
Abstract
Carbon-based adsorbents possess exceptional adsorption capability, making them an ideal platform for the remediation of environmental contaminants. Here, we demonstrate carbonized lignosulfonate (LS)-based porous nanocomposites with excellent adsorption performance towards heavy metal ions and cationic dye pollutants. Through microwave-assisted hydrothermal carbonization, a green approach was employed to carbonize lignosulfonate to carbon spheres. The LS-derived carbon spheres were then oxidized into nanographene oxide (nGO) carbon dots. A facile two-step procedure that involved the self-assembly of nGO and gelatin into a hydrogel precursor coupled with freeze-drying enabled the construction of three-dimensional (3D) free-standing porous composites without the use of organic solvents or chemical crosslinking agents. The favorable pore structure and abundance of surface functional groups on the nGO/gelatin porous composite proved to substantially facilitate the adsorption of Cu(II) in comparison to conventionally-used activated carbon. Further enhancement of adsorption performance was achieved by introducing additional surface functional groups through a non-covalent functionalization of the porous composite with lignosulfonate. The presence of negatively-charged sulfonate groups increased the Cu(II) equilibrium adsorption capacity (66 mg/g) by 24% in comparison to the non-functionalized nGO/gelatin counterpart. Both functionalized and non-functionalized composites exhibited significantly faster adsorption rates (40 min) compared to many graphene- or GO-based adsorbents reported in literature. In addition to the adsorption of heavy metal ions, the composites also demonstrated good adsorption capacity towards cationic dyes such as methylene blue. This paves the way for a high value-added application of lignin in environmental remediation and opens up new possibilities for the development of sustainable materials for adsorption and water purification.
Similar content being viewed by others

Introduction
Graphene oxide (GO)-based materials are highly effective adsorbents for environmental remediation, but their widespread use is hindered by the high cost involved in GO synthesis. Graphite, the primary source of graphene, is derived from non-renewable reserves, and even more so the production of graphene and GO via synthetic routes from commercial precursors requires high temperatures and harsh reaction conditions [1, 2]. In recent years, the use of biomass waste as a precursor to carbonaceous materials has emerged as an effective strategy towards both valorization of low-value resources, as well as an economical and sustainable pathway towards the preparation of valuable carbon-based functional materials [3,4,5,6]. Wood, the largest biomass resource on earth, is mainly composed of cellulose, hemicellulose, and lignin [7]. In the pulping industry, the extraction of cellulose from wood generates large amounts of lignin-rich residues [8]. In spite of the copious amounts available as a side stream, lignin has so far only few low-value applications and it is mostly burned for energy production [8,9,10].
Lignin is a complex heterogeneous polymer composed in large part of p-coumaryl, coniferyl, and sinapyl alcohol building blocks making up a molecular chain with a large number of phenyl rings [10]. Due to the high carbon content of over 60%, lignin is considered to be a very promising raw material for carbonaceous products [11,12,13,14]. In comparison to cellulose-rich biomass, lignin-rich biomass yield chars with higher thermal stability and lower reactivity as a result of their higher fixed carbon contents [5]. Several successful methods have been employed in the synthesis of carbonaceous materials from lignin-based precursors, including high temperature pyrolysis [5, 15] and hydrothermal carbonization (HTC) [10, 11]. In this work, we employ a green approach via microwave-assisted HTC as adapted from previous work on carbonization of various biopolymers [16,17,18,19]. In comparison to pyrolytic carbonization, HTC proceeds at much lower temperatures in a closed system, thus significantly reducing energy consumption and air pollution [11, 20]. Implemented in combination with microwave irradiation, which facilitates rapid volumetric heating, increased reaction rates, and shortened reaction times compared to conventional heating, a time-efficient, cost-effective, and ecofriendly method of lignin carbonization is enabled [21, 22].
Herein, we investigate the feasibility of utilizing carbonized lignosulfonate-based nGO as a building block for the development of porous nGO/gelatin nanocomposites as adsorbents for wastewater purification. First, we employ a two-step reaction that involves carbonization of lignosulfonate and oxidation of the resulting carbon product to derive nGO. Next, we construct three-dimensional freestanding porous composites through the self-assembly of nGO and gelatin into hydrogel precursors in the absence of any chemical crosslinking agent. To further enhance the adsorption performance of the composites, additional active sites for adsorption in the form of negatively-charged sulfonate groups are introduced via non-covalent functionalization with lignosulfonate. Hence, we explore the adsorption performance of sustainable and environment friendly lignosulfonate-based nanocomposites towards the remediation of heavy metal ions and cationic dye pollutants from wastewater.
Experimental
Reagents and materials
Sodium lignosulfonate (LS) (Mw = 20,300; PDI = 2.34; 5–7% methoxy content, < 6% water) was obtained from Tokyo Chemical Industry UK Ltd. Gelatin, sulfuric acid (H2SO4; 95–98%), nitric acid (HNO3; 70%), copper sulfate pentahydrate (CuSO4•5H2O), poly(ethyleneimine) (PEI) (Mw = 750,000, 50% w/v in H2O), and methylene blue (C16H18ClN3S; ≥82%) were obtained from Sigma-Aldrich. All chemicals were used as received. Commercially available activated carbon for aquarium filtration systems was obtained from Fluval Carbon, Hagen Inc.
Synthesis of nanographene oxide (nGO) from LS
nGO was synthesized by oxidation of lignosulfonate-derived carbon spheres (CS), that were obtained through an acid-catalyzed microwave-assisted carbonization process adapted from previous work [18, 19]. Here, LS was used instead of α-cellulose or starch as biopolymer, and the carbonization temperature was increased from 220 °C to 240 °C. Briefly, CS were produced through the carbonization of 2 g of LS in 20 mL of aqueous H2SO4 solution (0.01 g/mL) for 2 h with a ramp time of 20 min using a flexiWAVE microwave device (Milestone Inc.). The obtained CS were sonicated in a 70% HNO3 aqueous solution (1:100 w/w) for 30 min. Oxidation was performed at 90 °C with constant stirring for 30 min. Cold deionized water was poured into the solution to stop the reaction. The solvent was removed via rotary evaporation, after which the produced nGO was freeze-dried and stored in vacuum at room temperature for at least 48 h before use.
