
Abstract
Background
Chronic myeloid leukemia (CML) is a myeloproliferative disorder characterized by the expression of the BCR-ABL1 fusion gene. Tyrosine kinase inhibitors (TKI) are used to treat CML, but mutations in the tyrosine kinase domain contribute to CML chemo-resistance. Therefore, finding alternative molecular-targeted therapy is important for the comprehensive treatment of CML. MicroRNAs (miRNA) are small non-coding regulatory RNAs which suppress the expression of their target genes by binding to the 3′ untranslated region (3′UTR) of the target mRNA. Hypothetically, the miRNA-mRNA interaction would suppress BCR-ABL1 expression and consequently reduce and inhibit CML cell proliferation. Thus, our objective was to determine the target interaction of human and plant miRNAs targeting the 3′UTR region of BCR-ABL1 in terms of miRNA binding conformity, protein interaction network, and pathways using in silico analysis. The 3′UTR sequence of BCR-ABL1 is obtained from Ensembl Genome Browser while the binding conformity was determined using the PsRNATarget Analysis Server, RNA22, Target Rank Server, and DIANA TOOLS. Protein-protein interaction network and pathway analysis are determined using STRING, Cytoscape, and KEGG pathway analysis.
Results
Five plants and five human miRNAs show strong binding conformity with 3′UTR of BCR-ABL1. The strongest binding conformity was shown by Oryza sativa’s Osa-miR1858a and osa-miR1858b with −24.4 kcal/mol folding energy and a p value of 0.0077. Meanwhile, in human miRNA, the hsa-miR-891a-3p shows the highest miTG score of 0.99 with −12 kcal/mol folding energy and a p value of 0.037. Apart from ABL1, osa-miR1858a/osa-miR1858b and hsa-miR891a-3p also target other 720 and 645 genes, respectively. The interaction network of Osa-miR1858a/osa-miR1858b and hsa-miR891a-3p identifies nineteen and twelve ABL1’s immediate neighboring proteins, respectively. The pathways analysis focuses on the RAS, MAPK, CML, and hematopoietic cell lineage pathway.
Conclusion
Both plant and human miRNAs tested in this study could be a potential therapeutic prospect in CML treatment, but thermodynamically, osa-miR1858a/osa-miR1858b binding to ABL1 is more favorable. However, it is important to carry out more research in vitro and in vivo and clinical studies to assess its efficacy as a targeted therapy for CML.
Graphical abstract

Similar content being viewed by others

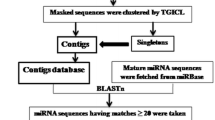
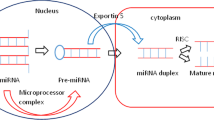
Background
Chronic myeloid leukemia (CML) is a type of cancer that affects blood and bone marrow cells. It is a clonal disorder of multipotential hematopoietic stem cells that excessively induces proliferation of immature and mature myeloid cells [1]. CML patients are characterized by the accumulation of Philadelphia chromosome-positive (Ph+) myeloid cells. Ph chromosome is the result of a reciprocal translocation between Abelson (ABL) proto-oncogene on chromosome 9 and the breakpoint cluster area (BCR) on chromosome 22, t(9;22) (q34;q11) [2, 3]. This translocation generates the BCR-ABL1 fusion gene, which is the key cause of CML by the development of highly active tyrosine kinase [4]. As the frontline treatment of CML, TKIs are used against the kinase activity of the BCR-ABL protein. However, because TKI resistance in some CML patients are due to tyrosine kinase domain (TKD) mutations, alternative treatments targeting other than TKD are needed [5,6,7]. Hence, miRNA is proposed as an alternative treatment for CML. This is due to the association between miRNA expression and the onset of TKI resistance [4]. MicroRNAs are small non-coding regulatory RNAs which suppress the expression of their target genes. It reduces the stability of messenger RNA (mRNA) and thus inhibits translation [8]. miRNA comprised 20 to 24 nucleotides that are involved in gene regulation and cell signaling. In cancer cells, miRNAs can modulate multiple dysregulated genes or signaling pathways. This is because gene regulation is controlled by the incomplete complementary binding between the seed sequence of miRNA with the 3′ untranslated region (3′UTR) of mRNA. This enables miRNA to simultaneously control the expression of many target genes [9]. Certain miRNAs like hsa-miR-21 and hsa-miR-17-92 clusters are elevated in cancer cells. Meanwhile, other cancer-associated miRNA families such as the hsa-let-7 and hsa-miR-34 with hsa-miR-34 are downregulated [10, 11]. These outcomes suggested that miRNAs play a very important