
Abstract
Background
The B cell chemoattractant CXCL13 is a promising biomarker in rheumatoid arthritis (RA), with a plausible role in supporting diagnosis, monitoring disease activity and as a prognostic value. It is a key chemokine driving the formation of lymphoid follicles within the inflamed synovium. The objective of this systematic review was to evaluate the role of CXCL13 as a viable biomarker in RA.
Methods
We conducted a systematic literature review of all published cohort and randomised controlled trials evaluating the role of CXCL13 in RA. The primary outcomes were; i) CXCL13 levels in RA patients compared to healthy controls, ii) the correlation between CXCL13 and markers of disease activity, and iii) the association between CXCL13 and treatment response.
Results
The search produced 278 articles, of which 31 met the inclusion criteria. Of the 12 studies evaluating CXCL13 expression in early or established RA, all reported higher levels than that seen in healthy controls. Twelve of sixteen studies reported a weakly positive correlation between CXCL13 and markers of disease activity including DAS28 and swollen joint count, with rho values between 0.20–0.67. In 2 studies, CXCL13 levels correlated with ultrasonographic evidence of synovitis. Eighteen studies assessed CXCL13 in response to therapeutic intervention. The majority signified a fall in levels in response to treatment including biologics and Janus kinase (JAK) inhibition. In some, this reduction was only seen in treatment responders. High CXCL13 levels predicted failure to achieve disease remission with csDMARDs. The evidence for treatment prediction with biologics was conflicting.
Conclusion
Despite evidence to suggest a role in diagnosing RA and in detecting synovitis, the heterogeneity of studies included in this review limit our ability to draw robust conclusions. At present there are inadequate results to justify the routine use of CXCL13 as a biomarker in RA routine clinical practice.
Similar content being viewed by others


Background
Rheumatoid arthritis (RA) is a chronic, systemic immune-mediated disease characterised by synovial inflammation and progressive joint destruction [1]. Although the classical paradigm of RA pathogenesis is centred on the role of the CD4+ T cells, it is clear that B cells also demonstrate a vital role including cytokine secretion, antigen presentation, and interaction with other inflammatory cells [2, 3]. Autoreactive B cells are also activated by T cells to produce IgG autoantibodies that may be directly involved in joint damage [2, 3].
Existing biomarkers in RA, including acute phase reactants have their limitations. C-reactive protein (CRP) is a sensitive measure of inflammation but lacks disease specificity for joint inflammation and elevated levels are seen in intercurrent illness or with obesity [4]. The erythrocyte sedimentation rate (ESR) is non-specific, influenced by confounding factors such as age, sex, anaemia, renal disease, immunoglobulin and fibrinogen levels [5]. It is not sensitive to short-term changes, with a slow response to the acute phase reaction leading to false negatives in an early inflammatory process [5]. Both CRP and ESR lack sensitivity in patients with limited small joint or predominantly lower limb involvement and in those receiving immunosuppression with steroids or drugs that target interleukin-6 (IL-6) e.g. tocilizumab [6]. IL-6 is a key driver of the acute phase response with an important role in the liver’s production of CRP. Rheumatoid factor (RF) and anti-citrullinated peptide (ACPA) have value in diagnostics, although may be negative in up to 30% of patients [7, 8]. They do have use as a prognostic marker of disease severity but not as markers of disease activity [9]. There is a current need for a biomarker with improved sensitivity which can aid diagnosis, monitor disease activity and provide prognostic value.
CXCL13, also known as B-lymphocyte chemoattractant (BLC) or C-X-C motif chemokine 13, is a key chemokine involved in the positioning and activation of cells at lymphoid and extra-lymphoid sites. It binds to its cognate receptor CXCR5, and attracts CXCR5-expressing B cells and follicular T-helper cells [10]. Overexpression of CXCL13 in extra-lymphoid structures enhances B cell migration and promotes ectopic lymphoid neogenesis [11]. In the RA inflammatory responses, TNF-alpha and IL-6 upregulate CXCL13 expression, driving the development of B cell follicles and germinal centre reactions within the synovium [10, 12]. The CXCL13-CXCR5 interaction enables B-cell maturation into plasma cells and the production of antibodies [13].