Preparation of LS-functionalized porous nGO/gelatin nanocomposites
Porous nGO-gelatin composites were first prepared via self-assembly forming hydrogels, followed by freeze-drying. First, aqueous suspensions of nGO (10 mg/mL) were prepared by stirring 100 mg of the LS-derived nGO in 10 mL of deionized H2O for 1 h, followed by sonication for 30 min. Different predetermined amounts of LS were then dispersed in the nGO suspensions via sonication for 10 min. Hydrogel self-assembly was initiated according to a previous procedure [23, 24], with the important difference that here, zero-dimensional nGO derived from LS was utilized instead of the (in lateral dimension) micro-sized GO from commercial graphite powder. Briefly, 1.5 mL of aqueous gelatin solution with varying concentrations was added drop-wise to the nGO/lignosulfonate suspension, prompting immediate hydrogel formation. The mixture was subsequently heated to 60 °C for 5 min to induce the formation of covalent and non-covalent bonds between nGO and gelatin, which acts as a reducing agent and can partially reduce nGO. The composition of the prepared nanocomposites are found in Table 1 and denoted as nGOX-GelY-LSZ where X:Y:Z indicate the nGO:gelatin:lignosulfonate mass ratio. The prepared hydrogels were washed in deionized water for 1 week with regular water changes. Finally, the hydrogels were freeze-dried for 48 h to obtain porous composite materials. All characterizations and adsorption tests were executed on granulated nGO/gelatin composites.
Characterization of LS-derived carbon products and LS-functionalized nGO/gelatin composites
Apparent porosity was determined using Eq. 1, where M1 is the mass of the dry sample, M2 is the mass of the saturated sample after immersion in deionized water, ρ is the density of water, and V is the volume of the saturated sample.
The analysis of functional groups was performed using Fourier transform infrared spectroscopy (FT-IR) and carried out by PerkinElmer Spectrum 2000 FTIR spectrometer equipped with attenuated total reflectance (ATR) accessory (Graseby Sperac). All spectra were recorded in the wavenumber range of 4000–600 cm− 1 using 16 scans at a resolution of 4 cm− 1.
Characterization of structural disorder in the lattice of the carbon products was performed via confocal Raman spectroscopy. Raman measurements were conducted on a HR800 UV Jobin Yvon Raman spectrometer (Horiba Scientific, Ltd.) with a solid-state laser set at an excitation wavelength of 514 nm. The laser was focused on the sample by passing through a 50x objective lens, and the Raman signal was collected by the same lens using a 600 groove mm− 1 density grating. Spectra were acquired at a resolution of 1 cm− 1 at 7 accumulations with an acquisition time of 40 s.
The thermal behavior and decomposition profiles of the carbon products and composites were investigated through thermogravimetric analysis (TGA), which was performed on a Mettler-Toledo TGA/SDTA 851e instrument. The samples (5–6 mg) were placed in 70 μL alumina cups and heated in a temperature range of 25–800 °C at a rate of 10 °C/min in N2 atmosphere set to a flow rate of 50 mL/min.
Transmission electron microscopy (TEM) images of the CS and nGO were acquired on a Hitachi HT7700 instrument. For TEM sample preparation, CS and nGO were dispersed in ethanol (0.2 mg/mL) via sonication for 30 min and drop casted on an EM-TEC formvar carbon support film on a copper 200 square mesh (Micro to Nano V.O.F.).
In the case of the composites, high-resolution images were taken on an ultrahigh resolution field emission scanning electron microscope, FE-SEM Hitachi S-4800. For SEM sample preparation, the samples were attached on an aluminum stub using carbon tape, and sputter coated with a platinum/palladium (Pt/Pd) coater target at 2 nm thickness using a Cressington 208HR Sputter Coater (Cressington Scientific Instruments, Ltd.).
Elemental analysis of the carbon products and the composites before and after heavy metal- or dye-adsorption were performed via energy-dispersive x-ray spectroscopy (EDX) measurements on Pt/Pd-coated samples using an X-MaxN 80 Silicon Drift Detector (Oxford Instruments) attached to the Hitachi SEM S-4800. Quantitative EDX measurements were performed by point analysis of randomly selected sites on the specimen using an electron beam accelerating voltage of 15 kV for all samples with the exception of the analysis of the composites after Cu-adsorption, for which 20 kV was used. Elemental compositions are given as an average of 10 point analysis measurements for each sample.
Batch adsorption test
Batch adsorption experiments were carried out to investigate the adsorption behavior of the composites towards Cu(II) and methylene blue (MB), respectively. A stock solution of Cu(II) (0.008 M) was prepared by dissolving 1.96 g of the CuSO4•5H2O in 1000 mL of deionized water. All subsequent solutions with varying Cu(II) concentrations were prepared by diluting the stock solution. The pH values of all solutions were adjusted to pH 5.2 via drop-wise additions of 0.01 M NaOH or HCl. For the adsorption of methylene blue, an aqueous solution (9.38 × 10− 5 M) was prepared. The pH of the MB solution was adjusted to pH 9 to maximize dye adsorption [25, 26] with drop-wise additions of 0.01 M NaOH. Ten milligram of the composite was added to 30 mL of Cu(II) solution or 20 mL of MB solution. The solution was then agitated at 240 rpm using a rotary shaker at room temperature until equilibrium adsorption was reached. Dye adsorption was carried out under dark conditions to prevent photocatalytic degradation of methylene blue.
A UV-vis absorption spectrophotometer (Shimadzu UV-2550) was used to determine the concentrations of Cu(II) at λ = 625 nm after complexation with poly(ethyleneimine) (PEI), which enabled detection of low concentrations of the ion (Supporting Information 1), or MB at λmax = 663 nm. Calibration curves were constructed to correlate the concentration of Cu(II) (R2 = 0.9965, Fig. S2) or MB (R2 = 0.9895, Fig. S3) with their respective maximum absorbances. All adsorption experiments were carried out in duplicate, and average values were used in data analysis. The adsorption capacity (qt) of the porous composite was calculated according to Eq. 2, where C0 (mg/L), Ct (mg/L), and Ce (mg/L) are the initial adsorbate concentration, the adsorbate concentration at time t, and the adsorbate concentration at the equilibrium time, respectively. V (L) stands for the volume of the adsorption solution used, and m (g) represents the dry weight of the sample.
Recyclability test
The reusability of the composite adsorbent was investigated through five cycles of Cu(II) adsorption and desorption. For every adsorption cycle, the composite material was immersed in fresh solutions of Cu(II) (0.006 M, 30 mL, pH 5.2) for 40 min with constant agitation at 240 rpm. After each adsorption test, the Cu(II)-loaded samples were washed with aqueous HCl solution (0.1 M) for 2 h at 240 rpm. The regenerated samples were then washed in deionized water and freeze-dried for the subsequent adsorption cycle. The recyclability test was performed in duplicate.
