
Abstract
Background
The knowledge about blood circulating microbiome and its functional relevance in healthy individuals remains limited. An assessment of changes in the circulating microbiome was performed by sequencing peripheral blood mononuclear cells (PBMC) bacterial DNA from goats supplemented or not in early life with rumen liquid transplantation.
Results
Most of the bacterial DNA associated to PBMC was identified predominantly as Proteobacteria (55%) followed by Firmicutes (24%), Bacteroidetes (11%) and Actinobacteria (8%). The predominant genera found in PBMC samples were Pseudomonas, Prevotella, Sphingomonas, Acinetobacter, Corynebacterium and Ruminococcus. Other genera such as Butyrivibrivio, Bifidobacterium, Dorea and Coprococcus were also present in lower proportions. Several species known as blood pathogens or others involved in gut homeostasis such as Faecalibacterium prausnitzii were also identified. However, the PBMC microbiome phylum composition differed from that in the colon of goats (P ≤ 0.001), where Firmicutes was the predominant phylum (83%). Although, rumen liquid administration in early-life altered bacterial community structure and increased Tlr5 expression (P = 0.020) in colon pointing to higher bacterial translocation, less than 8% of OTUs in colon were also observed in PBMCs.
Conclusions
Data suggest that in physiological conditions, PBMC microbiome differs from and is not affected by colon gut microbiota in small ruminants. Although, further studies with larger number of animals and covering other animal tissues are required, results point to a common circulating bacterial profile on mammals being phylum Proteobacteria, and genera Pseudomonas and Prevotella the most abundants. All suggest that PBMC microbiome in healthy ruminants could be implicated in homeostatic condition. This study expands our knowledge about PBMC microbiome contribution to health in farm animals.
Similar content being viewed by others


Introduction
The circulation is a closed system and the blood in healthy organism was first believed to be a sterile environment [1]. However, the principle of the presence of truly sterile blood in healthy individuals has been challenged, as operationally it does not mean that dormant or non-culturable forms of organisms are absent [2]. Over the last few years, an increasing number of bacteria blood culture isolates have been reported. Therefore the concept of blood microbiome has been emerging although is still under debate. Usually, the presence of a blood microbiome has also been associated with a variety of diseases. Indeed, the etiology of diabetes, cardiovascular disease, hematological disorders and cirrhosis has been ascribed to the translocation of bacteria from the intestinal tract, primarily via the intestinal epithelial mucosa [3,4,5,6,7]. Consequently, the gut microbiome was thought to be the main contributor of the blood microbiome, although the origin of these bacteria is still unknown. Moreover, blood microbial DNA (plasma derived, cell free microbial nucleic acids) was recently proposed as a potential tool to discriminate between numerous types of cancer and healthy individuals [8] providing more evidences of particular microbial DNA in relation to health status.
The first evidence of microbial presence in the blood was found nearly 20 years ago by Nikkari et al. [9] who reported that healthy blood specimens can contain bacterial 16S rRNA gene. Detection of 16S rRNA gene does not confirm the presence of viable microbes and external DNA contamination could account for bacterial DNA found in the hematic samples. Potgieter et al. [10], used transmission electron microscopy analysis to show the presence of bacteria internalized in erythrocytes providing further evidences of a blood microbiome. Later, the presence of comparable bacterial phyla in different studies appears to support the existence of a healthy human blood microbiome [2,3,4, 11,12,13,14,15,16]. Then, more research raised a new paradigm, proposing that healthy individuals harbour a rich microbiota in their blood, including known pathogens that can survive in a dormant form in blood and inside red blood cells [17, 18]. However, the distribution of microbial DNA among white blood cells is unknown although could be also relevant in the physiological transport of bacteria in circulation. According to recent studies from our group, symbiotic bacteria Lactobacillus plantarum used as probiotic and found in extraintestinal sites such as blood or milk, was able to survive inside healthy human’s monocytes up to 24 h [19]. Thus, it seems possible that bacteria from different origins may translocate into the systemic circulation, but not being detected through standard culture methods. Currently, the use of recently developed tools to minimize contributions of contaminants to microbial signatures could be applied to blood from healthy individuals to identify the presence of bacterial DNA [20,21,22,23]. Following specific protocols to control contaminants, Paϊssé et al. [12] showed that a diverse microbiota was present in the blood of healthy human individuals. Accordingly, it seems necessary to ascertain whether this is also a trait of other mammals to better understand the different microbiome and their relationships with health in farm animals. In this study, we aimed to evaluate the peripheral blood mononuclear cells (PBMC) bacterial microbiome in healthy goats. In order to promote substantial changes in the microbial diversity in colon content with potential to translocate into blood cells, two groups of ruminants were used. One group was orally inoculated during early life with rumen liquid from adults and the other without inoculation.
Material and methods
All management and experimental procedures involving animals were performed by trained personnel according to the Spanish guidelines (RD 53/2013). Experimental protocols were approved by the Ethical Committee for Animal Research (EEZ-CSIC) regional government (09/03/2017).