CXCL13 is of interest as a biomarker in RA, with a profound effect on shaping local synovial architecture and may provide a signature of a specific RA disease subset. Several studies have investigated the link between CXCL13, disease activity, serological markers (such as RF and ACPA) and change in response to treatment. To our knowledge, these studies have not been analysed collectively. Our objective was to evaluate the current existing literature in RA, to assess how CXCL13 correlates with disease activity, treatment response, and to determine its use as a potential biomarker in clinical practice.
Methods
Search strategy
The literature was searched systematically by two investigators (CS and AD) using MEDLINE and EMBASE databases. The search terms used were ‘rheumatoid arthritis’ OR ‘inflammatory arthritis’ AND ‘CXCL13’ OR ‘Chemokine Ligand 13’. The search was undertaken in February 2018 and re-run prior to the final analysis to identify new studies that could be retrieved for incorporation in the systematic review.
Study selection
We identified English language publications of cohort or randomised controlled trials (RCTs). Publications were included if they met the following eligibility criteria: 1) the study evaluated CXCL13 levels 2); the study included patients diagnosed with RA based on the American College of Rheumatology (ACR) criteria. Studies presenting duplicate data were excluded. Titles and abstracts were screened independently by two investigators (CS and AD). The full text of the potential studies for inclusion were retrieved and assessed for eligibility. Data was extracted independently (by CS and AD). Disagreements over study eligibility were resolved through discussion with a third reviewer (KB). The primary outcome of interest was i) the level of CXCL13 in RA patients compared to healthy control (HC) and ii) the correlation between CXCL13 levels and markers of disease activity and iii) the association between CXCL13 and treatment response.
Results
The search identified 278 articles. Of these, 228 were excluded based on title and abstract. A further 19 studies were excluded after full paper review. Figure 1 summarises the flowchart in accordance with the PRISMA statement [9]. In total, 31 studies were included, comprising 20 published articles and 11 conference abstracts. Of these studies, 8 were randomised controlled trials (RCTs) and 23 observational cohorts (Table 1). The total number of patients analysed were 5894, with a median population of 132 (IQR: 57–242) in the RCTs and 147 (IQR: 30–225) in the observational cohorts. Nearly all RCTs evaluated CXCL13 levels in patients with established RA (EstRA), whilst observational cohorts assessed patients with both early (ERA) and established disease.

Flow chart of studies included in the systematic review
CXCL13 assays
CXCL13 was measured in serum in 25 studies, in synovial tissue in 4 studies, and in both serum and synovial tissue in the remaining 2 studies (Tables 1 and 2). The majority of studies quantified CXCL13 levels using commercial enzyme-linked immunosorbent assay (ELISA) kits, although an assortment of manufacturers was sourced. The remaining studies used Luminex -based assays [35], electrochemiluminescent (ECLA) [40], or a combination of ELISA, Luminex or Meso Scale Discovery (MSD) [18, 41]. Six studies did not mention the immunoassay method used [19, 30, 33, 34, 37, 42]. The method of measurement is of importance as data generated with Luminex technology is sensitive to heterophilic antibodies in the serum, such as rheumatoid factor and may result in false positive cytokine levels. Some studies analysed values as continuous variables whilst other studies employed a predefined cut off or a level above the median.
Baseline levels of CXCL13 in patients with rheumatoid arthritis
Twelve studies reported baseline CXCL13 levels compared to healthy controls; 7 in early RA, 3 in established disease, and 2 studies analysed levels in both patient groups (Table 1). All of the studies reported higher levels of CXCL13 in RA compared to healthy controls. Moura et al. [15] compared levels in early RA to that seen in established disease and did not detect significant differences.
CXCL13 as a marker of disease activity and severity
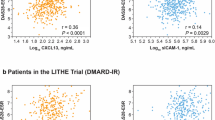
Sixteen studies evaluated how well levels of CXCL13 correlated with other measures of RA disease activity (Table 1). These included; tender joint counts (TJC), swollen joint counts (SJC), ESR, CRP and Disease Activity Score with ESR (DAS28-ESR) and CRP (DAS28-CRP) and ultrasound. Twelve of the sixteen studies reported a positive correlation between markers of disease activity and CXCL13. For the most part, these were weakly positive correlations, with rho values between 0.20–0.67. The strongest correlation was reported in association with DAS28-ESR [28]. Correlations were also noted with DAS28-CRP [17, 26, 27]. Six studies reported on the individual components of the DAS28 score. In half of these baseline CXCL13 weakly correlated with the swollen joint count with rho values between 0.23–0.34 [23, 31, 32]. The majority of studies used the 28-joint count. No significant correlation was observed with the tender joint count. One study reported a correlation with patient-derived measures (patient global assessment) [23]. Four studies examined CXCL13 as a marker of ultrasound synovitis, reporting a correlation with ultrasonographic scores for Grey Scale (r = 0.27) [24, 31] and Power Doppler signals (r = 0.26–0.69) [24, 21, 29, 31]. Four studies evaluated the association between CXCL13 level and seropositivity (RF and CCP). CXCL13 levels were found to be higher in RF and ACPA positive patients [16, 19, 32], with a positive correlation between synovial CXCL13 expression and ACPA titres [22]. Two studies assessed the association with CXCL13 and radiological evidence of erosive disease, demonstrating a strong association with progression of joint destruction in patients with high baseline CXCL13 levels [32].