Results and discussion
Porous nanographene oxide (nGO)/gelatin nanocomposites were developed by a self-assembly strategy utilizing lignosulfonate-derived carbon products as building blocks. First, sodium lignosulfonate (LS) was hydrothermally carbonized into carbon spheres (CS). The CS were then oxidized into nanographene oxide (nGO)-type carbon dots to introduce oxygen-containing functional groups that give rise to improved chemical reactivity and dispersibility in polar solvents [27, 28]. Oxygen functionalities also served as tethering sites onto which LS was noncovalently attached to further enhance adsorption properties. Self-assembly of a three-dimensional porous hydrogel was thereafter initiated by using gelatin as a low cost, highly available and biodegradable crosslinker. In a final step to obtain lightweight, free-standing materials in aqueous environments, the hydrogels were freeze-dried to fabricate porous nGO/gelatin nanocomposites. Adsorption tests were performed to evaluate the adsorption performance of the composites towards Cu(II) and methylene blue (MB).
Characterization of lignosulfonate (LS)-derived carbon products
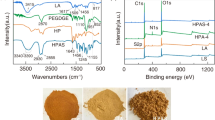
FT-IR spectroscopy (Fig. 1a) indicates that during the hydrothermal carbonization of sodium lignosulfonate, dehydration occurs and ether linkages are broken, while carboxyl and/or carbonyl groups are formed on the resulting CS product. Pure LS is characterized by a broad O-H band at 3600–3000 cm− 1, a stretching vibration peak of the aromatic ring skeleton C=C groups at 1606 cm− 1, a stretching vibration peak of C-O-C ether bonds at 1144 cm− 1, and an S=O stretching peak at 1040 cm− 1. After carbonization, CS depicted similar characteristic peaks with the exception of the disappearance of the C-O-C ether peak and the appearance of peaks attributed to the C=O (1724 cm− 1) and C-O (1225 cm− 1) stretching vibrations, which suggest the elimination of ether bonds [20, 29] and the formation carboxyl and/or carbonyl groups [14, 30]. Additionally, the decrease in the intensity of the O-H band indicates that dehydration of LS occurs during carbonization. The oxidation of carbonized LS to nGO, on the other hand, depicted a significant increase in oxygen-containing functional groups, that is the alcohol (O-H), carboxyl (C=O and C-O), and epoxide (C-O-C) groups as indicated by asymmetric ring deformation occurring at 920 cm− 1. This is similar to what was observed for nGO synthesized through the same oxidation process from other biopolymer-derived CS [16, 19]. Finally, both CS and nGO maintained some of the sulfonate groups present in lignosulfonate, which thus persisted both carbonization and the acid treatment during the oxidation process.

a FT-IR spectra, b Raman spectra, c TGA curves, and d SEM and TEM images and elemental compositions of lignosulfonate (LS) and LS-derived carbon products – carbon spheres (CS) and nanographene oxide (nGO)
An increase in the lattice defects of nGO compared to CS due to oxidation was confirmed via confocal Raman spectroscopy (Fig. 1b). D and G bands positioned at 1350–1400 cm− 1 and 1590 cm− 1, respectively, were observed for both carbon products CS and nGO, which is in agreement with what is typically observed for carbonaceous materials [31]. The D band corresponds to the presence of structural lattice defects, which arises from the cleavage of sp2 carbon bonds into sp3 bonds, while the G band represents graphitic domains [32, 33]. The amount of lattice defects thus correlates with the ID/IG ratio, which increases from 0.50 for CS to 0.71 for nGO (Supporting Information 2). This increase in ID/IG ratio suggests the introduction of defects in the form of carboxyl, hydroxyl, and epoxide groups to the planar carbon structure of nGO. Similar results were shown in previous studies by our group that employed the same microwave-assisted HTC approach to carbonize different biomass or biopolymers into carbon sphere precursors, followed by the oxidation into nGO dots. For example, carbonized and oxidized α-cellulose and carbonized and oxidized spent coffee grounds, which are of lignocellulosic nature and typically consits of 24% lignin [34], were revealed to possess X-ray powder diffraction (XRD) patterns featuring (002) facets of graphite and (001) lattice planes of graphene oxide, along with disordered carbons [19, 35]. The thickness of the graphene oxide sheets in both studies was approximately 1–3 nm, indicating a few graphene oxide layers. Carbonized but not oxidized sodium lignosulfonate, on the other hand, was found to possess an XRD pattern distinct to graphite-like structures, but with larger interplanar distance due to the presence of some defects in the form of oxygen-containing groups and hybridized sp3 carbon [36].
The thermal decomposition behavior of the carbon products was investigated by TGA as shown in Fig. 1c. LS decomposed in three stages, as depicted by an initial mass loss between 60 and 70 °C due to water removal, a second mass loss at 150–400 °C, previously ascribed to the pyrolysis of oxygen-containing groups released as aromatics (e.g., phenol, guaiacol, or syringol), alkyls, CO2, CO, and small molecules containing sulfur and sodium, and a final decomposition step at 630–760 °C attributed to the loss of the remaining oxygen-containing groups on carbon edges [37]. LS-derived CS exhibits the same initial mass loss due to water removal, as well as a second decomposition stage occurring at a much higher temperature range (230–600 °C). This decomposition step, with maximum mass loss occurring at 420 °C, was previously attributed to the cleavage of plasticizing side groups and formed functionalities from hydrothermal carbonization, which can result in a more cross-linked and thermally stable material [10]. The residue content of LS-derived CS was approximately 57 wt%, which is much higher than both that of LS and nGO. This is proposed to be due to the large amount of stable phenolic structures that contribute to char formation through a condensation reaction [20]. In the case of nGO, rapid decomposition due to the pyrolysis of labile oxygen-containing functional groups is observed between 110 and 275 °C, with maximum mass loss occurring at 150 °C. Compared to CS, nGO clearly consists of a much larger amount of oxygen groups as manifested by its significantly lower residual weight, with a carbon content of 24%.