Animals and experimental design
Sixteen male goat kids were used in this experiment. All kids were raised with milk-replacer (Sello azul, Lemansa, León, Spain) and randomly distributed into 2 treatments (n = 8): one group was orally inoculated with pooled fresh rumen liquid transplantation (RLT) from 4 adult goats whereas the control group (CTL) did not receive inoculation. Details of the inoculation process were previously published [24]. Briefly, rumen inocula were obtained from donor animals fitted with permanent rumen fistula which were fed a diet based on a 70% of concentrate and 30% of forage. Rumen fluid was daily collected, filtered through two layers of muslin and immediately inoculated to the RLT animals by oral drenching of 2.5 ml/animal during week 1 and 5 ml/animal thereafter. Inoculation was daily repeated from birth until 2.5 months of age when they were fully adapted to a solid diet. Both groups were separated from each other to avoid physical contact and potential microbial transfer. Animals were weaned at the age of 7 weeks and experimental groups remained separated throughout the entire experiment. At 6 months of age, blood samples were taken from the jugular vein, animals were slaughtered and colon content was sampled and snap frozen in liquid nitrogen. Samples of the colon tissue were washed immediately after collection with 0.01 M phosphate-buffered saline (PBS) buffer (pH 6.8). The samples of washed tissue were then transferred to RNA later solution (Qiagen Ltd., West Sussex, UK) and stored at − 80 °C until further analysis.
Gene expression analysis in colonic tissue
Total RNA was extracted from colon tissue. They were homogenized with 0.9 mm stainless steel bead and 1 ml of TRIzol Reagent (Invitrogen) using a bullet Blender homogenizer as previously described [25]. RNA integrity number was measured using Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA). The extracted total RNA was reversed transcribed using M-MLV reverse transcriptase (Thermo) and the yielded cDNA was used as template for real time quantitative PCR (RTqPCR) analysis to evaluate the expression of Tlr2, 4, 5 and 9, β-defensin and peptidoglycan recognition protein 1 (Pglrp1) genes. RTqPCR amplification and detection was performed on optical grade 384-well plates in a ViiA7 Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA, USA) with PerfeCTa qPCR Tough Mix (Quantabio, Beverly, MA, USA). Specific primers at their annealing temperature were used as previously reported [25]. To normalize mRNA expression, β-actin was used as housekeeping gene. The mRNA relative quantification was calculated using the ΔΔCt method. PCR efficiency was always between 90 and 110%, and a negative control was run for each set of primers.
Samples preparation and DNA extraction
Blood samples were collected into EDTA coated vacutainer. Peripheral blood mononuclear cells (PBMC: lymphocytes and monocytes) isolation was performance following ficoll density gradient method based on the principle of differential migration of blood cells through the media during the centrifugation (400×g for 40 min, at room temperature and without break). Cells were preserved in Trizol. The DNA extraction from PBMC was performed to minimize any risk of contamination according to TRIzol reagent protocol (15,596,026.PPS) in a laminar air-flow hood cleaned with 70% ethanol, and UV-irradiated for 20 min before execution of sample processsing. The quantity of extracted DNA was measured by Qubit (Thermofisher Scientific). DNA extracts were stored at − 20 °C until further processing. An empty vial was used as a template-free “negative blank” into which each reagent (new and filtrated) used was added and further processed with the same DNA extraction protocol and amplicon production method as the experimental PBMC samples, and sequenced on the same run to ensure the absence of artifacts such as bacterial DNA contaminants from reagents or nonspecific amplification of eukaryotic DNA.
In order to understand the translocation mechanism in ruminants, colon content was also analyzed. After freeze drying and homogenizing, DNA from colon content was extracted using QIAamp DNA Stool Mini Kit following manufacturer’s protocol and employing the 95 °C heating option. Concentrations of total bacteria in colon content were determined by quantitative PCR as previously described [25]. Primer sets used were as follows: forward GTGSTGCAYGGYTGTCGTCA and reverse ACGTCRTCCMCACCTTCCTC [26].
16S rRNA gene sequencing and data processing
Microbial community composition of samples taken from PBMC and colon was determined using amplicon sequencing of the 16S rRNA gene of experimental groups of kid (n = 3 for CTR and n = 4 RFT). Both, PBMCs and colon content sequencing libraries were prepared by an adapted procedure, based on the standard protocol “16S Metagenomic Sequencing Library Preparation from Illumina” (Part # 15044223 Rev. A). In this protocol, a first PCR to amplify template DNA using region of interest-specific primers with overhang adapters attached was carried out. Then, to add dual indexes and Illumina sequencing adapters a second PCR was performed using the Nextera XT Index Kit. Adaptation of this methodology consists of the substitution of the sequences targeting the amplification of the V3-V4 hypervariable region of the 16S rRNA gene proposed in this protocol by more specific ones (5′-TCCTACGGGAGGCAGCAGT-3′ and 5′-GGACTACCAGGGTATCTAATCCTGTT-3′ [27]). These sequences have been reported to have high sensitivity (targeting 95% of the bacterial sequences found in the Ribosomal Database Project) and 100% specificity (no eukaryotic, mitochondrial, or Archaea DNA targeted) [12]. Colon content libraries were obtained from 12.5 ng of DNA (as recommended) and PBMCs ones from 125 ng of DNA. Region of interest was amplified by PCR (25 cycles) and performed in 25 μl, containing 1x of KAPA HiFi HotStart ReadyMix (Roche Molecular Systems, Inc., Cat.# KK2601), 0.2 μM of specific primers and the mentioned quantities of genomic DNA. To verify the correct amplicon size, 1 μl of PCR product was visualized on a Bioanalyzer High Sensitivity DNA chip (Agilent Technologies, Cat. # 5067–4626) and after purification with AMPure beads (Beckman Coulter, Cat.# A63881), PCR product was eluted in 52.5 μl of elution buffer. Then, 5 μl of cleaned DNA were used in the second PCR (8 cycles), where Nextera-XT adapters (Illumina Inc., Cat.# (FC-131-1001 or FC-131-1002) with dual indexes were added. The second PCR was performed in 50 μl, containing 1x of KAPA HiFi HotStart ReadyMix (Roche Molecular Systems, Inc., Cat.# KK2601), 5 μl of each Illumina Inc.’s adapter and the 5 μl of the purified first PCR product. To verify the performance of the second PCR, 1 μl of each reaction was visualized on a Bioanalyzer High Sensitivity DNA chip. Finally, libraries were cleaned up using AMPure beads, eluted in 25 μl of water, and then quantified using Qubit dsDNA HS DNA Kit (Thermo Fisher Scientific, Cat. # Q32854). The amplicons sequencing was performed on a MiSeq (Illumina Inc.) platform following a standard protocol for paired-end reads of 300 nucleotides.