Baseline and change in CXCL13 levels with treatment
Nineteen studies evaluated changes in CXCL13 levels with treatment (Table 2). The therapies evaluated included conventional synthetic DMARDs (n = 4), TNF inhibitors including adalimumab, etanercept, infliximab and golimumab (n = 7), IL-6 cytokine and receptor blockers including tocilizumab, vobarilizumab, sarilumab (n = 6), anti-CD20 B cell depleting therapy rituximab (n = 2), JAK inhibitors tofacitinib and filgotinib (n = 2) and anti-lymphotoxin alpha agent pateclizumab (n = 2).
In csDMARD treated patients, high baseline levels of CXCL13 were associated with a reduced likelihood of achieving low disease activity. Bugatti reported a high negative predictive value of 80% for remission after 12 months of methotrexate therapy [19]. With anti-TNF therapy, the findings were not consistent across studies. Three studies reported reductions in CXCL13 levels with adalimumab and etanercept in treatment responders [16, 17, 42]. Kennedy et al. reported a significant reduction in CXCL13 with adalimumab compared to placebo, with levels returning to pre-dose values during the safety follow-up [41]. However, Moura et al. found no changes in CXCL13 levels before and after therapy although the authors did not stratify by therapeutic response [15]. The inconsistencies may be explained by Dennis et al. observation, that TNF response rates are influenced by the predominate RA synovial phenotype. High serum CXCL13 accompanied a lymphoid phenotype which associates with a lower response rate to TNF, compared to patients with a myeloid phenotype which associated with low CXC13 levels and high soluble intercellular adhesion molecule 1 levels [40]. Similar discrepancies were reported with anti-IL6 therapy. Rinaldi et al. reported a gradual reduction in CXCL13 levels with vobarilizumab and tocilizumab, with levels increasing back to pre-treatment levels on treatment discontinuation [33]. Gabay et al. reported a statistically significant reduction in CXCL13 levels with sarilumab, which was greatest in treatment responders [38], whilst Loza et al. did not document any change with sirukumab treatment [18].
Reduction in levels were also noted with rituximab, tofacitinib and filgotinib and pateclizumab. With rituximab, CXCL13 levels significantly decreased in all patients coinciding with B cell depletion irrespective of responder status [35, 36, 44]. Baseline levels did not predict response to therapy, however low levels predicted a lower B cell return at 6 months compared to high baseline level [44]. The JAK inhibitors (tofacitinib and filgotinib) reduced a range of RA associated tissue derived biomarker including CXCL13 [37, 39]. In the pateclizumab studies, CXCL13 were evaluated as a biomarker for target modulation. Levels decreased rapidly and significantly compared to treatment with placebo and returned to pre-dose levels during safety follow up. Despite reductions in CXCL13 levels, improvement in disease activity was not statistically significant [41, 43].
Discussion
To our knowledge, this is the first systematic review exploring whether CXCL13 is a clinically viable diagnostic marker in RA. CXCL13 is raised in RA, both in early and established disease. Serum levels may predict active joint inflammation and synovitis over current biomarkers. Although levels fall in response to certain RA therapies, the prognostic value of CXCL13 in treatment response remains unclear.
CXCL13 is a marker of synovial inflammation in patients with RA. Levels are significantly higher than seen in healthy controls, present in very early RA (even in cases of less than 6 weeks disease duration) [25] and in both seropositive and seronegative disease, signifying a promising role as a diagnostic marker. However CXCL13 is also expressed in other inflammatory diseases driven by lymphoid organization, for example Sjogren’s syndrome, where levels correlate with the extent of salivary gland inflammation and subsequent lymphoma development [28].
