results indicate that isolated compounds, i.e., stigmasta-5,22-dien-3-ol (44.12μg/ml), octadecenamide (35.65μg/ml), and umbelliferone (80.60μg/ml) showed antiproliferative activity, but it was low compared to positive control Adriamycin (10.09 μg/ml). Also, according to our knowledge, this study is the first report on the isolation and identification of octadecenamide and umbelliferone from the leaves of S. campanulata.
Graphical abstract

Similar content being viewed by others

Background
Plants have been used for the prevention and treatment of various diseases and ailments since ancient times and are considered an excellent source of bioactive components. The vast array of pharmacological properties associated with medicinal plants included antidiabetic, analgesic, anti-inflammatory, antimalarial, hypoglycemic, antimicrobial, stimulant, immune-modulator, and anticancer [1]. According to the World Health Organization (WHO), 80% of the world population uses herbal medicine in some aspects of primary health care. It is proved that plants have always been a useful and promising source for anticancer compounds. Approximately 60% of currently used anticancer agents are derived from plants [2]. The National Cancer Institute (NCI) has investigated approximately 35,000 different plant species for potential anticancer properties, and about 3000 species have demonstrated reproducible anticancer activity [3]. Vincristine, vinblastine, etoposide, colchicinamide, Taxol, 10-hydroxycamptothecin, elliptinium, curcumol, gossypol, lycobetaine, tetrandrine, homoharringtonine, monocrotaline, and curdione are remarkable anticancer phytochemicals derived from plant species [4].
Spathodea campanulata P. Beauv. belongs to the Bignoniaceae family commonly known as the African Tulip tree. It is a perennial plant native to the African continent commonly used as folkloric medicine in Nigeria [5]. Flowers of the plant show anti-inflammatory and diuretic properties, while the leaves are used in kidney diseases, urethra inflammations, and as an antidote against animal poison [6]. Numbers of phytochemicals have been isolated from S. campanulata such as triterpene acid, spathodic acid, sitosterol-3-β-D-glucopyranoside, steroids, ursolic acid, tomentosolic acid, and pectic substances [7, 8]. Spathodol, triterpenoids 3-β acetoxyoleanolic acid, siaresinolic acid, and oleanolic acid are reported from leaves [9], whereas the fruits contain polyphenols, tannins, saponins, and glycosides [10]. Flowers contain oleic acid, anthocyanin, 1, 2-benzenedicarboxylic acid, N-hexadecanoic acid, and diisooctyl ester [11]. The plant was reported to have molluscicidal [12], hypoglycemic, anti-complementary and anti-HIV [13], antimicrobial [14], analgesic and anti-inflammatory [15], antimalarial [16], and cytotoxic [17, 18] activity. In this study, we focused on the isolation and identification of phytochemicals from chloroform extracts of leaves of S. campanulata, and then we evaluated the cytotoxic activity of isolated compounds on HL-60 cell lines.
Methods
Plant material
The leaves of S. campanulata were collected from Nagarjuna School campus, Kautha, Nanded district, Maharashtra, India. The plant was authenticated by a botanist. The voucher specimen was submitted to the department (voucher specimen number: BSI/WRC/100-2/Tech/2017/39).
Preparation of extract
S. campanulata leaves were dried at room temperature and ground to a coarse powder prior to extraction using an electric grinder. One kilogram of powdered leaf material was defatted with n-hexane and then conducted extraction in the Soxhlet apparatus using chloroform. The extract was filtered, concentrated, and dried under vacuum using a rotary evaporator at 40 °C and stored in a refrigerator for further analysis.
Chemicals and reagents
Chloroform, n-hexane, ethyl acetate, methanol, cell lines (HL-60, From NCCS, Pune), cell culture medium, fetal bovine serum, sulforhodamine B, sodium bicarbonate, trichloroacetic acid, acetic acid 10mM unbuffered Tris base solution, dimethyl sulfoxide, and Adriamycin. All required chemicals and reagents were purchased from Merck Chemicals Ltd.