Quality control of the reads was carried out using FASTQC software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads were filtered from the adapter sequences and their quality score using trim_galore software (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) and only are retained those with at least 20 phred quality score. Trimmed sequences were merged via FLASH [28] to create amplicons of approximately 460 bp. Amplicons were analysed using QIIME: Quantitative Insights Into Microbial Ecology software package [29]. Sequences were clustered as operational taxonomic units (OTUs) of 97% similarity using uclust [30]. Taxonomic assignment of sequences was performed against the Greengenes database (version 13–8) [31]. The significances of grouping in the PCA plots were tested by analysis of similarity (ANOSIM) with 999 permutations using vegan R-package [32, 33]. The sequences obtained in this study were deposited in the European Nucleotide Archive (ENA) under the project number PRJEB39180.
Then, the relative abundance of each OTU present in the negative-blank was removed from each PBMC library results using R programming language (https://stackoverflow.com/questions/51514356/how-to-subtract-values-of-a-first-column-from-all-columns-by-function-in-r). In the absence of further studies, bioinformatic software (PICRUSt2) was used to investigate functional differences between PBMC and gastrointestinal microbial ecosystem. After normalizing the OTU table through Cumulative Sum Squares (CSS), KEGG Pathways were predicted by PICRUSt algorithm (level 3) [34]. Figure represents differential PICRUSt predicted KEGG pathways between bacterial communities found in PBMC samples compare to colon ones detected by STAMP software [35].
Statistical analysis
Statistical analyses represented in Fig. 1 (nonparametric Mann-Whitney’s tests) were conducted using PRISM (Version 8.05, GraphPad, Inc., La Jolla, CA). The significant fold change of OTU’s was performed using DESeq2 [36]. The STAMP [35] statistical comparison between groups was performed by two-sided Welch’s t-test within 95% confidence interval. Spearman’s correlation test was used to assess the relationships among bacterial composition from colon and PBMC. Only OTUs present in both type of samples, in more than 3 goats and which abundance was higher than 20 OTUs per animal were used for the analysis. Only correlations with a value of P < 0.05 were considered as significant and were represented.
Results
Early-life intervention with rumen liquid transplantation did not affect body weight (26 ± 3.21 Kg), colon weight (893.7 ± 97.95 g) or colon pH (6.29 ± 0.54) from the experimental groups.
Gene expression in colon
The expressions of genes related to host response to microbiota in the colon of goats was measured using RT-qPCR relative to β-actin expression using the ΔCt value. Expression of Pglypr1, βdefensin, Tlr2, Tlr4, and Tlr9 were not modified by rumen fluid transplant. However, Tlr5 used by the mucosal immune system of the gut to detect flagellin showed higher expression (P = 0.02) in the colon from goats transplanted with rumen fluid early in life (Fig. 1a).

a Gene expression level in colon tissue. b Total bacterial DNA in colon content determined by qPCR. Experimental groups: CTL (Control) and RLT (rumen liquid transplantation)
PBMC and colon content microbial profile
To evaluate the potential impact of colon microbiota on blood bacterial composition in goats, the taxonomic diversity and profile of the bacterial DNA present in PBMC and colon samples were analysed by high-throughput 16S rRNA gene amplicon sequencing. A total of 3,387,331 high-quality sequences (reads) were obtained ranging from 193,826 to 222,762 in colon and from 177,386 to 330,387 in PBMC samples. After clustering, 31,421 ± 900 OTUs in colon and 12,904 ± 1042 OTUs in PBMC samples were used for microbial analysis. Good’s coverage was over 99% in all experimental samples. Taxonomic assignment, observed species and Shannon diversity indices (illustrated at the OTU level in Fig. 2a) displayed that colon presents a higher bacterial diversity than PBMC (P = 0.001). Then, PCA showed that the supplementation with rumen fluid in early life affected (ANOSIM, p = 0.028) ecosystem structure in colon samples (Fig. 2b). In addition, the quantity of bacterial DNA present in the colon was also affected with lower numbers (P = 0.03) in goats supplemented with rumen fluid (Fig. 1b). However, in agreement with alpha diversity, when all samples were compared together, PBMC ones were significantly different (ANOSIM, P ≤ 0.001) from colon samples (Fig. 2c).