The morphologies and elemental composition of the LS-derived carbon products were investigated using SEM-EDX and TEM (Fig. 1d). The original LS appeared invariably as spherical particles of a size up to several micrometers having needle-shaped filaments decorating its surface. During the HTC of LS, solid-solid conversion takes place as the preferred mechanism for carbon product formation, wherein only the dissolved lignin fragments and the surface fragments of non-dissolved lignin undergo carbonization [38]. It is reported that these decomposed hydrolyzed fragments are then converted to phenolic hydrochar, which finally settle on the surface of non-dissolved lignin to form the eventual structure of the carbon product [38]. The TEM micrographs of the carbonized product, CS, revealed that it has the same spherical structure and needle-like protrusions on its outer surface, but with a much smaller diameter of approximately one micrometer or less. The surface area of CS is expected to be very limited, as is typical of lignocellulosic biomass-derived hydrothermally carbonized products [39] and as demonstrated by a previous study on α-cellulose similarly carbonized via microwave-assisted HTC [40]. Although the product of carbonization primarily consisted of the aforementioned spherically shaped particles, other carbon structures were also identified, such as nanotubes and nanodots (Fig. S7) similar to what has been uncovered before [36, 41]. Apart from elongated structures, plate-like structures such as carbon flakes have also been reported [36]. The non-ordered and heterogeneous structure of LS-derived CS could be explained by the heterogeneous nature of lignin and, specifically, by its lack of uniform repeating unit [10, 41, 42]. LS-derived nGO carbon dots formed by oxidation of CS, on the other hand, appeared to be more homogeneous and composed of 10–100 nm sized nGO dots. EDX analysis confirmed the successful introduction of oxygen-containing functional groups during the oxidation step. It also confirmed, in agreement with FTIR analysis, that some of the sulfonate groups of the LS starting material were preserved during carbonization and oxidation.
Characterization of LS-derived nGO/gelatin porous nanocomposites
nGO/gelatin porous nanocomposites were fabricated through self-assembly of LS-derived nGO and gelatin into hydrogel precursors, which were subsequently freeze-dried. The apparent porosities of the adsorbents did not differ significantly among the different material compositions, and were within the range of 57–60% (Table S3). The structural stability of the porous materials, however, was highly dependent on the nGO/gelatin ratio. Hydrogel self-assembly did not occur in the absence of nGO or when the nGO content was lower than in nGO1-Gel2 or higher than in nGO1-Gel1. Similarly, gel formation also did not occur at low gelatin mass ratios (1:0.5 nGO:gelatin).
The main type of interaction between nGO and gelatin was elucidated via FT-IR spectroscopy (Fig. 2a). The evident reduction of oxygen-containing functional groups in the nGO/gelatin composites is exemplified by nGO1-Gel1-LS0.1 suggesting covalent bond formation between nGO and gelatin. More specifically, a significant decrease in the peaks for carboxyl groups (C=O at 1724 cm− 1 and C-O at 1225 cm− 1) and epoxy groups (C-O-C at 920 cm− 1) indicates partial reduction of the oxygen-containing functional groups of nGO [24]. The weakening of peaks originating from carboxyl groups is most likely due to the amidation of the carboxyl groups of nGO by the amino groups of gelatin, and the disappearance of the epoxy peak is possibly caused by the ring-opening of the epoxy groups of nGO via nucleophilic substitution reaction with amino groups of gelatin [43,44,45].

a FT-IR spectra and b TGA curves of lignosulfonate (LS)-functionalized nGO1-Gel1-LS0.1 composite and its gelatin and nanographene oxide (nGO) building blocks
TGA was performed to determine any changes in the thermal stability of the composites related to the composition of nGO, gelatin, and LS building blocks (Fig. 2b). The significant increase in decomposition temperature of the non-functionalized (nGO1-Gel1) and LS-functionalized (nGO1-Gel1-LS0.1) composites in comparison to that of LS nGO suggests enhancement of thermal stability, likely due to the strong interfacial interactions between nGO and the polar groups of gelatin [24, 46, 47]. Apart from the pyrolysis of oxygen-containing functional groups, the weight loss of approximately 50 wt% between 270 and 460 °C could also be attributed to the pyrolysis of grafted gelatin, which has a decomposition temperature between 275 and 390 °C.
The non-functionalized (nGO1-Gel1) and LS-functionalized (nGO1-Gel1-LS0.1) composites both featured open and interconnected porous networks (Fig. 3) with equal apparent porosities of 57%. The LS-functionalized composite, however, possessed more uniformly sized pores in comparison to its non-functionalized counterpart (Fig. S8a,b). This could be explained by the dispersant effect of lignosulfonate, which, due to its amphiphilic character and negatively-charged functional groups, could have facilitated the efficient dispersion of nGO within the porous composites via electrostatic repulsion [48]. Elemental maps of the materials obtained via EDX demonstrated the presence and even distribution of sulfur across the surfaces of both non-functionalized and LS-functionalized composites, whereas a higher amount of sulfonate groups were present in the latter. The notable increase in sulfur content from 1.1 to 4.6 wt% on the surface of the composites after functionalization with LS indicates successful addition of LS via noncovalent bonds.

SEM images and elemental maps (where carbon is represented in red, oxygen in green, and sulfur in blue) of non-functionalized nGO1-Gel1 (1–6) and lignosulfonate (LS)-functionalized nGO1-Gel1-LS0.1 (7–12) composites and their surface and cross-sectional elemental compositions
Adsorption of copper and methylene blue
Compared to plain nGO/gelatin, the LS-functionalized nGO/gelatin composites demonstrated enhanced adsorption capacity towards Cu(II) due to the additional active sites from lignosulfonate. Figure 4a and b show the real-time monitoring Cu(II) adsorption curves and pseudo-second order sorption kinetics (Fig. S5) of all samples, as well as that of commercial activated carbon. The adsorption capacities at equilibrium time are presented in Table 2, while the maximum adsorption capacity, 69 mg/g, was determined from the Langmuir model (Fig. S6) shown in Fig. 4c. All samples, except nGO1-Gel1-LS0.2, showed higher adsorption capacities than activated carbon. This could be due to the larger pore size and better interconnectivity of nGO/gelatin composites compared to activated carbon, which possessed a poorly connected network structure that possibly inhibited the efficient diffusion of pollutant ions or molecules (Fig. S9). The effect of pore structure on adsorption behavior was further demonstrated by the decreased adsorption capacity once the maximum gelatin and/or lignosulfonate content was reached. Apart from reduced porosity and inhomogeneous pore structure at high gelatin ratio (e.g. 1:2 nGO:gelatin), the reduction in adsorption capacity may also be due to the consumption of functional groups that act as active sites for adsorption, as a result of increased crosslink density between gelatin and nGO via covalent and noncovalent interactions [49]. As a result of this, nGO1-Gel2 and nGO1-Gel2-LS0.1 both exhibited lower adsorption capacities compared to their lower gelatin content counterparts. Lignosulfonate, on the other hand, has the tendency to self-crosslink when present in excess amount [48], thereby resulting in blockage of pores, which can reduce the adsorption performance of the composites as observed for nGO1-Gel1-LS0.2, in comparison those functionalized with lower amount of lignosulfonate.