role in promoting or preventing cancer. miRNAs can be manipulated as cancer therapeutic agents using miRNA restoration agents known as miRNA mimics. These mimics are used to compensate for the loss of some miRNA [12, 13]. Meanwhile, another approach is by using the miRNA-inhibiting agents, the antagomiR which functions in blocking the aberrantly overexpressed miRNA. Despite the controversy of cross-kingdom inhibition of cancer cells by plant miRNA, there is a report which indicated that synthetic mimic of plant miRNAs of ath-miR159a (from Arabidopsis thaliana), gma-miR159a-3p, and gma-miR159e-3p (from Glycine max) was able to inhibit the proliferation of breast cancer cells [14]. Meanwhile, another study confirmed the ability of mol-miR168 (from Moringa oleifera) to downregulate the protein expression of SIRT1, when its mimic was transfected into the human hepatoma cell line G2 (HEPG2) cells [15]. Therefore, to understand the RNA silencing mechanism, this study utilized in silico analysis in predicting the miRNA-mRNA interactions. The current study aimed to predict miRNAs that targeted the 3′UTR of the BCR-ABL1 gene and compare their binding affinity, target genes annotation, pathways involved, and protein-protein interaction in relation to CML pathogenesis.
Method
Sequence of BCR-ABL1 and miRNAs
The 3′UTR of BCR-ABL1 gene sequence was obtained from the Ensembl Genome Browser (https://www.ensembl.org) [16]. miRNA sequences of the plants were obtained from the Plant Small RNA Target (PsRNATarget) Database (http:// plantgrn.noble.org/psRNATarget/) [17]. Human miRNA sequences were obtained from miRbase (http://www.mirbase.org/) [18].
Predicting miRNA-mRNA target interaction in plants
The PsRNATarget and Schema V2 criteria were used in predicting the interaction. The 3′UTR of the BCR-ABL1 sequence was imported into the software, and miRNAs were selected based on expectation equal to or less than 5. Subsequently, RNA22 (https://cm.jefferson.edu/rna22/Interactive/) was used to determine miRNA folding energy, p value, and heteroduplex structure by inserting miRNA sequences and single target sequence [19]. Next, miRNA sequences were inserted into Target Rank (http://genes.mit.edu/targetrank/) to obtain the list of target genes [20].
Predicting miRNA-mRNA target interaction in human
Screening of human miRNAs targeting 3′UTR of ABL1 was performed using DIANA TOOLS, with the microT-CDS [21] and miTG scores being set at more than 0.9. Target genes of each selected miRNA were also identified using microT-CDS with the threshold set at 0.7. Predicted miRNAs were later assessed based on their folding energy, p value, and predicted target site using the RNA22 software.
Protein-protein interaction (PPI) network
The interactions between genes could be analyzed quickly by constructing the protein interaction network. STRING which is an online website (http://string-db.org) was used to build the interactive networks of target genes. By restricting the condition to “human species,” networks were constructed, and the “TSV” format file of the gene interaction relationship was downloaded, and then the file is imported into the Cytoscape software (http://www.cytoscape.org/) for further analysis and clustering [22]. The Cytoscape MCODE plug-in was employed to perform module analysis of the target network and protein clustering. The module selection criteria were as follows: degree cutoff ≥ 2, node score cutoff ≥ 0.2, K-score ≥ 2, and max depth = 100. The first neighbors of selected nodes used the “directed: outgoing” feature to further analyzed the neighboring protein of ABL1.
Gene Ontology and pathway enrichment analysis
Selected genes from Cytoscape networks were further analyzed in STRING using the Cytoscape plug-in for their pathway enrichment of the nodes. The roles of these genes in the metabolic pathway were elucidated based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway.
Results
Plant miRNA-mRNA target interaction
Five plant miRNAs, namely cpa-miR8154, osa-miR1858a, osa-miR1858b, mtr-miR2654, and sof-miR159e, were predicted to show binding conformation with 3′UTR of BCR-ABL1. The minimum folding energy (MFE) levels needed for the binding of each miRNA to the 3′UTR of BCR-ABL1 were identified. The best MFE score was observed in osa-miR1858a and osa-miR1858b at −24.4 kcal/mol whereas the lowest MFE score was shown by sof-miR159e with −14.4 kcal/mol which is still considered as a strong and energetic binding affinity. The results are summarized in Table 1.