a Alpha diversity measurement, observed species and shannon indices in PBMC and colon content samples. b PCA plot from control (CTL) and rumen fluid transplantation (RFT) groups (ANOSIM; p = 0.028) colon content. c PCA plots comparing PBMC and colon microbiome (ANOSIM, p = 0.0005) from experimental goats
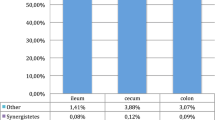
The distribution of reads assigned at the phylum level (Fig. 3) reveals that the PBMC contain bacterial DNA mostly from the Proteobacteria phylum, which represents on average 55% of the reads, followed by Firmicutes phylum (24%) and a lower proportion of Bacteroidetes (11%) and Actinobacteria (8%) phyla without differences due to rumen fluid supplementation. At a deeper taxonomic level, the predominant family was Oxalobacteracea followed by Pseudomonadaceae, Lachnospiraceae, Ruminococcaceae, Sinobacteraceae and Enterobacteriaceae (Fig. 4a). Although Oxalobacteraceae, Pseudomonadaceae and Sinobacteraceae were also observed in negative blank, their relative abundances were removed from all PBMC samples. Microbial distribution at family level in PBMC and colon content is fully described in supplementary figure 1.

a PBMC and colon content phylum distribution in experimental goats

a Twenty most abundant families present in PBMC and colon content microbiome. b Twenty most abundant genera detected in PBMC and colon content microbial community in healthy goats
At genus level, the 20 most abundant OTUs in colon and PBMC microbiome were represented in Fig. 4b. Predominant genera found in PBMC were Pseudomonas, Prevotella, Ruminococcus, Sphingomonas and Acinetobacter. Other genera such as Corynebacterium, Butyrivibrio, Bifidobacterium, Dorea, Coprococcus or Blautia, were also present in both microbial environments. Among the taxa present in higher abundance in almost all PBMC samples, it was observed that several genera (Acinetobacter, Corynebacterium, and Pseudomonas) containing species that are pathogens with known capacity to infect blood. On the other side, other leading players in the maintenance of the homeostasis in the gut such as Faecalibacterium prausnitzii (Firmicutes phylum) considered one of the main butyrate producer in the gut and Lachnospiraceae members (Butyrivibrio, Dorea, Coprococcus, Blautia and Ruminococcus) among the main producers of short chain fatty acids, were also observed. In general, less than 8% of OTUs detected were shared between PBMC and colon environment. In this regard, colon content was mainly composed of Firmicutes (83%), followed by Actinobacteria (8.2%), Bacteroidetes (4.8%) and Proteobacteria (1.3%).
To further investigate how differences in the microbiome composition could impact the functionality of microbiota, we performed predictive analysis of functional pathways and compared the pathway representation between colon content and PBMC. The mean proportion of predicted KEGG pathways (at level 3) of the metagenome functional content (bacterial genes) and significantly augmented in the PBMC of goats compared to colon content were illustrated in Supplementary Figure 1. Most of them were associated to anti-inflammatory profile of immune cells.
In order to understand whether the wide range of OTUs found in colon microbial composition were related to circulating microbiota, Pearson correlation analyses were performed. Variation in bacterial community composition detected in colon and PBMC samples showed a significant correlation (Fig. 5). The presence of Acinetobacter and Streptococcus in colon microbiome correlated (P ≤ 0.01) with the same OTU in circulating microbiome. A positive correlation between Butyrivibrio in colon and Bifidobacterium, Campylobacter, Faecalibacterium and Parabacteroides in PBMC samples was observed. On the other hand, the presence of Bifidobacterium in colon showed a negative correlation with Butyrivibrio, Prevotella and Ruminococcus in PBMC bacterial environment. Parabacteroides also showed a negative correlation with Butyrivibrio, Coprococcus and Pseudomonas.

Pearson correlation of microbial community present in colon and PBMC microbiome. Pearson correlation analysis indicated that the variation in bacterial community composition in both environment had significant correlations (p = 0.05). The size and intensity of color for each circle represents the strength of the correlation (the larger, darker circles demonstrate a strong correlation), blue colors illustrate positive correlations and red colors illustrate negative correlation coefficient
Discussion
Many studies have reported the successful culture and microscopic observation of numerous bacteria from blood of healthy individuals, especially in humans [12]. However, no exhaustive analysis has been performed of the blood microbiome in ruminants and even less in their circulating immune cells. Thanks to the evolution of sequencing technologies and the optimization of the processes, we have successfully characterized the taxonomic profile of the PBMC present in young healthy ruminants in the absence of any pathological condition. As expected, colon gut microbiota was modified by the rumen fluid transplantation, showing different microbial community according to experimental groups. However, its impact in dry matter colon content was not observed. Differences in the colon microbiota could rely on two aspects: i) a change in the microbial colonization pattern in early life with some persistency later in life [37,38,39] and ii) an indirect effect driven by a higher rumen microbial fermentation of the feed in RLT animals, mostly associated with the presence of rumen protozoa, which could limit the availability of fermentable substrate in the hindgut [38]. In a recent study we have demonstrated that inoculation of young goat kids with rumen fluid from adult goats accelerated the rumen microbial development favouring the presence of rumen protozoa, a higher bacterial diversity and increased abundances of certain bacterial taxa (e.g. Firmicutes and Fibrobacteres) [40]. Our results are in agreement with previous studies [25] where gene expression levels at the rumen epithelium of newborn goats were not affected although feeding management (maternal versus milk replacer) during the first month of life promoted different rumen microbial colonization. In that case, only Tlr5 expression in rumen varied. In the present study, a higher translocation of bacteria in supplemented goats was suggested as Tlr5 is expressed mostly on the basolateral side of intestinal epithelial cells for detecting whether bacteria have crossed the gut epithelia. Although, in general it is accepted that bacteria can pass into tissues originating from the gut, in this study, it was revealed that in standard physiological conditions, only a low proportion of microbiota was shared between colon and PBMC. Results in this study suggest non-intestinal sources as main contributors to microbial DNA in circulating PBMC.