Equipment
FTIR (BRUKER FTIR, ALPHA), ESI-TOF MS, (Q-TOF MICROMASS), 1H NMR (BRUKER AVANCE NEO 500MHz), 13C NMR (BRUKER AVANCE NEO 500MHz), melting point apparatus (METTLER TOLEDO).
Isolation of phytochemicals from S. campanulata chloroform extract by chromatography
In this study, we attempted to isolate compounds from the chloroform extract of S. campanulata. The chloroform extract of leaves (15 g) was subjected to column chromatography on silica gel (60–120 mesh, Merck) eluted using a solvent mixture of increasing order of polarity of chloroform, ethyl acetate, and methanol mixture. By using chloroform (F1-20), chloroform: ethyl acetate (F21-30), and chloroform: ethyl acetate: methanol (F31-50) fractions were collected. Fifty fractions (F1-50) were collected and analyzed on TLC. Fractions with similar TLC patterns were pulled together, and fractions with inseparable compounds were rejected. Crude fractions, i.e., CF1 (F9-19), CF2 (F21-25), and CF3 (F30-34) were identified based on their response to spraying reagent and produced considerable yield. CF1 (F9-19) was processed by column chromatography with chloroform and obtained a compound known as compound 1. Preparative thin layer chromatography of CF2 (F21-25) was performed using silica gel H as adsorbent and chloroform: ethyl acetate (9.5:0.5) as a mobile phase. It was further purified by column chromatography to get compound 2. Compound 3 was recovered from CF3 (F30-34) by column chromatography using chloroform: ethyl acetate: methanol (8.5:1:0.5).
Identification of phytochemicals by spectroscopy
The isolated compounds were identified by using spectral data of Fourier transform infrared (FTIR), ESI-TOF MS, and nuclear magnetic resonance (1H & 13C NMR) spectroscopy and compared with available reference data.
The melting point of the isolated compounds was determined using the Digital Melting Point apparatus. A small amount of samples was filled in the capillary and kept in the apparatus. The melting point of the samples was recorded which was displayed on the screen FTIR spectra of isolated compounds recorded on the BRUKER ALPHA FTIR instrument. Moisture-free samples were directly kept on the sample holder plate and recorded the spectra. 1H NMR and 13C NMR of the isolated compounds were recorded on BRUKER AVANCE NEO 500 MHz NMR spectrometer using Tetramethylsilane (TMS) as internal standard, and all chemical shifts were measured in δ ppm. Mass spectra of the isolated compounds were recorded on WATERS, Q-TOF MICROMASS (ESI-MS) at SAIF, Panjab University, Chandigarh.
Cell growth inhibition assay
In vitro cytotoxic activities of the isolated compounds were carried out by SRB assay against HL-60 cell lines [19, 20]. The SRB assay carried out by the cells were seeded in 96 well-plate at the density of 20,000 cells per well in culture medium and incubated overnight (24 h) at CO2 incubator.
Compounds were solubilized in dimethyl sulfoxide (DMSO) and diluted with the medium to get different concentrations (10, 20, 40, 80, 100 μg/ml) before they were added to the cell culture medium. Plates were incubated for 24 h at 37 °C in a 5% CO2 atmosphere. After the incubation period, cells were fixed by the gentle addition of 50 μl of ice-cold 30% (w/v) TCA (final concentration, 1% TCA) and incubated again for 60 min at 4 °C. The supernatant was discarded, and the plates were washed five times with distilled water and air-dried. Fifty microliters SRB solution was added to each well and incubated at room temperature for 20 min. After the staining process, the residual dye was removed by washing five times with 1% acetic acid, and the unbound dye was recovered. The plates were air-dried. The bound stain was subsequently eluted by using 10 mM trizma base, and the absorbance was read on a plate reader at a wavelength of 510 nm with 690 nm reference wavelength. In this assay, negative control (medium with cells without compounds) and positive control (medium with cells and Adriamycin) were used.