Human miRNA-mRNA target interaction
DIANA TOOLS predicted five miRNAs which are the hsa-miR891a-3p, hsa-miR3131, hsa-miR6847-5p, hsa-miR1185-1-3p, and hsa-miR1185-2-3p that interacted with ABL1. Hsa-miR891a-3p showed the highest miTG score of 0.99 with −12 kcal/mol folding energy and a p value of 0.037 (Table 1).
Target gene annotation
Osa-miR1858a and osa-miR1858b were found to have 720 target genes while hsa-miR891a-3p had 645 target genes apart from ABL1 (Table 1). All these genes were imported into STRING analysis for the protein-protein interaction and pathway analysis.
Protein interaction networks, key candidate targets, and pathways analysis
The results showed that the protein network of hsa-miR891a-3p produced 550 nodes and 1550 edges (Fig. 1a). The first neighbors of selected nodes of the ABL1 module were then chosen for further analysis. This module had 12 nodes and 16 edges (Fig. 1b). Pathway enrichment analysis revealed that these nodes were primarily enriched in pathways such as RAS and MAPK signaling pathways. In addition, the encoded proteins were involved mainly in multiple cancer pathways specific to various signaling pathways, including the ErbB signaling pathway (hsa04012), CML (hsa05220), and microRNAs in cancer pathways (hsa05206). The genes that were involved in the pathways were NTRK2, RIN1, NRAS, PPM1B, MAPT, and ABL1. The Gene Ontology of biological processes is emphasized on the cell population proliferation, regulation of apoptotic process, and MAPK cascade (Table 2). Meanwhile, Osa-miR1858a and osa-miR1858b protein networks showed 588 nodes and 1925 edges (Fig. 2a) from 721 genes. The first neighbors of selected nodes of ABL1 displayed a module of 19 nodes and 39 edges (Fig. 2b). The pathway analysis exhibited an enriched pathway of hematopoietic cell lineage and CML. Furthermore, these nodes were also involved with pathways such as ErbB, RAS (hsa04014), Jak-Stat (hsa04630), and Wnt signaling pathways (hsa04310). The genes that were included in the pathways are CDL, STAT5B, BCR, SHC4, ABL1, RAC2, CCND2, PDGFB, XIAP, PAK2, CD5, and TFRC. Moreover, Gene Ontology of biological process focused on the cell population proliferation, hemopoiesis, regulation of apoptotic process, and regulation of MAPK cascade.

Protein interaction network construction and module analysis of identified genes from hsa-miR891a-3p. a The protein interaction network of 646 identified genes; the color depth of the gene deepens with the “degree” of the node, and the size of the node and edges increases with the “degree.” b The first neighbors of selected nodes (ABL1) outgoing module in the network, including 12 nodes and 16 edges; edge line intensity increased as higher interaction. The module function is closely related to the regulation of the apoptotic process, positive regulation of cell population proliferation, and MAPK signaling cascade

Protein interaction network construction and module analysis of identified genes from osa-miR1858a and osa-miR1858b. a The protein interaction network of 721 identified genes; the color depth of the gene deepens with the “degree” of the node, and the size of the node and edges increases with the “degree.” b The first neighbors of selected nodes (ABL1) outgoing module in the network, including 19 nodes and 39 edges; the module function is closely related to the hemopoiesis, regulation of apoptotic process, regulation of cell population proliferation, and regulation of MAPK cascade
Discussion
CML is a myeloid clonal proliferation characterized by the translocation of the BCR-ABL1 gene in the Philadelphia chromosome. TKI is preferred as the frontline treatment to treat CML. However, in approximately 30% of CML patients, TKI resistance has become a challenging problem [23]. Therefore, finding the molecular targets for CML treatment is of primary importance to solve this problem. Thus, the current research aimed to determine and evaluate miRNAs from plants and humans that targeted the 3′UTR of the BCR-ABL1 gene in CML in terms of folding energy, gene annotation, and gene enrichment analysis. It is known that miRNA expression is often altered in diseases, especially in cancers. Either acting as an oncogenic or a tumor-suppressive agent, miRNA had gained the researcher’s interest to be used as an alternative therapeutic agent to enhance the current treatment for cancer. Furthermore, cross-kingdom studies of miRNAs between plant and animal kingdoms are intensely explored in recent days. Therefore, this research employed the in silico analysis to predict the specific plant and human miRNAs that can target 3′UTR of the human BCR-ABL1 gene in CML. This is because a cross-kingdom interaction is possible due to the presence of various conserved regions in the human and plant genome [14, 24]. In addition, due to the highly conserved region in the composition of miRNAs processing machinery, the biogenesis and working mechanism of miRNAs in plants showed a similar molecular and development roles as in humans [25]. Besides, it was found that sequences of diverse endogenous plant miRNAs were complementary to the components of the genomes and transcriptomes of humans and animals [26].