a Real-time monitoring adsorption curves, b pseudo-second order adsorption kinetics, and c Langmuir adsorption isotherm (for nGO1-Gel1-LS0.1) for Cu(II) adsorption
The LS-functionalized composites with LS:nGO mass ratio of 1:10 all exhibited enhanced adsorption capacities towards Cu(II) in comparison to their non-functionalized counterparts. This demonstrates that the increased number of negatively-charged oxygen-containing functional groups facilitates the adsorption of positively-charged Cu(II) ions via electrostatic attraction. Furthermore, along with carboxyl groups of nGO, sulfonate groups from LS have been shown to possess greater capability to capture pollutants than the epoxy and hydroxy groups because of their probable position on the sp3-hybridized edge of nGO, thereby acting as a “netting twine” that could extend to a longer range and increase the probability of interaction with ionic pollutants [50]. The maximum adsorption capacity of the LS-functionalized nGO1-Gel1-LS0.1 was 69 mg/g, which is comparable with other carbonaceous adsorbents (Table 3). Furthermore, the time to reach equilibrium adsorption-desorption was only 40 min, which is significantly faster than equilibrium times of many adsorbents [51, 53, 55]. It should be noted, however, that a direct comparison between the different adsorbents is difficult because adsorption behavior is affected by several factors, such as adsorbent dosage, initial adsorbate concentration, and volume of adsorbate solution, to name a few.
Electrostatic interactions were assumed to be the mechanism that governed the adsorption of positively charged Cu(II) ions by nGO and LS with negatively-charged oxygen-containing functional groups and sulfonate groups. The potential of the composites for cationic dye adsorption was, therefore, also investigated. Methylene blue (MB) was chosen as the model cationic dye for adsorption on LS-functionalized nGO1-Gel1-LS0.1 composite. The material exhibited an adsorption capacity of 38 mg/g after 130 min. Compared to the adsorption of Cu(II), the adsorption of methylene blue occurred at a much slower pace; after 130 min, a removal efficiency of 35% was achieved. It should be noted that adsorption equilibrium had not been reached yet. Apart from electrostatic forces, the adsorption of MB by graphene-based carbon materials has been reported to also be governed by π-π stacking interactions and hydrogen bonding [57]. The differences in the adsorption performance towards Cu(II) and MB may be due to the large size of the dye compared to the heavy metal ion [58].
FT-IR spectroscopy was performed in order to elucidate the chemical alterations after adsorption of Cu(II) and MB on the nGO1-Gel1-LS0.1 composite (Fig. 2a). Several additional bands appeared after MB-adsorption, specifically at 885 cm− 1 and within the range 1390–1450 cm− 1. These bands are associated with the aromatic group C=C bend and C=C stretch, respectively. In addition, an increase in the intensity of the peak at 1331 cm− 1 was observed, which indicated the presence of additional aromatic amine C-N groups besides those provided by gelatin. These results confirmed the adsorption of MB. As expected, the spectrum of the composite after Cu-adsorption exhibited no new peaks compared to pristine sample.
Morphological alterations and elemental compositions of nGO1-Gel1-LS0.1 after Cu(II) and MB-adsorption were investigated using SEM and EDX. Figure 5 shows the SEM images of the sample before and after the adsorption of Cu(II) and MB. The adsorbed Cu(II) is revealed as crystal-like structures, which suggests that besides the adsorption of the heavy metal ion, the sulfate anion of the salt was also simultaneously adsorbed and formed a chelate complex [59]. After MB adsorption, spherical methylene blue particulates were found on the surface. To confirm the compositions of the surfaces and observed structures, elemental mapping and multipoint analysis via EDX were performed. Elemental maps after Cu(II) adsorption confirmed that the aforementioned crystal structures are mainly composed of copper. Up to 25 wt% of elemental copper was detected on the surface, while only 13 wt% was found in the cross-section. Similarly, a higher amount of sulfur, which indicated MB adsorption, was present on the surface after MB adsorption compared to the cross-section. This suggests the preferential adsorption of both Cu(II) and MB onto the surface of the composite, possibly due to the larger number of active sites from functionalized LS or to the limited diffusivity of the adsorbates through the pores.

SEM images and elemental maps of (a) Cu-adsorbed (1–5) and (b) methylene blue (MB)-adsorbed (6–10) nGO1-Gel1-LS0.1 composite and their surface and cross-sectional elemental compositions
Recyclability test
The reusability of the adsorbent is one of the key factors for potential use in industrial applications. Five cycles of adsorption-desorption were performed for the recyclability test (Fig. 6). After each Cu(II)-adsorption cycle, the composite was regenerated via immersion in 0.1 M aqueous HCl solution for 2 h prior to next re-adsorption. The sample was found to retain 79% of its adsorption capacity after the fifth cycle. This demonstrates that in spite of the use of a low concentration of acid and short regeneration time, the composite is adequately able to sustain its adsorption capacity. Additionally, 100% of the composite is reclaimed after the recyclability test. Other work reported in literature generally required a regeneration step with a stronger acid (e.g. HNO3) [60] at higher concentration (e.g. 0.3 M HCl) [61] and longer regeneration time (e.g. 24 h) [62]. It is possible that a longer regeneration time would further improve the reusability.

Recyclability test results involving five cycles of Cu(II) adsorption-desorption for lignosulfonate (LS)-functionalized nGO1-Gel1-LS0.1 composites
Conclusions
Free-standing three-dimensional (3D) lignin-derived porous nGO-based nanocomposites with excellent adsorption capacity for heavy metal ions and cationic dyes were successfully designed. nGO was derived through a two-step reaction, including carbonization and oxidation of lignosulfonate. The abundant oxygen functionalities on nGO conferred the construction of porous composites possessing three-dimensional (3D) interconnected network structure through the self-assembly of hydrogels using only gelatin as a crosslinker, coupled by freeze-drying method. Functionalization with lignosulfonate (LS) provided additional negatively-charged functional groups that served as active sites for adsorption and as dispersants for nGO via electrostatic repulsion. The dual role of LS enhanced the equilibrium adsorption capacity of the composites, which increased from 50 mg/g to 66 mg/g for the non-functionalized and LS-functionalized nGO1-Gel1-LS0.1 composites, respectively. Furthermore, the LS-functionalized composites demonstrated good reusability through a mild regeneration step, with 79% retention of the adsorption capacity after five adsorption-desorption cycles. Apart from heavy metal ions, the potential for cationic dye adsorption was also demonstrated as the LS-functionalized composite exhibited an adsorption capacity of 38 mg/g for methylene blue. The 3D porous constructs fabricated from lignosulfonate-derived functional carbon products were demonstrated as promising fully biobased materials for adsorption of environmental contaminants.