Statistical analysis
Each experiment was conducted in triplicate, and the obtained results were given as mean values. IC50 values were calculated using a mathematical formula and applying statistics [19, 20].
Result
In this study, stigmasta-5, 22-dien-3-ol, octadecenamide, and umbelliferone were isolated from chloroform extract of S. campanulata. All isolated compounds were analyzed by spectroscopic methods (FTIR, ESI-TOF MS, 1H NMR, and 13C NMR) and compared with those reported literature data. The compounds identified were as follows:
Stigmasta-5, 22-dien-3-ol (67 mg): (C29H48O3) white crystalline powder, melting point 160 °C; FTIR spectra showed characteristic band at 3666.18 cm−1, 2933.57 cm−1, 1453.21 cm−1, 1376.23 cm−1, and 1054.57 cm−1. Mass spectra were taken and molecular ion peak observed at 412.25 [M]+ and other ion peak appeared at 409.42, 393.52, 380.21, 301.48, 302.52, 274.65, 254.52, and 149.42. 1H NMR spectra (CDCl3) showed peaks at δ 0.6 (3H, s, CH3-29), 0.7 (3H, s, CH3-18), 0.8 (3H, d, CH3-26, and CH3-27), 3.5(1H, -OH, H-3), and 5.3 (olefinic protons H-6). 13C NMR spectrum showed presence of 29 carbon atoms in the structure. The spectrum showed different ppm, viz, δ 31.67 (C-1), 37.26 (C-2), 71.83 (C-3), 42.23 (C-4), 140.75(C-5), 121.73 (C-6), 31.91 (C-7), 31.91 (C-8), 50.16 (C-9), 36.53 (C-10), 21.10 (C-11), 39.69 (C-12), 42.31 (C-13), 56.88 (C-14), 24.37 (C-15), 28.93 (C-16), 55.96 (C-17), 12.26 (C-18), 19.41 (C-19), 40.50 (C-20), 21.08 (C-21), 138.33 (C-22), 129.20 (C-23), 51.25 (C-24), 31.91 (C-25), 18.99 (C-26), 21.22 (C-27), 21.22 (C-27), 25.42 (C-28), 12.06 (C-29) (Fig. 1).

Spectra of compound 1 a FTIR, b MS, c 1H NMR, d 13C NMR
Octadecenamide (70 mg): (C18H35NO) white, wax like solid, melting point 95 °C. The FTIR spectra showed frequency bands at 3300 cm−1, 2935.41 cm−1, 1690.85 cm−1, 1516.77 cm−1, 1457.50 cm−1, 1375.55 cm−1, and 1053.32 cm−1. Mass spectra showed molecular ion peak at m/z 282.60 [M]+, and other ion peak appeared in the mass spectra at 265.73, 247.63, 223.65, and 209.23. The 1H NMR exhibited signals at δ 0.8 (3H, t, CH3-18), 1.3–1.4 (20H, m, methylene), 1.6 (2H, m, H-3), 2.0 (4H, m, H-8, H-11), 2.2 (2H, CH2CONH2, H-2), and 5.3 (2H, m, H-9, H-10). 13C NMR (CDCl3) showed peaks at different ppm, viz., δ 176.45 (C-1), 35.78 (C-2), 31.74 (C-3), 27.76-27.36 (C-4 to C-7) and (C-12 to C-15), 25.54 (C-8 and C-11), 127.77 (C-9), 127.72 (C-10), 22.63 (C-17), and 14.11 (C-18) (Fig. 2).