Several sources can potentially contribute to add external contamination: bacterial contaminants collected during the sampling in the experimental farm, the manipulation of the samples and the reagents in the laboratory during extraction and sequencing library preparation pipeline could lead to the artificial identification of PBMC microbiome which add a background to the analysis of the blood microbiome and can be misinterpreted as bacteria present in the samples [20, 21, 23, 41]. Hence, in this study negative controls were run all over the process to track potential contamination during the manipulation and sequencing of the samples. In this sense, Poore et al. [8] reported that in silico decontamination did not appear to differentially affect the types of samples under study, validating gold-standard microbiology practices for low biomass studies.
While evidence for the existence of a blood-microbiome in various domesticated mammals and birds do exist [42,43,44], this study was the first to track the major part of PBMC microbiome in healthy ruminants by 16S rRNA gene amplicon sequencing. Here, higher abundance of OTUs assigned to Proteobacteria phylum (more than 50%) was found in PBMC compared to the colon microbiome (Firmicutes more than 80%). In addition, rumen composition also differed from circulating bacterial profile, being Bacteroidetes the predominant phylum showing significant differences according to treatment, representing 41 and 60% in CTL and RLT groups, respectively (p = 0,01) [45]. Relative abundance was followed by Proteobacteria and Firmicutes although they did not show differences between groups. Other potential source of bacteria to circulating microbiome would be oral cavity as daily activities including chewing or when the barriers between oral environment and the circulatory system are compromised, result in the translocation of oral bacteria into the bloodstream [11, 46]. However, results described in oral secretion did not agree between both ecosystems being the most abundant phylum reported Firmicutes (50%), followed by Proteobacteria (29%), Actinobacteria (3.5%) and Fusobacteria (1%) [47]. Our results are in agreement with different studies aiming to investigate blood microbiome in healthy human donors where Proteobacteria was found over 80%, followed by Actinobacteria (between 7 and 10%) and Firmicutes (from 3 to 6%) [12,13,14,15,16, 48, 49]. However, the characterization of blood bacterial diversity varies between studies [50]. The abundance of Proteobacteria in PBMC comprises both obligate and facultative anaerobic bacteria genera therefore could be related to their ability to tolerate a range of oxic conditions. They are highly present in the gut of neonatal goats [25], specifically in rumen, and other mammals, which are abundant in oxygen immediately post-partum contributing to the homeostasis of the anaerobic environment. In general, these results suggest a common circulating bacterial profile on mammals.
Bacterial translocation from gut was described several years ago but mostly related to pathological conditions. However, nowadays more evidence supports bacterial translocation in physiological context. The difference of bacterial profiles between the gut and PBMC could be explained by the role of filter played by the intestinal barrier and immune cells, which limits the translocation of a specific portion of the gut microbiota to the periphery [51,52,53]. Other compartments of the digestive tract, tissues and organs, such as skin, oral cavity, nasal, and lung mucosa probably are making a significant contribution to the bacterial DNA present in PBMC. It appears that maternal origin would be also accepted. In agreement, Whittle et al. [15] demonstrated that blood-microbiome closely resembles the skin and oral microbiomes and differs substantially from the intestinal microbiome. Due to the low number of animals used in this experiment, it was not sufficient to assert whether PBMC bacterial composition was affected by rumen fluid transplantation. However, results indicated that the bacterial environment inside PBMC is quite stable in time and comprise a core set instead of a dynamic and adaptive group of microorganisms. Probably the specificity and stability of the bacterial DNA is linked to the function that has to perform in such location and only the ones that play a role are the only ones allowed to use PBMC as “vehicles”. As previously mentioned, Poore et al. [8] could discriminate among samples from healthy, cancer-free individuals and those from patients with multiple types of cancer (prostate, lung, and melanoma) using only plasma-derived, cell-free microbial nucleic acids exhibiting a relevant role of blood microbiome in health and disease. Although its biological significance remains to be further explored, the discovery of a PBMC microbiome in ruminants might represent an important step toward a better understanding of the microbial relationships with health.
The information provided here opens the possibility to support different hypothesis in ruminants such as entero-mammary route. However, living, nonviable, or dormant bacteria were not addressed in this study. Therefore, the understanding of the PBMC microbiota will require further investigations in the future. Nevertheless, to provide evidence to the theory of travellers’ microorganism in the blood immune cells, we have recently described the survival capacity of Akkermansia muciniphila [54] in healthy human monocytes for more than 3 h.
To conclude, our results demonstrate the presence of a highly diversified PBMC microbiome in healthy ruminants that differs from that in the colon community. The rumen fluid transplantation in early life modified the bacterial community structure in the colon providing a wide range of microbes. However, these microbial differences were not observed in the PBMC. Our results suggest that in healthy physiological conditions, the intestinal barrier plays a critical role limiting the bacterial groups able to reach circulating immune cells. Proteobacteria was the most abundant phylum, and Pseudomonas and Prevotella were the dominant genera in the PBMC microbiome. In this study, a positive correlation between Butyrivibrio found in the colon and genera such as Bifidobacterium and Faecalibacterium detected in PBMC samples was observed. However, this observation does not necessarily implies causality since the origin and role of the physiological PBMC microbiome, and its interaction with the host remains to be elucidated.