Availability of data and materials
Most of the data generated or analyzed during this study are included in this published article and its supplementary information files. Remaining data is available from the corresponding author on reasonable request.
Abbreviations
- ATR:
-
Attenuated total reflectance
- CS:
-
Carbon spheres
- EDX:
-
Energy-dispersive x-ray spectroscopy
- FE-SEM:
-
Field emission scanning electron microscopy
- FT-IR:
-
Fourier transform infrared spectroscopy
- GO:
-
Graphene oxide
- HTC:
-
Hydrothermal carbonization
- LS:
-
Lignosulfonate
- MB:
-
Methylene blue
- MWCNT:
-
Multi-walled carbon nanotubes
- nGO:
-
Nanographene oxide
- PDI:
-
Polydispersity index
- PEI:
-
Polyethylene imine
- TEM:
-
Transmission electron microscopy
- TGA:
-
Thermogravimetric analysis
- XRD:
-
X-ray diffraction
References
U. Alvarez, C. Granda, R. Santamaria, R. Menendez, New alternatives to graphite for producing graphene materials. Carbon. 93, 812–818 (2015). https://doi.org/10.1016/j.carbon.2015.05.105
S.H. Jung, Y. Myung, B.N. Kim, I.G. Kim, I.K. You, T.Y. Kim, Activated biomass-derived graphene-based carbons for supercapacitors with high energy and power density. Sci. Rep. (2018). https://doi.org/10.1038/s41598-018-20096-8
Z. Gao, Y. Zhang, N. Song, X. Li, Biomass-derived renewable carbon materials for electrochemical energy storage. Mat Res Lett 5, 69–88 (2017). https://doi.org/10.1080/21663831.2016.1250834
Q. Ma, Y. Yu, M. Sindoro, A.G. Fane, R. Wang, H. Zhang, Carbon-based functional materials derived from waste for water remediation and energy storage. Adv. Mater. (2017). https://doi.org/10.1002/adma.201605361
C.R. Correa, M. Stollovsky, T. Hehr, Y. Rauscher, B. Rolli, A. Kruse, Influence of the carbonization process on activated carbon properties from lignin and lignin-rich biomasses. ACS Sustain Chem Eng 5(9), 8222–8233 (2017). https://doi.org/10.1021/acssuschemeng.7b01895
L. Guardia, L. Suarez, N. Querejeta, V. Vretenar, P. Kotrusz, V. Skakalova, T. Centeno, Biomass waste-carbon/reduced graphene oxide composite electrodes for enhanced supercapacitors. Electrochim. Acta 298, 910–917 (2019). https://doi.org/10.1016/j.electacta.2018.12.160
C. Xu, M. Stromme, Sustainable porous carbon materials derived from wood-based biopolymers for CO2 capture. Nanomaterials (2019). https://doi.org/10.3390/nano9010103
W. Lan, J.S. Luterbacher, A road to profitability from lignin via the production of bioactive molecules. ACS Cent Sci 5(10), 1642–1644 (2019). https://doi.org/10.1021/acscentsci.9b00954
S.P. Leitner, G. Gratzl, C. Paulik, H.K. Weber, Carbon materials from lignin and sodium lignosulfonate via Diisocyanate cross-linking and subsequent carbonization. C. 1(1), 43–57 (2015). https://doi.org/10.3390/c1010043
H. Wikberg, T. Ohra-aho, F. Pileidis, M.M. Titirici, Structural and morphological changes in Kraft lignin during hydrothermal carbonization. ACS Sustain. Chem. Eng. 3(11), 2737–2745 (2015). https://doi.org/10.1021/acssuschemeng.5b00925
H. Mao, X. Chen, R. Huang, M. Chen, R. Yang, P. Lan, M. Zhou, F. Zhang, Y. Yang, X. Zhou, Fast preparation of carbon spheres from enzymatic hydrolysis lignin: Effects of hydrothermal carbonization conditions. Sci. Rep. (2018). https://doi.org/10.1038/s41598-018-27777-4
D. Saha, Y. Li, Z. Bi, J. Chen, J. Keum, D. Hensley, H. Grappe, H. Meyer, S. Dai, P. Paranthaman, A. Naskar, Studies on supercapacitor electrode material from activated lignin-derived mesoporous carbon. Langmuir 30(3), 900–910 (2014). https://doi.org/10.1021/la404112m
S. Hu, Y.L. Hsieh, Ultrafine microporous and mesoporous activated carbon fibers from alkali lignin. J. Mater. Chem. A 1, 11279–11288 (2013). https://doi.org/10.1039/C3TA12538F
M. Snowdon, A. Mohanty, M. Misra, A study of carbonized lignin as an alternative to carbon black. ACS Sustain. Chem. Eng. 2(5), 1257–1263 (2014). https://doi.org/10.1021/sc500086v
C.I. Contescu, S.P. Adhikari, N.C. Gallego, N.D. Evans, B.E. Biss, Activated Carbons Derived from High-Temperature Pyrolysis of Lignocellulosic Biomass. C (2018). https://doi.org/10.3390/c4030051
D. Wu, H. Xu, M. Hakkarainen, From starch to polylactide and nano-graphene oxide: Fully starch derived high performance composites. RSC Adv. 6, 54336–54345 (2016). https://doi.org/10.1039/C6RA08194K
S. Hassanzadeh, N. Aminlashgari, M. Hakkarainen, Chemo-selective high yield microwave assisted reaction turns cellulose to green chemicals. Carbohydr. Polym. 112, 448–457 (2014). https://doi.org/10.1016/j.carbpol.2014.06.011
K.H. Adolfsson, S. Hassanzadeh, M. Hakkarainen, Valorization of cellulose and waste paper to graphene oxide quantum dots. RSC Adv. 5, 26550–26558 (2015). https://doi.org/10.1039/C5RA01805F
N.B. Erdal, K.H. Adolfsson, T. Pettersson, M. Hakkarainen, Green strategy to reduced Nanographene oxide through microwave assisted transformation of cellulose. ACS Sustain. Chem. Eng. 6, 1246–1255 (2018). https://doi.org/10.1021/acssuschemeng.7b03566
S. Kang, X. Li, J. Fan, J. Chang, Classified separation of lignin hydrothermal liquefied products. Ind Eng Chem Res 50(19), 11288–11296 (2011). https://doi.org/10.1021/ie2011356