The present study predicted plant miRNAs by using the psRNATarget. psRNATarget is a platform that was developed to identify plant small RNA (sRNA) targets by analyzing complementary matching between sRNA and target mRNA sequences. It employed a predefined grading schema that evaluated the target site accessibility between miRNA and mRNA [27]. The analysis was continued by exporting miRNA sequences into another platform including RNA22 and Target Rank for further validation. These platforms were specifically designed to predict the interaction between miRNA and mRNA in humans. Positive results from RNA22 and Target Rank indicated that the selected plant miRNAs were capable of binding with 3′UTR of BCR-ABL1 in humans. From the in silico analysis, the current study predicted five plants and five human miRNAs that have binding conformity with 3′UTR of the BCR-ABL1 gene. These miRNAs were selected based on their folding energy, p value, psRNATarget score, and miTG score. For plant miRNAs, osa-mir1858a and osa-mir1858b showed the most promising results. They have the lowest folding energy of −24.4 kcal/mol and a significant p value of 0.007. Thus, the binding between osa-miR1858a and osa-miR1858b and 3′UTR of BCR-ABL1 was much favorable. Nevertheless, in human miRNAs, it was observed that hsa-miR891a-3p showed the highest miTG score of 0.99 with −12kcal/mol folding energy and a p value of 0.037. Therefore, binding of hsa-miR891a-3p towards 3′UTR of BCR-ABL1 was less favorable compared to osa-miR1858a and osa-miR1858b towards 3′UTR of BCR-ABL1. A miRNA can have multiple targets, and one mRNA can be targeted by many miRNAs [28]. This is observed in Table 1, showing that hsa-miR891a-3p, osa-miR1858a, and osa-miR1858b have numerous target genes despite ABL1. miRNAs can have multiple targets mainly due to the partial complementary between miRNA and its target mRNA which is known as the seed pairing [29, 30]. Because of this feature, in silico tools can predict thousands of miRNA-regulated genes. Recently, studies have found that hsa-miR-891a and hsa-miR-891b can be used as prognostic biomarker and therapeutic target in different types of cancer such as lung cancer [31], breast cancer [32], colorectal cancer [33], and gastrointestinal stromal tumor [34]. In concordance with the present study, hsa-miR891a-3p, osa-miR1858a, and osa-miR1858b were also predicted to have another 645 and 720 targets genes, respectively. Thus, it is important to analyze all the target genes of hsa-miR891a-3p, osa-miR1858a, and osa-miR1858b. Further analysis on the protein-protein interaction network and KEGG pathway showed that hsa-miR891a-3p, osa-miR1858a, and osa-miR1858b were all involved in the MAPK signaling pathway. The MAPK signaling pathway is very critical for cell existence, and its signaling cascade plays a central role in the pathogenesis of CML [35]. In addition, fundamental cellular processes such as cell proliferation and cell survival are regulated by this pathway [36, 37]. Moreover, the RAS/MAPK signaling can also lead to the survival of positive BCR-ABL cells under imatinib pressure [38]. Previous findings suggested that apoptosis could be triggered by the inhibition of MAPK signaling in the K562 cell line, and the use of MAPK-specific inhibitors showed cooperative effects with imatinib in CML cells [39]. Furthermore, among the targeted genes of osa-miR1858a and osa-miR1858b, the STAT5 gene was one of the important targets. This is because STAT5 promoted imatinib resistance by significantly being involved in the activation of hematopoietic cells transformation by p210Bcr/Abl [40]. The activation of this gene would speed up cell division and inhibit DNA repair, which thus causes genomic instability such as the blast crisis of CML [40, 41]. In addition, the current Gene Ontology analysis also showed