Availability of data and materials
Raw sequences were made available at European Nucleotide Archive (ENA) under the project number PRJEB39180.
References
Proal AD, Albert PJ, Marshall TG, Blaney GP, Lindseth IA. Immunostimulation in the treatment for chronic fatigue syndrome/myalgic encephalomyelitis. Immunol Res. 2013;56(2-3):398–412. https://doi.org/10.1007/s12026-013-8413-z.
McLaughlin RW, Vali H, Lau PCK, Palfree RG, De Ciccio A, Sirois M, et al. Are there naturally occurring pleomorphic bacteria in the blood of healthy humans? J Clin Microbiol. 2002;40(12):4771–5. https://doi.org/10.1128/JCM.40.12.4771-4775.2002.
Amar J, Lange C, Payros G, Garret C, Chabo C, Lantieri O, et al. Blood microbiota dysbiosis is associated with the onset of cardiovascular events in a large general population: the DESIR study. PLoS One. 2013;8(1):e54461. https://doi.org/10.1371/journal.pone.0054461.
Dinakaran V, Rathinavel A, Pushpanathan M, Sivakumar R, Gunasekaran P, Rajendhran J. Elevated levels of circulating DNA in cardiovascular disease patients: metagenomic profiling of microbiome in the circulation. PLoS One. 2014;9(8):e105221. https://doi.org/10.1371/journal.pone.0105221.
Sato J, Kanazawa A, Ikeda F, Yoshihara T, Goto H, Abe H, et al. Gut dysbiosis and detection of “live gut bacteria” in blood of Japanese patients with type 2 diabetes. Diabetes Care. 2014;37(8):2343–50. https://doi.org/10.2337/dc13-2817.
Manzo VE, Bhatt AS. The human microbiome in hematopoiesis and hematologic disorders. Blood. 2015;126(3):311–8. https://doi.org/10.1182/blood-2015-04-574392.
Traykova D, Schneider B, Chojkier M, Buck M. Blood microbiome quantity and the hyperdynamic circulation in decompensated cirrhotic patients. PLoS One. 2017;12(2):e0169310. https://doi.org/10.1371/journal.pone.0169310.
Poore GD, Kopylova E, Zhu Q, Carpenter C, Fraraccio S, Wandro S, et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature. 2020;579(7800):567–74. https://doi.org/10.1038/s41586-020-2095-1.
Nikkari S, McLaughlin IJ, Bi W, Dodge DE, Relman DA. Does blood of healthy subjects contain bacterial ribosomal DNA? J Clin Microbiol. 2001;39(5):1956–9. https://doi.org/10.1128/JCM.39.5.1956-1959.2001.
Potgieter M, Bester J, Kell DB, Pretorius E. The dormant blood microbiome in chronic, inflammatory diseases. FEMS Microbiol Rev. 2015;39(4):567–91. https://doi.org/10.1093/femsre/fuv013.
Amar J, Lange C, Payros G, Garret C, Chabo C, Lantieri O, et al. Blood microbiota dysbiosis is associated with the onset of cardiovascular events in a large general population: the D.E.S.I.R. study. PLoS One. 2013;8:e54461.
Païssé S, Valle C, Servant F, Courtney M, Burcelin R, Amar J, et al. Comprehensive description of blood microbiome from healthy donors assessed by 16S targeted metagenomic sequencing. Transfusion. 2016;56(5):1138–4. https://doi.org/10.1111/trf.13477.
Li Q, Wang C, Tang C, Zhao X, He Q, Li J. Identification and characterization of blood and neutrophil-associated microbiomes in patients with severe acute pancreatitis using next-generation sequencing. Front Cell Infect Microbiol. 2018;8:5. https://doi.org/10.3389/fcimb.2018.00005.
Olde Loohuis LM, Mangul S, Ori APS, Jospin G, Koslicki D, Yang HT, et al. Transcriptome analysis in whole blood reveals increased microbial diversity in schizophrenia. Transl Psychiatry. 2018;8(1):96. https://doi.org/10.1038/s41398-018-0107-9.
Whittle E, Leonard MO, Harrison R, Gant TW, Tonge DP. Multi-method characterization of the human circulating microbiome. Front Microbiol. 2018;9:3266.
Qiu J, Zhou H, Jing Y, Dong C. Association between blood microbiome and type 2 diabetes mellitus: a nested case-control study. J Clin Lab Anal. 2019;33(4):e22842. https://doi.org/10.1002/jcla.22842.
Minasyan H. Erythrocyte and blood antibacterial defense. Eur J Microbiol Immunol. 2014;4(2):138–43. https://doi.org/10.1556/EuJMI.4.2014.2.7.
Pretorius E, Bester J, Vermeulen N, Lipinski B, Gericke GS, Kell DB. Profound morphological changes in the erythrocytes and fibrin networks of patients with hemochromatosis or with hyperferritinemia, and their normalization by iron chelators and other agents. PLoS One. 2014;9(1):e85271. https://doi.org/10.1371/journal.pone.0085271.