Spectra of compound 2 a FTIR, b MS, c 1H NMR, d 13C NMR
Umbelliferone (60 mg): (C9H6O3) white, crystalline solid, melting point 232 °C. FTIR spectra showed frequency band at 3204.27 cm−1, 1683.15 cm−1, 1606.53 cm−1, 1581.25 cm−1, and 831.08 cm−1. The mass spectra showed peak at m/z 162.32 [M]+, 149.42, 134.63, 105.25, and 85.43. 1H NMR spectra showed signals at δ 6.7 (1H, d, H-4), 6.8 (1H, d, H-8), 7.3 (1H, d, H-6), 7.4 (1H, d, H-5), 7.5(1H, d, H-3), and 7.6 (1H, s, at OH). 13CNMR showed the presence of nine carbon atoms in the structure and showed peaks at different ppm, viz., δ 162.40 (C-2), 113.41 (C-3), 153.74 (C-4), 111.30 (C-4a), 125.99 (C-5), 113.42 (C-6), 162.12(C-7), 103.35 (C-8), and 155.03 (C-8a) (Fig. 3).

Spectra of compound 3 a FTIR, b MS, c 1H NMR, d 13C NMR
The IC50 value of isolated compounds (Table 1) was calculated in Microsoft excel by linear regression equation and found to be 44.12 μg/ml (stigmasta-5, 22-dien-3-ol), 35.65μg/ml (octadecenamide), and 80.60μg/ml (umbelliferone).
Discussion
Cancer is one of the most life-threatening and dreadful diseases. It is the second leading cause of death in developed and developing countries and represents a major public health problem [22]. Cancer is a complicated genetic disease defined as uncontrolled growth, angiogenesis, invasion, and metastasis [23].
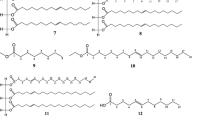
The present study aimed to isolate phytochemicals from chloroform extract by column chromatography and preparative TLC which led to the isolation of three compounds (Fig. 4). The structures of the isolated compounds were determined using spectroscopic methods including FTIR, ESI-TOF MS, 1HNMR, and 13 C NMR.

Chemical structures of the isolated compounds from leaves of S. campanulata
Compound 1 was obtained as a white crystalline solid. Its molecular formula was established to be (C29H48O) from its ESI-TOF MS spectral data. The IR spectrum exhibited frequencies at 3666.18 cm−1 that is characteristic for –OH stretching. The stretching and bending vibration of the methyl part was indicated by an intense peak at 2933.57 cm−1 and the vibration of the methylene part was shown by medium intensity peak at 1453.21 cm−1. The peak at 1376.23 cm−1 is due to CH2(CH3)2. The corresponding C–C vibration was shown as a weak intense peak at 1054.57 cm−1. The 1H-NMR spectrum of compound 1 showed methyl proton peaks at δ 0.6 (3H, CH3-29), 0.7 (3H, CH3-18), and 0.8 (3H, CH3-26, and CH3-27). The peak at δ 3.5 indicates the proton of hydroxyl group attached at the C-3 position (i.e., 1H, −OH on C-3). 1H-NMR spectrum also showed peaks at δ 5.3 indicating the presence of olefinic protons in the structure. The compound was further confirmed by 13C NMR spectroscopy. In 13C NMR spectrum, the peaks at δ 140.75, 121.73, 138.33, and 129.20 are assigned to C-5, C-6, C-22, and C-23 of C=C double bonds, respectively. The peaks at δ 140.75 and 121.73 (C-5 and C-6, respectively) are assigned to C=C double bond carbons in the cyclic structure of the compound. Reports showed that the existence of unsaturation between C-5 and C-6 introduces easily recognizable signals at δ 140.75 and 121.73. The peaks at δ 138.33 and 129.20 are assignable to the external C=C double bond carbon atoms, and the peak at δ 71.83 is associated with the β hydroxyl carbon of C-3. The spectrum of compound 1 also confirmed that the peaks at δ 140.75, δ 36.53, and δ 42.31 indicate quaternary carbon atoms. Thus, by comparing spectral data with literature reports, compound 1 was found to be identical with stigmasta-5, 22-dien-3-ol [24–26].