Serum CXCL13 levels can be used as a surrogate marker for synovitis. CXCL13 is produced by multiple cell types within the synovium, including T cells, monocytes/macrophages, endothelial cells and fibroblast [25]. A number of synovial inflammatory markers correlate with CXCL13 synthesis [31, 44] and levels reflect the extent of synovial inflammation in clinical and imaging evaluation [31]. The inflamed rheumatoid synovium significantly contributes to serum CXCL13 levels, with a strong relationship between synovial expression and serum level [24, 26]. This is in contrast to acute phase proteins, which are primarily produced by hepatocytes released during inflammation, or other cytokines expressed in the synovium which are low or undetectable in the serum. In this study we found that CXCL13 levels correlated with swollen joint counts and ultrasound findings of synovitis. It is biologically plausible that serum CXCL13 levels directly reflect synovial production and synovial inflammation. Measurement of CXCL13 may help to predict active joint inflammation, yielding this biomarker a valuable surrogate in the objective assessment of the synovitis.
However, the evidence surrounding the correlation between CXCL13 and markers of disease activity is conflicting. Some studies report associations with DAS28, CRP, ESR and swollen joint count, whilst other studies have found no such correlation. There are several possible reasons for this. The DAS28 score does not solely reflect active disease driven by inflammation. The more subjective components of the score (tender joint count and patient global assessment) could be driven by non-inflammatory mediators, which may explain the lack of association with CXCL13. Unlike serum CXCL13 which is primarily produced by the inflamed synovial tissue, CRP is produced by hepatocytes in response to interleukins released during inflammation and may not directly reflect synovial inflammation. This may explain the absence of correlation between these two markers [31]. There is also disagreement regarding the association between CXCL13 and radiographic joint destruction. Associations with erosive disease were seen in patients with established RA [22, 32] but not in those with early disease [31]. This may relate to the homogeneousness in radiographic disease seen within the early RA cohort.
RA is a heterogeneous disease with a variety of clinical presentations and differences in responsiveness to treatment modalities. Considerable molecular heterogeneity has been reported and different patient subgroups proposed on the basis of gene expression patterns and predominant synovial cell infiltrates [45]. The lymphoid phenotype had a high expression of B cell-associated genes and synovial CXCL13 levels whilst the myeloid phenotype demonstrates higher levels of other important synovial cytokines such as TNFα [40]. This may explain the conflicting evidence regarding the association with CXCL13 and disease activity, especially as studies have not defined their populations by predominant synovial infiltrates. In studies that did examine cellular infiltrates, synovial CXCL13 expression associated with an active lymphoid cell and a more severe erosive disease pattern [22].
Multiple studies reported that CXCL13 levels reduced in response to treatment with biologics including anti-TNF, anti-IL6, B cell depletion and small molecule JAK inhibitor. In some studies, the reduction in CXCL13 was only seen in treatment responders. These finding were not duplicated in all studies. Three studies found no change in levels with treatment. With methotrexate initiation, in contrast to the early reduction in acute phase markers, CXCL13 levels did not significantly change, reflecting a more persistent pattern of synovial inflammation [24].
As a predictor of treatment response, evidence is also conflicting. In patients receiving csDMARDs, CXCL13 levels predicted failure for disease remission, especially when combined with autoantibody positivity [19, 34]. With anti-TNF therapy, two studies reported high baseline levels associated with greater treatment response [16, 23]. In contrast, Dennis et al. reported low baseline levels associated with anti-TNF treatment response, whilst high baseline levels associated with a greater response to tocilizumab [40], a finding duplicated in an earlier study [42]. A biologically plausible mechanism was suggested to be linked to the predominant synovial phenotype. Patients with low baseline CXCL13 (and high soluble intercellular adhesion molecule) were more likely to have a myeloid inflammatory phenotype, with an overrepresentation of TNFα-associated gene expression, and hence a better response to TNFα blockade [40]. When considering these results, it is important to note variation in measurements of CXCL13 between studies, with some employing a predefined cut off of > 100 pg/ml, and others adopting a level above the median, or even a ratio computed with sICAM1 levels. It is conceivable that different assays are confounders.
There are limitations to this review. The most significant of these is the heterogeneity of the research questions and results between the studies reviewed. Each employed a different study design, with diverse study populations and analyses of an assortment RA therapies. The samples size across the studies ranged from 18 to 1135. Without a meta-analytical approach, it is impossible to incorporate the impact from a large study into the results. Disease activity, severity and treatment response were measured by a variety of different outcomes, limiting the ability to make direct comparisons.