that STAT5, CCND2, and PDGFB were involved in the JAK/STAT pathway. This finding is supported by a previous study which indicated that STAT5 could regulate the transcription factor and modulate cytokine receptors and growth factors [42]. Therefore, this pathway is crucial in mediating cytokine signaling by promoting the growth of leukemic cells [40]. According to the KEGG pathway enrichment analysis of osa-miR1858a and osa-miR1858b, the pathways are concentrated on the hematopoietic cell lineage and CML pathway. miRNAs are important regulators involved in many cell functions including the hematopoietic cell lineages. It has been reported that the miRNA expression is regulated during hematopoietic differentiation, suggesting miRNA involvement in the process [43]. Thirteen miRNAs with differential expression in peripheral blood cells have been documented in a previous study, and these hematopoietic-specific miRNAs are essential for blood cells to function properly. In addition, another study also observed that the differently expressed miRNAs allowed the cell-specific transgene expression in human hematopoietic cell lineages [44]. Apart from that, osa-miR1858a and osa-miR1858b additionally involved in the ErbB, Jak-Stat, and Wnt pathways. The Wnt pathway plays an important function during the growth of normal hematopoietic stem cells (HSC) and leukemic stem cells (LSC). Activation of the Wnt/beta-catenin pathway plays a pivotal part in maintaining the self-renewal potential in HSCs. In CML, the BCR-ABL1 oncogene stimulates the β-catenin within the blast stage thus encouraging the LSC reestablishment and repressing imatinib’s activity [45]. In addition, another study demonstrated that the Wnt-3a pathway could promote the proliferation and survival of acute lymphoid leukemia cells [46]. Nevertheless, target gene annotation results observed that osa-miR1858a and osa-miR1858b targeted both the BCR and ABL1 genes which perhaps might help in further reduction of CML cell proliferation. Therefore, the determination of miRNA’s target pathways and genes that are involved in imatinib resistance could be a crucial data that might perhaps be manipulated as alternative treatments for CML therapy.
Conclusion
In summary, Osa-miR1858a/Osa-miR1858b showed the strongest binding conformity to 3′UTR of BCR-ABL1 with −24.4 kcal/mol folding energy. Nevertheless, hsa-miR891a-3p was chosen based on an excellent miTG score of 0.99 with −12 kcal/mol folding energy. The interaction network of Osa-miR1858a/osa-miR1858b and hsa-miR891a-3p identified 19 and 12 ABL1’s immediate neighboring protein, respectively. These proteins were involved in the RAS, MAPK, CML, and hematopoietic cell lineage pathway. miRNA application as a biomarker is being widely used. Therefore, the determination of plants and human miRNA determination using in silico analysis that could be used as the molecular targeted therapy for CML was our main objective in the current study. By using in silico analysis, this study successfully predicted miRNAs from plants and humans that were able to bind with 3′UTR of BCR-ABL1. Theoretically, these miRNAs are capable of suppressing BCR-ABL1 expression, thus reducing CML cell proliferation. Therefore, it is predicted that these miRNAs could inhibit the BCR-ABL1 by binding to the 3′UTR and would be the best potential molecular targeted therapy for CML. In silico study is an informative and powerful tool in helping the researcher to form an estimation of miRNA-mRNA target interaction. For future study, in vitro work could be carried out using CML cells to validate the Osa-miR1858a/osa-miR1858b and hsa-miR891a-3p capability in inhibiting CML cell proliferation by targeting 3′UTR of the BCR-ABL1 gene.
Availability of data and materials
All data analyzed during this study are included in this article.