Compound 2 was obtained as a white solid substance. ESI-TOFMS spectra gave molecular ion peak at m/z at 282.60, compatible with the molecular formula (C18H35NO). The IR spectra of compound 2 revealed the characteristic absorption bands at 3300 cm−1 due to N–H stretching that predicts the presence of amide NH2 group, and amide C=O stretching was observed at 1690.85 cm−1. The stretching and bending vibration of the methyl part was indicated by an intense peak at 2935.41 cm−1, and the vibration of the methylene part was shown by a medium intensity peak at 1457.50 cm−1. The peak at 1375.55 cm−1 is due to C–H bending of CH3. The corresponding C–C vibration was shown as a weak intense peak at 1053.32 cm−1. 1H NMR spectra showed the resonance signals, olefinic protons –CH=CH at δ 5.3 (2H, m), protons α to carbonyl and adjacent to –C–O at 2.2 (2H, t), protons β to carbonyl at δ 1.6 (2H, m), methylene group –CH2- adjacent to –CH=CH at δ 2.0 (4H, m), and others at δ1.3–1.4 (20H, m), terminal –CH3 δ 0.8 (3H). 13C NMR spectra indicated the presence of a total of 20 carbon atoms in the structure and confirmed the presence of –C=O at C-1 (176.45), two olefinic carbons at 127.77, 127.72 for C-9 and C-10 respectively, 8 –CH2 ranging from 27.76 to 27.36, and one –CH3 at 14.11. Based on the spectral data and comparison of the data given in the previous literature, compound 2 was identified as octadecenamide [27].
Compound 3 obtained as yellowish-white, crystalline solid, ESI-TOFMS spectra gave molecular ion peak at m/z at 162.32, compatible with the molecular formula (C9H6O3). FTIR spectra of compound 3 showed bands at 3204.27 cm−1 for the phenolic aromatic ring (Ar–OH) and characteristic band at 1683.15 to 1606.53 cm−1 for lactone ring. The corresponding C=C vibration was shown by peaks at 1581.25 cm−1, and the C–H vibrations of the unsaturated part were observed at 831.08 cm−1. The NMR spectrum showed signals displayed by three protons on the benzene ring at δ 7.4 (1 H, d), 6.8 (1 H, d), and 6.7 (l H, s) probably due to the presence of 1, 2, 4-trisubstituted benzene ring. The NMR spectrum also showed the presence of 1, 2-disubstituted cis-olefin protons at 7.3 (1H, d) and 7.4 (1H, d). The 13C NMR spectral data showed eleven signals. The C-2 of the structure indicative of signs of carbon α-carbonyl, β-unsaturated at δ 162.40 characteristic feature of lactone ring without substituent in the C-3 and C-4 carbon atom, and δ 155.03 relates to =C–O. Based on spectral data and comparison with published data in previous literature, compound 3 was confirmed as umbelliferone [21, 28].
Numerous studies have reported the cytotoxic activity of isolated compounds against different human cancer cell lines, viz., stigmasta-5, 22-dien-3-ol, octadecenamide, and umbelliferone through various mechanisms and pathways. The anticancer activity of stigmasterol against Ehrlich ascites carcinoma [21], breast cancer cells [30], liver cancer cells [31], and Hela cells [32] were reported in the literature. The anticancer activity of umbelliferone against HepG2 hepatocellular carcinoma cells via the induction of apoptosis and cell cycle [33] and human renal cell carcinoma [34] were reported. In previous reports, antibacterial and anticancer activities of 9-octadecenamide (Z) present in biosurfactants from halophilic Halomonas sp. was shown [35]. Antibacterial and anticancer activity of a sediment-derived fungus fraction that contains oleamide was also identified [36].
In the present study, the cytotoxic activity of the isolated compounds was evaluated against HL-60 cell lines, and IC50 values were found to be 44.12 μg/ml (stigmasta-5, 22-dien-3-ol), 35.65 μg/ml (octadecenamide), and 80.60μg/ml (umbelliferone). Among isolated compounds, octadecenamide showed more cytotoxicity but less than Adriamycin against HL-60 cell lines.