A further limitation relates to the variability in CXCL13 assays. It is important to recognise that CXCL13 assays are primarily used in the research setting and are not accredited clinical laboratory assays. There is significant potential variation across technologies (ELISA, Luminex etc) and laboratories. We have presented the differences in methodology across the studies to make sure this aspect of variation is fully appreciated. The timing of biomarker collection, e.g. at time of assessment or at disease flare, was also not matched across the studies. In addition, as mentioned earlier, a number of studies used binary thresholds for CXCL13 rather than presenting absolute values which needs to be considered when interpreting the results presented.
Conclusion
In summary, CXCL13 expression is raised in a subset of patients with early and established RA, and levels may fall in response to therapy. It may have a discrete role as a biomarker in selected synovial pathotypes as a surrogate indicator of lymphoid organogenesis and active joint inflammation. In our opinion, there is not yet evidence for CXCL13 to predict disease progression or treatment response when used alone, partly because of the heterogeneity of the research questions and results from the studies reviewed here. We conclude that at present, the results are insufficient to justify the routine use of CXCL13 as a biomarker in the clinic setting. More research is required. For example, a study designed to recruit patients with new onset inflammatory arthritis, defined phenotypically and stratified to receive therapy based on serum CXCL13 levels would be of considerable interest. Without such a trial to test the hypothesis that CXCL13 is a useful biomarker, it is difficult to make a firm recommendation regarding its use in RA.
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Abbreviations
- RA:
-
Rheumatoid arthritis
- ERA:
-
Early RA
- EstRA:
-
Established RA
- HC:
-
Healthy control
- csDMARDs:
-
Conventional synthetic disease modifying anti-rheumatic drugs
- bDMARDs:
-
Biologic disease-modifying anti-rheumatic drugs
- TNFi:
-
Tumour Necrosis Factor inhibitor drug
- ADA:
-
Adalimumab
- ETN:
-
Etanercept
- antiIL-6R:
-
Anti-interleukin 6 receptor
- ELISA:
-
Enzyme-linked immunosorbent assay
- ECLA:
-
Electrochemiluminescent (ECLA)
- MSD:
-
Meso Scale Discovery
- qPCR:
-
Quantitative polymerase chain reaction (qPCR)
- DAS28:
-
Disease activity score for 28 joint count
- SJC:
-
Swollen joint count
- TJC:
-
Tender joint count
- PGA:
-
Patient global assessment
- RF:
-
Rheumatoid factor
- ACPA:
-
Anti–citrullinated peptide antibodies
References
Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. 2016;388(10055):2023–38.
Wolfe F. The prognosis of rheumatoid arthritis: assessment of disease activity and disease severity in the clinic. Am J Med. 1997;103(6a):12s–8s.
Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, et al. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350(25):2572–81.
Visser M, Bouter LM, McQuillan GM, Wener MH, Harris TB. Elevated C-reactive protein levels in overweight and obese adults. Jama. 1999;282(22):2131–5.
Matsui T, Kuga Y, Kaneko A, Nishino J, Eto Y, Chiba N, et al. Disease activity score 28 (DAS28) using C-reactive protein underestimates disease activity and overestimates EULAR response criteria compared with DAS28 using erythrocyte sedimentation rate in a large observational cohort of rheumatoid arthritis patients in Japan. Ann Rheum Dis. 2007;66(9):1221–6.
Nishimoto N, Yoshizaki K, Miyasaka N, Yamamoto K, Kawai S, Takeuchi T, et al. Treatment of rheumatoid arthritis with humanized anti-interleukin-6 receptor antibody: a multicenter, double-blind, placebo-controlled trial. Arthritis Rheum. 2004;50(6):1761–9.
Nell VP, Machold KP, Stamm TA, Eberl G, Heinzl H, Uffmann M, et al. Autoantibody profiling as early diagnostic and prognostic tool for rheumatoid arthritis. Ann Rheum Dis. 2005;64(12):1731–6.
Schellekens GA, Visser H, de Jong BA, van den Hoogen FH, Hazes JM, Breedveld FC, et al. The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheum. 2000;43(1):155–63.
van Gaalen FA, Linn-Rasker SP, van Venrooij WJ, de Jong BA, Breedveld FC, Verweij CL, et al. Autoantibodies to cyclic citrullinated peptides predict progression to rheumatoid arthritis in patients with undifferentiated arthritis: a prospective cohort study. Arthritis Rheum. 2004;50(3):709–15.