Abbreviations
- 3′UTR:
-
3′ untranslated region
- CML:
-
Chronic myeloid leukemia
- HSc:
-
Hematopoietic stem cells
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- LSC:
-
Leukemic stem cells
- MAPK:
-
Mitogen-activated protein kinase
- miRNA:
-
MicroRNA
- mRNA:
-
Messenger RNA
- sRNA:
-
Small RNA
- TKD:
-
Tyrosine kinase domain
- TKI:
-
Tyrosine kinase inhibitor
References
Honda H, Ushijima T, Wakazono K, Oda H, Tanaka Y, Aizawa S, Ishikawa T, Yazaki Y, Hirai H (2000) Acquired loss of p53 induces blastic transformation in p210(bcr/abl)-expressing hematopoietic cells: a transgenic study for blast crisis of human CML. Blood. 95(4):1144–1150. https://doi.org/10.1182/blood.V95.4.1144.004k04_1144_1150
Cilloni D, Saglio G (2012) Molecular pathways: BCR-ABL. Clin Cancer Res. 18(4):930–937. https://doi.org/10.1158/1078-0432.CCR-10-1613
Zamecnikova A (2010) Targeting the BCR-ABL tyrosine kinase in chronic myeloid leukemia as a model of rational drug design in cancer. Expert Rev Hematol. 3(1):45–56. https://doi.org/10.1586/ehm.09.66
Di Stefano C, Mirone G, Perna S, Marfe G (2016) The roles of microRNAs in the pathogenesis and drug resistance of chronic myelogenous leukemia. Oncol Rep. 35(2):614–624. https://doi.org/10.3892/or.2015.4456
Elias MH, Baba AA, Husin A, Abdullah AD, Hassan R, Sim GA, Wahid SF, Ankathil R (2012) Contribution of BCR-ABL kinase domain mutations to imatinib mesylate resistance in Philadelphia chromosome positive Malaysian chronic myeloid leukemia patients. Hematol Rep. 4(4):e23. https://doi.org/10.4081/hr.2012.e23
Walz C, Sattler M (2006) Novel targeted therapies to overcome imatinib mesylate resistance in chronic myeloid leukemia (CML). Crit Rev Oncol Hematol. 57(2):145–164. https://doi.org/10.1016/j.critrevonc.2005.06.007
Yap E, Tumian NR, Azma RZ, Sharifah NA, Salwati S, Hamidah NH, Elias MH, Wong CL (2017) Primary imatinib resistance in chronic myeloid leukemia patients in a developing country: BCR-ABL kinase domain mutations or BCR-ABL independent mechanisms? Malays J Pathol. 39(2):107–113
To KKW, Fong W, Tong CWS, Wu M, Yan W, Cho WCS (2020) Advances in the discovery of microRNA-based anticancer therapeutics: latest tools and developments. Expert Opin Drug Discov. 15(1):63–83. https://doi.org/10.1080/17460441.2020.1690449
Hosseinahli N, Aghapour M, Duijf PH, Baradaran B (2018) Treating cancer with microRNA replacement therapy: a literature review. J Cell Physiol. 233(8):5574–5588. https://doi.org/10.1002/jcp.26514
Esquela-Kerscher A, Slack FJ (2006) Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 6(4):259–269. https://doi.org/10.1038/nrc1840
He L, He X, Lowe SW, Hannon GJ (2007) microRNAs join the p53 network—another piece in the tumour-suppression puzzle. Nat Rev Cancer. 7(11):819–822. https://doi.org/10.1038/nrc2232
Hanna J, Hossain GS, Kocerha J (2019) The potential for microRNA therapeutics and clinical research. Front Genet. 10:478. https://doi.org/10.3389/fgene.2019.00478
Rupaimoole R, Slack FJ (2017) MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 16(3):203–222. https://doi.org/10.1038/nrd.2016.246
Chin AR, Fong MY, Somlo G, Wu J, Swiderski P, Wu X, Wang SE (2016) Cross-kingdom inhibition of breast cancer growth by plant miR159. Cell Res. 26(2):217–228. https://doi.org/10.1038/cr.2016.13
Pirrò S, Minutolo A, Galgani A, Potestà M, Colizzi V, Montesano C (2016) Bioinformatics prediction and experimental validation of microRNAs involved in cross-kingdom interaction. J Comput Biol. 23(12):976–989. https://doi.org/10.1089/cmb.2016.0059
Newman V, Moore B, Sparrow H, Perry E (2018) The Ensembl Genome Browser: strategies for accessing eukaryotic genome data. Methods Mol Biol. 1757:115–139
Dai X, Zhuang Z, Zhao PX (2018) psRNATarget: a plant small RNA target analysis server (2017 release). Nucleic Acids Res. 46(W1):W49–W54. https://doi.org/10.1093/nar/gky316