Conclusion
In conclusion, the phytochemical study of chloroform extract of leaves of Spathodea campanulata led to the isolation of three compounds, i.e., stigmasta-5, 22-dien-3-ol, octadecenamide, and umbelliferone. The identification of isolated compounds was carried out by using spectroscopic data. All isolated compounds exhibited dose-dependent cytotoxicity against HL-60 cell lines, which was found to be less than positive control Adriamycin. This is the first time that octadecenamide and umbelliferone were isolated from this plant. This traditional plant could be considered as a potential source of bioactive phytochemicals in the drug discovery process.
Availability of data and materials
All data provided in the manuscript are available upon request.
Abbreviations
- S. campanulata :
-
Spathodea campanulata
- SRB:
-
Sulforhodamine B assay
- FTIR:
-
Fourier transform infrared spectroscopy
- 1H NMR:
-
Proton nuclear magnetic resonance
- 13C NMR:
-
Carbon nuclear magnetic resonance
- ESI-TOF MS:
-
Electrospray ionization time-of-flight mass spectrometer
- IC50 :
-
50% maximal inhibitory concentration
- CDCl3 :
-
Deuterated chloroform
- OD:
-
Optical density
References
Aye MM, Aung HT, Sein MM, Armijos C (2019) A review on the phytochemistry, medicinal properties and pharmacological activities of 15 selected Myanmar medicinal plants. Molecules 24:293. https://doi.org/10.3390/molecules24020293
Shankara BR, Ramachandra YL, Rajan SS, Ganapathy PS, Yarla NS, Richard SA, Dhananjaya BL (2016) Evaluating the anticancer potential of ethanolic gall extract of Terminalia chebula (Gaertn.) Retz. (Combretaceae). Pharmacognosy Res 8(3):209. https://doi.org/10.4103/0974-8490.182919
Desai AG, Qazi GN, Ganju RK, El-Tamer M, Singh J, Saxena AK, Bedi YS, Taneja SC, Bhat HK (2008) Medicinal plants and cancer chemoprevention. Curr Drug Metab 9:581–591. https://doi.org/10.2174/138920008785821657
Singh S, Sharma B, Kanwar SS, Kumar A (2016) Lead phytochemicals for anticancer drug development. Front Plant Sci 8:1667. https://doi.org/10.3389/fpls.2016.01667
Joly AB (1985) Introduction to taxonomic vegetation. Ed. National, Sao Paulo
Mensah AY, Houghton PJ, Dickson RA, Fleischer TC, Heinrich M, Bremner P (2016) In vitro evaluation of effects of two Ghanaian plants relevant to wound healing. Phytother Res 11:941–944. https://doi.org/10.1002/ptr.1978
Ngouela S, Nyasse B, Tsamo E, Sondengam BL, Connolly JD (1990) Spathodic acid: a triterpene acid from the stem bark of Spathodea campanulata. Phytochemistry 29:3959–3961
Ngouela S, Tsamo E, Sondengam BL (1988) Extractives from Bignoniaceae: constituents of the stem bark of Spathodea campanulata. Planta Med 54:476–476
Ngouela S, Tsamo E, Sondengam BL, Connolly JD (1991) Spathodol, a new polyhydroxysterol from the leaves of Spathodea campanulata. J Nat Prod 54:873–876
Amusan OG, Msonthi JD, Makhubu LP (1995) Molluscicidal activity of Spathodea campanulata, Andrachneovalis, Phytolacca dodecandra and Hypoxis rooperi. Fitoterapia 66:113–116
Wagh AS, Butle SR (2019) Plant profile, phytochemistry and pharmacology of Spathodea campanulata P. Beauvais (African Tulip Tree): a review. Int J Pharm Pharm Sci 10:1–6. https://doi.org/10.22159/ijpps.2018v10i5.24096
Mendes NM, Souza CP, Araujo N, Pereira JP, Katz N (1986) Molluscicidal activity of some natural products on Biomphalaria glabrata. Mem Inst Oswaldo Cruz 81:87–91. https://doi.org/10.1590/s0074-02761986000100012