Armas-Gonzalez E, Dominguez-Luis MJ, Diaz-Martin A, Arce-Franco M, Castro-Hernandez J, Danelon G, et al. Role of CXCL13 and CCL20 in the recruitment of B cells to inflammatory foci in chronic arthritis. Arthritis Res Ther. 2018;20(1):114.
Humby F, Bombardieri M, Manzo A, Kelly S, Blades MC, Kirkham B, et al. Ectopic lymphoid structures support ongoing production of class-switched autoantibodies in rheumatoid Synovium. PLoS Med. 2009;6(1):e1.
Mandik-Nayak L, Huang G, Sheehan KC, Erikson J, Chaplin DD. Signaling through TNF receptor p55 in TNF-alpha-deficient mice alters the CXCL13/CCL19/CCL21 ratio in the spleen and induces maturation and migration of anergic B cells into the B cell follicle. J Immunol. 2001;167(4):1920–8.
Wengner AM, Hopken UE, Petrow PK, Hartmann S, Schurigt U, Brauer R, et al. CXCR5- and CCR7-dependent lymphoid neogenesis in a murine model of chronic antigen-induced arthritis. Arthritis Rheum. 2007;56(10):3271–83.
Pandya JM, Lundell AC, Andersson K, Nordstrom I, Theander E, Rudin A. Blood chemokine profile in untreated early rheumatoid arthritis: CXCL10 as a disease activity marker. Arthritis Res Ther. 2017;19(1):20.
Moura RA, Quaresma C, Vieira AR, Goncalves MJ, Polido-Pereira J, Romao VC, et al. B-cell phenotype and IgD-CD27- memory B cells are affected by TNF-inhibitors and tocilizumab treatment in rheumatoid arthritis. PLoS One. 2017;12(9):e0182927.
Han BK, Kuzin I, Gaughan JP, Olsen NJ, Bottaro A. Baseline CXCL10 and CXCL13 levels are predictive biomarkers for tumor necrosis factor inhibitor therapy in patients with moderate to severe rheumatoid arthritis: a pilot, prospective study. Arthritis Res Ther. 2016;18:93.
Kumagai S, Uemura Y, Saito T, Umeda R, Muta A, Izumi M, et al. AB0240 MMP-3 as A Biomarker of Disease Activity of Rheumatoid Arthritis. Ann Rheum Dis. 2016;75(Suppl 2):980.
Loza M, Sweet K, Peffer N, Franks C, Ma K, Campbell K, Sims M, Dasgupta B. Consistent Pharmacodynamic Effects of Sirukumab, an Anti–IL-6 Cytokine Monoclonal Antibody, on Serum Analytes Across Four Phase 3 Clinical Trials in Rheumatoid Arthritis [abstract]. Arthritis Rheumatol. 2016;68(suppl 10). https://acrabstracts.org/abstract/consistent-pharmacodynamic-effects-of-sirukumab-an-anti-il-6-cytokine-monoclonal-antibody-on-serumanalytes-across-four-phase-3-clinical-trials-in-rheumatoid-arthritis/. Accessed Aug 20 2020.
Bugatti S, Vitolo B, Benaglio F, Montecucco C, Caporali R, Manzo A. A5.14 serum CXCL13 is a non-invasive synovitis marker holding non-redundant information compared with acute phase reactants and autoantibodies in patients with rheumatoid arthritis. Ann Rheum Dis. 2016;75(Suppl 1):A46–A7.
Han JH, Suh CH, Jung JY, Nam JY, Kwon JE, Yim H, et al. Association of CXCL10 and CXCL13 levels with disease activity and cutaneous manifestation in active adult-onset Still's disease. Arthritis Res Ther. 2015;17:260.
Yeo L, Scheel-Toellner D, Ludwig C, Sahbudin I, Juarez M, Buckley C, Raza K, Filer A. Synovial Ultrasound Power Doppler Enhancement Reflects Multiple Cytokine Expression in Synovial Tissue, Distinguishing Distinct High Macrophage-Associated and Low Inflammation Profiles [abstract]. Arthritis Rheumatol. 2015;67(suppl 10). https://acrabstracts.org/abstract/synovial-ultrasound-power-doppler-enhancement-reflects-multiple-cytokine-expression-insynovial-tissue-distinguishing-distinct-high-macrophage-associated-and-low-inflammation-profiles/. Accessed Aug 20 2020.