Pellon A, Atondo E, Montesinos-Polledo M, Pascual-Itoiz MA, Peña-Cearra A, Carreras-Gonzalez A, et al. Intracellular lifestyle of the probiotic bacteria Lactobacillus plantarum; implications for its extraintestinal dissemination. Eur Cong Immunol. 2018;WS.D4.07.05:110.
Salter SJ, Cox MJ, Turek EM, Calus ST, Cookson WO, Moffatt MF, et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014;12(1):87. https://doi.org/10.1186/s12915-014-0087-z.
Glassing A, Dowd SE, Galandiuk S, Davis B, Chiodini RJ. Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathog. 2016;8(1):24. https://doi.org/10.1186/s13099-016-0103-7.
Davis NM, Proctor DM, Holmes SP, Relman DA, Callahan BJ. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome. 2018;6(1):226. https://doi.org/10.1186/s40168-018-0605-2.
Eisenhofer R, Minich JJ, Marotz C, Cooper A, Knight R, Weyrich LS. Contamination in low microbial biomass microbiome studies: issues and recommendations. Trends Microbiol. 2019;27(2):105–17. https://doi.org/10.1016/j.tim.2018.11.003.
Belanche A, Palma-Hidalgo JM, Nejjam I, Jiménez E, Martín-García AI, Yáñez-Ruiz DR. Inoculation with rumen fluid in early life as a strategy to optimize the weaning process in intensive dairy goat systems. Int J Dairy Sci. 2020;103(6):5047–60. https://doi.org/10.3168/jds.2019-18002.
Abecia L, Jiménez E, Martínez-Fernandez G, Martín-García AI, Ramos-Morales E, Pinloche E, et al. Natural and artificial feeding management before weaning promote different rumen microbial colonization but not differences in gene expression levels at the rumen epithelium of newborn goats. PLoS One. 2017;12(8):e0182235. https://doi.org/10.1371/journal.pone.0182235.
Maeda H, Fujimoto C, Haruki Y, Maeda T, Kokeguchi S, Petelin M, et al. Quantitative real-time PCR using TaqMan and SYBR Green for Actinobacillus actinomycetemcomitans, Porphyromonas gingivalis, Prevotella intermedia, tetQ gene and total bacteria. FEMS Immunol Med Mic. 2003;39(1):81–6. https://doi.org/10.1016/S0928-8244(03)00224-4.
Nadkarni MA, Martin FE, Jacques NA, Hunter N. Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology. 2002;148(1):257–66. https://doi.org/10.1099/00221287-148-1-257.
Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27(21):2957–63. https://doi.org/10.1093/bioinformatics/btr507.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–6. https://doi.org/10.1038/nmeth.f.303.
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460–1. https://doi.org/10.1093/bioinformatics/btq461.
McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012;6(3):610–8. https://doi.org/10.1038/ismej.2011.139.
R Core Team. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2020. https://www.R-project.org. Accessed 11 Aug 2020.
Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’hara RB, et al. Package Vegan: community ecology package, version 2.5–6. https://onlinelibrary.wiley.com/doi/abs/10.1111/j.1654-1103.2003.tb02228.x
Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31(9):814–21. https://doi.org/10.1038/nbt.2676.
Parks DH, Tyson GW, Hugenholtz P, Beiko RG. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics. 2014;30(21):3123–4. https://doi.org/10.1093/bioinformatics/btu494.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. https://doi.org/10.1186/s13059-014-0550-8.
Yáñez-Ruiz DR, Abecia L, Newbold CJ. Manipulating rumen microbiome and fermentation through interventions during early life: a review. Front Microbiol. 2015;6:1133.
Belanche A, Yáñez-Ruiz DR, Detheridge AP, Griffith GW, Kingston-Smith AH, Newbold CJ. Maternal versus artificial rearing shapes the rumen microbiome having minor long-term physiological implications. Environ Microbiol. 2019;21(11):4360–77. https://doi.org/10.1111/1462-2920.14801.
Furma O, Shenhav L, Sasson G, Kokou F, Honig H, Jacoby S, et al. Stochasticity constrained by deterministic effects of diet and age drive rumen microbiome assembly dynamics. Nat Commun. 2020;11(1):1904. https://doi.org/10.1038/s41467-020-15652-8.
Palma-Hidalgo JM, Jiménez E, Popova M, Morgavi DP, Martín-García AI, Yáñez-Ruiz DR, et al. Inoculation with rumen fluid in early life accelerates the rumen microbial development and favours the weaning process in goats. Anim Microbiome. 2021;3(1):11. https://doi.org/10.1186/s42523-021-00073-9.
Tichopad A, Didier A, Pfaffl MW. Inhibition of real-time RTPCR quantification due to tissue-specific contaminants. Mol Cell Probes. 2004;18(1):45–50. https://doi.org/10.1016/j.mcp.2003.09.001.
Sze MA, Tsuruta M, Yang SW, Oh Y, Man SF, Hogg JC, et al. Changes in the bacterial microbiota in gut, blood, and lungs following acute LPS instillation into mice lungs. PLoS One. 2014;9(10):e111228. https://doi.org/10.1371/journal.pone.0111228.
Mandal RK, Jiang T, Al-Rubaye AA, Rhoads DD, Wideman RF, Zhao J, et al. An investigation into blood microbiota and its potential association with Bacterial Chondronecrosis with Osteomyelitis (BCO) in broilers. Sci Rep. 2016;6(1):25882. https://doi.org/10.1038/srep25882.