Niyonzima G, Laekernan G, Witvrouw M, Van Poel B, Pieters L (1999) Hypoglycemic, anti-complement and anti-HIV activities of Spathodea campanulata stem bark. Phytomedicine 6:45–49. https://doi.org/10.1016/s0944-7113(99)80034-8
Ofori-Kwakye K, Kwapong AA, Adu F (2009) Antimicrobial activity of extracts and topical products of the stem bark of Spathodea campanulata for wound healing. Afr J Tradit Complement Altern Med 6:168–174
Emmanuel EI, Peter AA (2009) Spathodea campanulata: an experimental evaluation of the analgesic and anti-inflammatory properties of a traditional remedy. Asian J Med Sci 1:35–38
Makinde JM, Amusan OO, Adesogan EK (1988) The antimalarial activity of Spathodea campanulata stems bark extract on Plasmodium berghei in mice. Planta Med 54:122–125. https://doi.org/10.1055/s-2006-962367
Victor K, Cedric T, Flora TM, Veronique PB, Thomas E (2016) Cytotoxicity of methanolic extracts of 10 Cameroonian medicinal plants towards multi-factorial drug-resistant cancer cell lines. BMC Complementary Altern Med 16:1–18. https://doi.org/10.1186/s12906-016-1253-3
Wagh AS, Butle SR (2019) Preliminary phytochemical and in vitro anticancer activity of Spathodea campanulata P. Beauv. Asian J Pharm Pharmacol 5:37. https://doi.org/10.31024/ajpp.2019.5.s1.3
Vanicha V, Kanyawim K (2006) Sulforhodamine B colorimetric assay for cytotoxicity screening. Nature Protocols:1112–1116. https://doi.org/10.1038/nprot.2006.179
Skehn P, Storeng R, Scudiero A, Monks J, McMohan D, Vistica D, Jonathan TW, Bokesch H, Kenney S, Boyd MR (1990) New colorimetric cytotoxicity assay for anticancer drug screening. J Natl Cancer Inst:1082–1007. https://doi.org/10.1093/jnci/82.13.1107
Ghosh T, Maity TK, Singh J (2011) Evaluation of antitumor activity of stigmasterol, a constituent isolated from Bacopa monnieri Linn aerial parts against Ehrlich Ascites carcinoma in mice. Orient Pharm Exp Med 11:41–49. https://doi.org/10.4103/0974-8490.1472
Akindele AJ, Wani ZA, Sharma S, Mahajan G, Satti NK, Adeyemi OO, Mondhe DM, Saxena AK (2015) In vitro and in vivo anticancer activity of root extracts of Sansevieria liberica Gerome and Labroy (Agavaceae). Evid Based Complementary Alter Med 201:1–11. https://doi.org/10.1155/2015/560404
Sen A, OzbasTuran S, Bitis L (2017) Bioactivity-guided isolation of anti-proliferative compounds from endemic Centaurea kilaea. Pharm Biol 55:541–546. https://doi.org/10.1080/13880209.2016.1255980
Tripti M, Rakesh KA, Sanjeev M, Pushpa J, Mahesh P, Baleshwar M, Upreti DK, Rana TS, Dipak D (2016) Isolation, characterization and anticancer potential of cytotoxic triterpenes from Betula utilis Bark. PLoS One 1:1–14. https://doi.org/10.1371/journal.pone.0159430
Rajput AP, Rajput TA (2012) Isolation of β-sitosterol and stigmasterol from chloroform extract of leaves of Corchorus fascularis Lam. Int J Biol Chem 6:130–135. https://doi.org/10.3923/ijbc.2012.130.135
Edilu A, Adane L, Woyessa D (2015) In vitro antibacterial activities of compounds isolated from roots of Caylusea abyssinica. Ann Clin Microbiol Antimicrob 14:15. https://doi.org/10.1186/s12941-015-0072-6
Cheng MC, Ker YB, Yu TH, Lin LY, Peng RY, Peng CH (2010) Chemical synthesis of 9 (Z)-octadecenamide and its hypolipidemic effect: a bioactive agent found in the essential oil of mountain celery seeds. J Agric Food Chem 58:1502–1508. https://doi.org/10.1021/jf903573g