M. Si, J. Zhang, Y. He, Z. Yang, X. Yan, M. Liu, S. Zhuo, S. Wang, X. Min, C. Gao, L. Chai, Y. Shi, Synchronous and rapid preparation of lignin nanoparticles and carbon quantum dots from natural lignocellulose. Green Chem. 20, 3414–3419 (2018). https://doi.org/10.1039/C8GC00744F
S. Elaigwu, G. Greenway, Microwave-assisted and conventional hydrothermal carbonization of lignocellulosic waste material: Comparison of the chemical and structural properties of the hydrochars. J. Anal. Appl. Pyrolysis 118, 1–8 (2016). https://doi.org/10.1016/j.jaap.2015.12.013
Y. Piao, B. Chen, Self-assembled graphene oxide-gelatin nanocomposite hydrogels: Characterization, formation mechanisms, and pH-sensitive drug release behavior. J. Polym. Sci. B 53, 356–367 (2014). https://doi.org/10.1002/polb.23636
Y. Piao, B. Chen, One-pot synthesis and characterization of reduced graphene oxide-gelatin nanocomposite hydrogels. RSC Adv. 6(8), 6171–6181 (2016). https://doi.org/10.1039/C5RA20674J
S. Shaibu, F. Adekola, H. Adegoke, O. Ayanda, A. Comparative, Study of the adsorption of methylene blue onto synthesized nanoscale zero-valent iron-bamboo and manganese-bamboo composites. Materials. 7, 4493–4507 (2014). https://doi.org/10.3390/ma7064493
L. Zhang, L. Tu, Y. Liang, Q. Chen, Z. Li, C. Li, Z. Wang, W. Li, Coconut-based activated carbon fibers for efficient adsorption of various organic dyes. RSC Adv. 8, 42280–42291 (2018). https://doi.org/10.1039/C8RA08990F
S. Nardecchia, D. Carriazo, M.L. Ferrer, M. Gutierrez, F. del Monte, Three dimensional macroporous architectures and aerogels built of carbon nanotubes and/or graphene: Synthesis and applications. Chem. Soc. Rev. 42, 794–830 (2013). https://doi.org/10.1039/C2CS35353A
Q. Fang, B. Chen, Self-assembly of graphene oxide aerogels by layered double hydroxides cross-linking and their application in water purification. J. Mater. Chem. A 2, 8941–8951 (2014). https://doi.org/10.1039/C4TA00321G
Z. Ding, F. Li, J. Wen, X. Wang, R. Sun, Gram-scale synthesis of single-crystalline graphene quantum dots derived from lignin biomass. Green Chem. 20, 1383–1390 (2018). https://doi.org/10.1039/C7GC03218H
D. Kim, K. Lee, K. Park, Upgrading the characteristics of biochar from cellulose, lignin, and xylan for solid biofuel production from biomass by hydrothermal carbonization. J Indust Eng Chem 42, 95–100 (2016). https://doi.org/10.1016/j.jiec.2016.07.037
L. Bokobza, J. Bruneel, M. Couzi, Raman spectra of carbon-based materials (from graphite to carbon black) and of some silicone composites. C. 1, 77–94 (2015). https://doi.org/10.3390/c1010077
E. Biru, H. Iovu, Graphene Nanocomposites Studied by Raman Spectroscopy (IntechOpen, 2018). https://doi.org/10.5772/intechopen.73487
K. Tsirka, A. Katsiki, N. Chalmpes, D. Gournis, A. Paipetis, Mapping of graphene oxide and single layer graphene flakes – Defects annealing and healing. Front Mater (2018). https://doi.org/10.3389/fmats.2018.00037
L. Ballesteros, J. Teixeira, S. Mussatto, Chemical, functional, and structural properties of spent coffee grounds and coffee Silverskin. Food Bioproc. Tech. 7, 3493–3503 (2014). https://doi.org/10.1007/s11947-014-1349-z
H. Xu, L. Xie, M. Hakkarainen, Coffee-ground-derived quantum dots for aqueous Processable Nanoporous graphene membranes. ACS Sustain. Chem. Eng. 5, 5360–5367 (2017). https://doi.org/10.1021/acssuschemeng.7b00663
G. Melilli, K. Adolfsson, A. Impagnatello, G. Rizza, M. Hakkarainen, Intriguing Carbon Flake Formation during Microwave-Assisted Hydrothermal Carbonization of Sodium Lignosulfonate, Global Challenges (2020), p. 1900111. https://doi.org/10.1002/gch2.201900111
J. Pang, W. Zhang, J. Zhang, G. Cao, M. Han, Y. Yang, Facile and sustainable synthesis of sodium lignosulfonate derived hierarchical porous carbons for supercapacitors with high volumetric energy density. Green Chem. 19, 3916–3926 (2017). https://doi.org/10.1039/C7GC01434A
K. Shimin, X. Li, J. Fan, J. Chang, Characterization of Hydrochars produced by hydrothermal carbonization of lignin, cellulose, D-xylose, and wood meal. Ind. Eng. Chem. Res. 51, 9023–9031 (2012). https://doi.org/10.1021/ie300565d
M.M. Titirici, R. White, C. Falco, M. Sevilla, Black perspectives for a green future: Hydrothermal carbons for environment protection and energy storage. Energ. Environ. Sci. 5, 6796–6822 (2012). https://doi.org/10.1039/C2EE21166A
K. Adolfsson, M. Golda-Cepa, N. Erdal, J. Duch, A. Kotarba, M. Hakkarainen, Importance of surface functionalities for antibacterial properties of carbon spheres. Adv Sustainable Syst 3, 1800148 (2019). https://doi.org/10.1002/adsu.201800148
W. Gindl-Altmutter, J. Köhnke, C. Unterweger, N. Gierlinger, J. Keckes, J. Zalesak, O. Rojas, Lignin-based multiwall carbon nanotubes. Composites Part A 121, 175–179 (2019). https://doi.org/10.1016/j.compositesa.2019.03.026
W. Sagues, A. Jain, D. Brown, S. Aggarwal, A. Suarez, M. Kollman, S. Park, D. Argyropoulos, Are lignin-derived carbon fibers graphitic enough? Green Chem. 21, 4253–4265 (2019). https://doi.org/10.1039/C9GC01806A
V. Luan, J. Chung, E. Kim, S. Hur, The molecular level control of three-dimensional graphene oxide hydrogel structure by using various diamines. Chem. Eng. J. 246, 64–70 (2014). https://doi.org/10.1016/j.cej.2014.01.105