Kozomara A, Griffiths-Jones S (2011) miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 39(Database issue):D152–D157. https://doi.org/10.1093/nar/gkq1027
Loher P, Rigoutsos I (2012) Interactive exploration of RNA22 microRNA target predictions. Bioinformatics. 28(24):3322–3323. https://doi.org/10.1093/bioinformatics/bts615
Nielsen CB, Shomron N, Sandberg R, Hornstein E, Kitzman J, Burge CB (2007) Determinants of targeting by endogenous and exogenous microRNAs and siRNAs. RNA. 13(11):1894–1910. https://doi.org/10.1261/rna.768207
Paraskevopoulou MD, Georgakilas G, Kostoulas N, Vlachos IS, Vergoulis T, Reczko M, Filippidis C, Dalamagas T, Hatzigeorgiou AG (2013) DIANA-microT web server v5. 0: service integration into miRNA functional analysis workflows. Nucleic acids Res. 41(W1):W169–WW73. https://doi.org/10.1093/nar/gkt393
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13(11):2498–2504. https://doi.org/10.1101/gr.1239303
Bhutra S, Lenkala D, LaCroix B, Ye M, Huang RS (2014) Identifying and validating a combined mRNA and microRNA signature in response to imatinib treatment in a chronic myeloid leukemia cell line. PLoS One. 9(12):e115003. https://doi.org/10.1371/journal.pone.0115003
Lee C-T, Risom T, Strauss WM (2007) Evolutionary conservation of microRNA regulatory circuits: an examination of microRNA gene complexity and conserved microRNA-target interactions through metazoan phylogeny. DNA Cell Biol. 26(4):209–218. https://doi.org/10.1089/dna.2006.0545
Chen X (2012) Small RNAs in development - insights from plants. Curr Opin Genet Dev. 22(4):361–367. https://doi.org/10.1016/j.gde.2012.04.004
Ivashuta SI, Petrick JS, Heisel SE, Zhang Y, Guo L, Reynolds TL, Rice JF, Allen E, Roberts JK (2009) Endogenous small RNAs in grain: semi-quantification and sequence homology to human and animal genes. Food Chem Toxicol. 47(2):353–360. https://doi.org/10.1016/j.fct.2008.11.025
Elias MH, Nordin N, Abdul HN (2020) In silico study of potential cross-kingdom plant microRNA based regulation in chronic myeloid leukemia. Curr Pharmacogenomics Personalized Med. 2020;17(2):125-132. https://doi.org/10.2174/1875692118666200106113610
Li Y, Wang H, Tao K, Xiao Q, Huang Z, Zhong L, Cao W, Wen J, Feng W (2013) miR-29b suppresses CML cell proliferation and induces apoptosis via regulation of BCR/ABL1 protein. Exp Cell Res. 319(8):1094–1101. https://doi.org/10.1016/j.yexcr.2013.02.002
Awan HM, Shah A, Rashid F, Wei S, Chen L, Shan G (2018) Comparing two approaches of miR-34a target identification, biotinylated-miRNA pulldown vs miRNA overexpression. RNA Biol. 15(1):55–61. https://doi.org/10.1080/15476286.2017.1391441
Subramanian M, Li XL, Hara T, Lal A (2015) A biochemical approach to identify direct microRNA targets. Methods Mol Biol. 1206:29–37. https://doi.org/10.1007/978-1-4939-1369-5_3
Lee HY, Han SS, Rhee H, Park JH, Lee JS, Oh YM et al (2015) Differential expression of microRNAs and their target genes in non-small-cell lung cancer. Mol Med Rep. 11(3):2034–2040. https://doi.org/10.3892/mmr.2014.2890
Zhang Z, Xu L, He L, Wang J, Shi X, Li Z, Shi S, Hou K, Teng Y, Qu X (2020) MiR-891a-5p as a prognostic marker and therapeutic target for hormone receptor-positive breast cancer. J Cancer. 11(13):3771–3782. https://doi.org/10.7150/jca.40750
Qian J, Zeng L, Jiang X, Zhang Z, Luo X (2019) Novel multiple miRNA-based signatures for predicting overall survival and recurrence-free survival of colorectal cancer patients. Med Sci Monit. 25:7258–7271. https://doi.org/10.12659/MSM.916948
Tong HX, Zhou YH, Hou YY, Zhang Y, Huang Y, Xie B, Wang JY, Jiang Q, He JY, Shao YB, Han WM, Tan RY, Zhu J, Lu WQ (2015) Expression profile of microRNAs in gastrointestinal stromal tumors revealed by high throughput quantitative RT-PCR microarray. World J Gastroenterol. 21(19):5843–5855. https://doi.org/10.3748/wjg.v21.i19.5843