Vientós-Plotts AI, Ericsson AC, Rindt H, Grobman ME, Graham A, Bishop K, et al. Dynamic changes of the respiratory microbiota and its relationship to fecal and blood microbiota in healthy young cats. PLoS One. 2017;12(3):e0173818. https://doi.org/10.1371/journal.pone.0173818.
Belanche A, Palma-Hidalgo JM, Neijam I, Jiménez E, Martín-García AI, Yáñez-Ruiz DR. Inoculation with rumen fluid in early-life as a strategy to optimize the weaning process in intensive dairy goat farms. J Dairy Sci. 2020;103(6):5047–60. https://doi.org/10.3168/jds.2019-18002.
Forner L, Larsen T, Kilian M, Holmstrup P. Incidence of bacteremia after chewing, tooth brushing and scaling in individuals with periodontal inflammation. J Clin Periodontol. 2006;33(6):401–7. https://doi.org/10.1111/j.1600-051X.2006.00924.x.
Fouhse JM, Smiegielski L, Tuplin M, Guan LL, Willing BP. Host immune selection of rumen bacteria through salivary secretory IgA. Front Microbiol. 2017;8:848.
Lelouvier B, Servant F, Païssé S, Brunet AC, Benyahya S, Serino M, et al. Changes in blood microbiota profiles associated with liver fibrosis in obese patients: a pilot analysis. Hepatology. 2016;64(6):2015–27. https://doi.org/10.1002/hep.28829.
Gosiewski T, Ludwig-Galezowska AH, Huminska K, Sroka-Oleksiak A, Radkowski P, Salamon D, et al. Comprehensive detection and identification of bacterial DNA in the blood of patients with sepsis and healthy volunteers using next-generation sequencing method - the observation of DNAemia. Eur J Clin Microbiol Infect Dis. 2017;36(2):329–36. https://doi.org/10.1007/s10096-016-2805-7.
Castillo DJ, Rifkin RF, Cowan DA, Potgieter M. The healthy human blood microbiome: fact or fiction? Front Cell Infect Microbiol. 2019;9:148.
Bellot P, García-Pagán JC, Francés R, Abraldes JG, Navasa M, Pérez-Mateo M, et al. Bacterial DNA translocation is associated with systemic circulatory abnormalities and intrahepatic endothelial dysfunction in patients with cirrhosis. Hepatology. 2010;52(6):2044–52. https://doi.org/10.1002/hep.23918.
Garidou L, Pomié C, Klopp P, Waget A, Charpentier J, Aloulou M, et al. The gut microbiota regulates intestinal CD4 T cells expressing RORct and controls metabolic disease. Cell Metab. 2015;22(1):100–12. https://doi.org/10.1016/j.cmet.2015.06.001.
Balmer ML, Slack E, de Gottardi A, Lawson MA, Hapfelmeier S, Miele L, et al. The liver may act as a firewall mediating mutualism between the host and its gut commensal microbiota. Sci Transl Med. 2014;6:237–66.
Peña-Cearra A, Pascual-Itoiz MÁ, Gutiérrez T, Pellón A, Atondo E, Palacios A, et al. Supervivencia de Akkermansia muciniphila en monocitos humanos. Anales de Microbiota, Probióticos & Prebióticos. Sumario XI Workshop Sociedad Española de Microbiota, Probióticos y Prebióticos. SEMiPyP. 2020;1:101.
Acknowledgements
We thank Isabel Jimenez and Rosa Serrano (Estación Experimental del Zaidín, Granada) for their assistance with the animal care and sample collection, and Laura Barcena for technical support.
Funding
This work was supported by grants from the Spanish Ministry of Science and Innovation (MCI) co-financed with FEDER funds [AGL2017-86757- to LA, AGL2017-86938-R to DRY]. Other contributions were SAF2015-65327-R to JA and SAF2015-73549-JIN to HR. LA is a Ramón y Cajal fellow [RYC-2013-13666] from the Spanish Ministry of Science and Innovation. APC is a recipient of a fellowship from the University of the Basque Country. We thank the MCI for the Severo Ochoa Excellence accreditation (SEV-2016-0644) and the Basque Department of Industry, Tourism and Trade (Etortek and Elkartek programs).
Author information
Authors and Affiliations
Contributions
APC and AB contributed equally. Conception and design of the study (LA), data collection (APC, AB, EJ), data analysis (APC, MAP, MG and AMA), bioinformatic analysis (APC and JLL), drafting the manuscript (LA), manuscript revision (APC, AB, HR, JA and DRY), statistical analysis (LA and JLL), obtained funding (DRY, JA, and LA), technical support (EJ). All authors approved the final version for publication.
Corresponding author
Ethics declarations
Ethics approval
Animal protocols were approved by the Ethical Committee for Animal Research (EEZ-CSIC) regional government (09/03/2017).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Figure 1.
Microbial distribution at family level in PBMC and colon content in healthy goats.
Additional file 2: Supplementary Figure 2.
Predicted pathways (level 3) with the most significant differences (p-value corrected) augmented in PBMC (orange bars) compared to colon (blue bars) samples.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Peña-Cearra, A., Belanche, A., Gonzalez-Lopez, M. et al. Peripheral blood mononuclear cells (PBMC) microbiome is not affected by colon microbiota in healthy goats. anim microbiome 3, 28 (2021). https://doi.org/10.1186/s42523-021-00091-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42523-021-00091-7