A. Bourlinos, D. Gournis, D. Petridis, T. Szabó, A. Szeri, I. Dékány, Graphite oxide: Chemical reduction to graphite and surface modification with primary aliphatic amines and amino acids. Langmuir. 19, 6050–6055 (2003). https://doi.org/10.1021/la026525h
O. Compton, D. Dikin, K. Putz, C. Brinson, S. Nguyen, Electrically conductive “alkylated” graphene paper via chemical reduction of amine-functionalized graphene oxide paper. Adv. Mater. 22, 892–896 (2010). https://doi.org/10.1002/adma.200902069
C. Wan, M. Frydrych, B. Chen, Strong and bioactive gelatin-graphene oxide nanocomposites. Soft Matter 7, 6159–6166 (2011). https://doi.org/10.1039/C1SM05321C
E.M. Zadeh, A. Yu, L. Fu, M. Dehgan, I. Sbarski, I. Harding, Physical and thermal characterization of graphene oxide modified gelatin-based thin films. Polym. Compos. 35, 2043–2049 (2014). https://doi.org/10.1002/pc.22865
F. Li, X. Wang, T. Yuan, R. Sun, A lignosulfonate-modified graphene hydrogel with ultrahigh adsorption capacity for Pb(II) removal. J. Mater. Chem. A 4, 11888–11896 (2016). https://doi.org/10.1039/C6TA03779H
Z. Feng, T. Danjo, K. Odelius, M. Hakkarainen, T. Iwata, A.-C. Albertsson, Recyclable fully biobased chitosan adsorbents spray-dried in one-pot to microscopic size and enhanced adsorption capacity. Biomacromolecules. 20, 1956–1964 (2019). https://doi.org/10.1021/acs.biomac.9b00186
Y. Shen, B. Chen, Sulfonated graphene Nanosheets as a superb adsorbent for various environmental pollutants in water. Environ. Sci. Technol. 49, 7364–7372 (2015). https://doi.org/10.1021/acs.est.5b01057
R. Xie, Y. Jin, Y. Chen, W. Jiang, The importance of surface functional groups in the adsorption of copper onto walnut shell derived activated carbon. Water Sci. Technol. 76(11), 3022–3034 (2017). https://doi.org/10.2166/wst.2017.471
X. Mi, G. Huang, W. Xie, W. Wang, Y. Liu, J. Gao, Preparation of graphene oxide aerogel and its adsorption for Cu2+ ions. Carbon. 50, 4856–4864 (2012). https://doi.org/10.1016/j.carbon.2012.06.013
C. Jiao, J. Xiong, J. Tao, S. Xu, D. Zhang, H. Lin, Y. Chen, Sodium alginate/graphene oxide aerogel with enhanced strength-toughness and its heavy metal adsorption study. Int. J. Biol. Macromol. 83, 133–141 (2016). https://doi.org/10.1016/j.ijbiomac.2015.11.061
L. Pan, Z. Wang, Q. Yang, R. Huang, Efficient removal of Lead, copper and cadmium ions from water by a porous calcium alginate/graphene oxide composite aerogel. Nanomaterials 8, 957–997 (2018). https://doi.org/10.3390/nano8110957
Y. Chen, L. Chen, H. Bai, L. Li, Graphene oxide-chitosan composite hydrogels as broad-spectrum adsorbents for water purification. J. Mater. Chem. A 1, 1992–2001 (2013). https://doi.org/10.1039/C2TA00406B
Z. Sui, Q. Meng, X. Zhang, R. Ma, B. Cao, Green synthesis of carbon nanotube-graphene hybrid aerogels and their use as versatile agents for water purification. J. Mater. Chem. 22, 8767–8771 (2012). https://doi.org/10.1039/C2JM00055E
J. Gong, J. Liu, X. Chen, Z. Jiang, X. Wen, E. Mijowska, T. Tang, Converting real-world mixed waste plastics into porous carbon nanosheets with excellent performance in the adsorption of an organic dye from wastewater. J. Mater. Chem. A 3, 341–351 (2015). https://doi.org/10.1039/C4TA05118A
R.R. Elmorsi, S.T. El-Wakeel, W.A. El-Dein, H.R. Lotfy, W.E. Rashwan, M. Nagah, S.A. Shaaban, S.A. Ahmed, I.Y. El-Sherif, K.S. Abou-El-Sherbini, Adsorption of methylene blue and Pb2+ by using acid-activated Posidonia oceanica waste. Sci. Rep. (2019). https://doi.org/10.1038/s41598-019-39945-1
M. Mende, D. Schwarz, C. Steinbach, R. Boldt, S. Schwarz, Simultaneous adsorption of heavy metal ions and anions from aqueous solutions on chitosan-investigated by spectrophotometry and SEM-EDX analysis. Colloids and Surfaces A: Physicochem Eng Aspects 510, 275–282 (2016). https://doi.org/10.1016/j.colsurfa.2016.08.033
X. Liu, L. Huang, L. Wang, C. Wang, X. Wu, G. Dong, Y. Liu, Preparation, adsorptive properties and chemical regeneration studies of high-porous activated carbon derived from Platanus orientalis leaves for Cr(VI) removal. J. Water Health 16.5, 814–826 (2018). https://doi.org/10.2166/wh.2018.068
X. Yi, F. Sun, Z. Han, F. Han, J. He, M. Ou, J. Gu, X. Xu, Graphene oxide encapsulated polyvinyl alcohol/sodium alginate hydrogel microspheres for cu (II) and U (VI) removal. Ecotoxicol. Environ. Saf. 158, 309–318 (2018). https://doi.org/10.1016/j.ecoenv.2018.04.039
Y. He, S. Li, X. Li, Y. Yang, A. Tang, L. Du, Z. Tan, D. Zhang, H. Chen, Graphene (rGO) hydrogel: A promising material for facile removal of uranium from aqueous solution. Chem. Eng. J. 338, 333–340 (2018). https://doi.org/10.1016/j.cej.2018.01.037
Acknowledgements
Not applicable.
Funding
The Swedish Research Council, VR, (Contract Grant 2018–03451) is gratefully acknowledged for financial support. Open access funding provided by Royal Institute of Technology
Author information
Authors and Affiliations
Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yao, J., Odelius, K. & Hakkarainen, M. Carbonized lignosulfonate-based porous nanocomposites for adsorption of environmental contaminants. Functional Composite Mater 1, 5 (2020). https://doi.org/10.1186/s42252-020-00008-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42252-020-00008-8