Chakraborty C, Sharma AR, Patra BC, Bhattacharya M, Sharma G, Lee SS (2016) MicroRNAs mediated regulation of MAPK signaling pathways in chronic myeloid leukemia. Oncotarget. 7(27):42683–42697. https://doi.org/10.18632/oncotarget.7977
Kim EK, Choi EJ (2010) Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 1802(4):396–405. https://doi.org/10.1016/j.bbadis.2009.12.009
Torii S, Yamamoto T, Tsuchiya Y, Nishida E (2006) ERK MAP kinase in G cell cycle progression and cancer. Cancer Sci 97(8):697–702. https://doi.org/10.1111/j.1349-7006.2006.00244.x
Chu S, Holtz M, Gupta M, Bhatia R (2004) BCR/ABL kinase inhibition by imatinib mesylate enhances MAP kinase activity in chronic myelogenous leukemia CD34+ cells. Blood. 103(8):3167–3174. https://doi.org/10.1182/blood-2003-04-1271
Kang CD, Yoo SD, Hwang BW, Kim KW, Kim DW, Kim CM, Kim SH, Chung BS (2000) The inhibition of ERK/MAPK not the activation of JNK/SAPK is primarily required to induce apoptosis in chronic myelogenous leukemic K562 cells. Leuk Res. 24(6):527–534. https://doi.org/10.1016/S0145-2126(00)00010-2
Sillaber C, Gesbert F, Frank DA, Sattler M, Griffin JD (2000) STAT5 activation contributes to growth and viability in Bcr/Abl-transformed cells. Blood. 95(6):2118–2125. https://doi.org/10.1182/blood.V95.6.2118
Kaymaz BT, Gunel NS, Ceyhan M, Cetintas VB, Ozel B, Yandim MK et al (2015) Revealing genome-wide mRNA and microRNA expression patterns in leukemic cells highlighted “hsa-miR-2278” as a tumor suppressor for regain of chemotherapeutic imatinib response due to targeting STAT5A. Tumour Biol. 36(10):7915–7927. https://doi.org/10.1007/s13277-015-3509-9
Turhan AG (2011) STAT5 as a CML target: STATinib therapies? Blood 117(12):3252–3253
Chen CZ, Li L, Lodish HF, Bartel DP (2004) MicroRNAs modulate hematopoietic lineage differentiation. Science. 303(5654):83–86. https://doi.org/10.1126/science.1091903
Teruel-Montoya R, Kong X, Abraham S, Ma L, Kunapuli SP, Holinstat M, Shaw CA, McKenzie SE, Edelstein LC, Bray PF (2014) MicroRNA expression differences in human hematopoietic cell lineages enable regulated transgene expression. PLoS One. 9(7):e102259. https://doi.org/10.1371/journal.pone.0102259
Ruan Y, Kim HN, Ogana H, Kim Y-M (2020) Wnt Signaling in leukemia and its bone marrow microenvironment. Int J Mol Sci. 21(17):6247. https://doi.org/10.3390/ijms21176247
Khan NI, Bradstock KF, Bendall LJ (2007) Activation of Wnt/β-catenin pathway mediates growth and survival in B-cell progenitor acute lymphoblastic leukaemia. Br J Haematol. 138(3):338–348. https://doi.org/10.1111/j.1365-2141.2007.06667.x
Acknowledgements
The authors would like to thank the Faculty of Medicine and Health Sciences, Universiti Sains Islam Malaysia, for providing the equipment and facilities. We would also like to thank Malaysia Ministry of Higher Education for the Fundamental Research Grant Scheme (FRGS/1/2018/SKK08/USIM/03/1.)
Funding
This study was funded by the Malaysia Ministry of Higher Education via Fundamental Research Grant Scheme (FRGS/1/2018/SKK08/USIM/03/1). The funder has no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
All authors have contributed substantially and equally to the analysis and reporting of this study. SFSM analyzed and interpreted the data regarding the miRNA binding site and was the major contributor in writing the manuscript. MHE performed the plant miRNA analysis and revised and finalized the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mohamad, S.F.S., Elias, M.H. Potential treatment for chronic myeloid leukemia using microRNA: in silico comparison between plants and human microRNAs in targeting BCR-ABL1 gene. Egypt J Med Hum Genet 22, 35 (2021). https://doi.org/10.1186/s43042-021-00156-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-021-00156-x