Kudumela RG, Mazimba O, Masoko P (2019) Isolation and characterization of sesquiterpene lactones from Schkuhria pinnata and their antibacterial and anti-inflammatory activities. S Afr J Bot 126:340–344. https://doi.org/10.1016/j.sajb.2019.04.002
Ofentse M (2017) Umbelliferone: source, chemistry and bioactive review. Bull Fac Pharm Cairo Univ 55:223–232. https://doi.org/10.1016/j.bfopcu.2017.05.001
Sugappriya M, Sudarsanam D, Bhaskaran R (2017) Druggability and binding site interaction studies of potential metabolites isolated from marine sponge Aurora globostellata against human epidermal growth factor receptor-2. Bioinformation 3:261. https://doi.org/10.6026/97320630013261
Atheer Abbas YF (2019) Stigmasterol inhibits proliferation of cancer cells via apoptotic genes. Biosci Res 16:695–702
Karnati R, Bhaskara T, Sharma GV, Murali Krishna R (2018) Anti-cancer activity of stigmasterol of Xylocarpus granatum in cytotoxicity studies using Hela and MCF-7 cells. OMCIJ 7:555719. https://doi.org/10.19080/OMCIJ.2018.07.555719
Yu SM, Hu DH, Zhang JJ (2015) Umbelliferone exhibits anticancer activity via the induction of apoptosis and cell cycle arrest in HepG2 hepatocellular carcinoma cells. Mole Med Rep 12:3869–3873. https://doi.org/10.3892/mmr.2015.3797
Wang X, Huang S, Xin X, Ren Y, Weng G, Wang P (2019) The antitumor activity of umbelliferone in human renal cell carcinoma via regulation of the p110γ catalytic subunit of PI3Kγ. Acta Pharm 69:111–119. https://doi.org/10.2478/acph-2019-0004
Donio MB, Ronica FA, Viji VT, Velmurugan S, Jenifer JS, Michaelbabu M, Dhar P, Citarasu T (2013) Halomonas sp. BS4, a biosurfactant producing halophilic bacterium isolated from solar salt works in India and their biomedical importance. Springer Plus 2:149. https://doi.org/10.1186/2193-1801-2-149
Farha AK, Hatha AM (2019) Bioprospecting potential and secondary metabolite profile of a novel sediment-derived fungus Penicillium sp. ArCSPf from continental slope of Eastern Arabian Sea. Mycology 10:109–117. https://doi.org/10.1080/21501203.2019.1572034
Acknowledgements
Authors would like to thank Director, School of Pharmacy, S R T M University Nanded, for providing necessary facilities for this research and Director, SAIF, Panjab University, Chandigarh, for spectral analysis.
Plant authentication
The plant was authenticated by Dr. Priyanka Ingale, Botanist, Botanical Survey of India, Pune. The voucher specimen was submitted to the department. The voucher specimen number is BSI/WRC/100-2/Tech/2017/39.
Funding
No funding was received for this research
Author information
Authors and Affiliations
Contributions
SB and AW were involved in conceptualization, design of experiment, and supervision; AW carried out the experiment, and SB, AW, and DR analyzed data and did structural elucidation. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The authors declare that the work did not involve humans or animals which required ethical approval or consent to participate.
Consent for publication
Not applicable.
Competing interests
The authors declare that there are no conflicts of interest regarding the publication of this manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wagh, A., Butle, S. & Raut, D. Isolation, identification, and cytotoxicity evaluation of phytochemicals from chloroform extract of Spathodea campanulata. Futur J Pharm Sci 7, 58 (2021). https://doi.org/10.1186/s43094-021-00205-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43094-021-00205-7