Bugatti S, Manzo A, Vitolo B, Benaglio F, Binda E, Scarabelli M, et al. High expression levels of the B cell chemoattractant CXCL13 in rheumatoid synovium are a marker of severe disease. Rheumatology (Oxford). 2014;53(10):1886–95.
Greisen SR, Schelde KK, Rasmussen TK, Kragstrup TW, Stengaard-Pedersen K, Hetland ML, et al. CXCL13 predicts disease activity in early rheumatoid arthritis and could be an indicator of the therapeutic 'window of opportunity. Arthritis Res Ther. 2014;16(5):434.
Bugatti S, Manzo A, Vitolo B, Benaglio F, Binda E, Scarabelli M, et al. THU0546 Serum CXCL13 as A Biomarker of Disease Activity and Severity in IN Rheumatoid Arthritis. Comparison with Acute Phase Reactants and the Autoantibody Profile. Ann Rheum Dis. 2014;73(Suppl 2):371.
Moura RA, Canhão H, Polido-Pereira J, Navalho M, Leandro MJ, Cambridge G, et al. AB0085 Serum Levels of CXCL13 Are Increased in Untreated Patients with Rheumatoid Arthritis from the First Weeks of Disease Development. Ann Rheum Dis. 2014;73(Suppl 2):832.
Jones JD, Hamilton BJ, Challener GJ, de Brum-Fernandes AJ, Cossette P, Liang P, et al. Serum C-X-C motif chemokine 13 is elevated in early and established rheumatoid arthritis and correlates with rheumatoid factor levels. Arthritis Res Ther. 2014;16(2):R103.
Sellam J, Rouanet S, Hendel-Chavez H, Miceli-Richard C, Combe B, Sibilia J, et al. CCL19, a B cell chemokine, is related to the decrease of blood memory B cells and predicts the clinical response to rituximab in patients with rheumatoid arthritis. Arthritis Rheum. 2013;65(9):2253–61.
Sherif NM, Arafa MM, Ibrahim SE, Moussa SG. CXC ligand 13 in rheumatoid arthritis and its relation to secondary Sjögren’s syndrome. Egypt Rheumatol. 2013;35(3):121–6.
Fathi Ahmed S, Badr T, Hosny SM, Hamayed HFA. Assessment of synovitis in early rheumatoid arthritis by CXCL13 serum levels and power Doppler ultrasonography: correlation with disease activity. Egypt Rheumatol. 2013;35(1):21–7.
Setiadi FL-K, N. Kummerfeld S, Choy DF, CTJ H, Fischer S, et al. Synovial Subset–Derived Baseline Serum Biomarkers Segregate Rheumatoid Arthritis Patients Into Subgroups With Distinct Serum Protein and Clinical Characteristics. Arthritis Rheum. 2013;65:S552–3 2013.
Bugatti S, Manzo A, Benaglio F, Klersy C, Vitolo B, Todoerti M, et al. Serum levels of CXCL13 are associated with ultrasonographic synovitis and predict power Doppler persistence in early rheumatoid arthritis treated with non-biological disease-modifying anti-rheumatic drugs. Arthritis Res Ther. 2012;14(1):R34.
Meeuwisse CM, van der Linden MP, Rullmann TA, Allaart CF, Nelissen R, Huizinga TW, et al. Identification of CXCL13 as a marker for rheumatoid arthritis outcome using an in silico model of the rheumatic joint. Arthritis Rheum. 2011;63(5):1265–73.
Rinaldi MVBT, Van Roy M, Bontinck L, Hohlbaum A, Snoeck V, Dombrecht E, Van Beneden K, Schoen P, Ulrichts H. Assessment of dose dependent effects of Vobarilizumab, an anti-IL6 receptor (IL-6R) Nanobody®, on systemic biomarkers in patients with moderate-to-severe rheumatoid arthritis (RA): results of two phase 2b studies [abstract]. Arthritis Rheum. 2017;69(suppl 10):2017.
Bugatti S, Manzo A, Vitolo B, Benaglio F, Caporali R, Montecucco C. SAT0087 Serum Levels of CXCL13 Refine The Predictive Ability of Autoantibodies To Identify Unstable Remission in Early Rheumatoid Arthritis. Ann Rheum Dis. 2016;75(Suppl 2):696.
de Jong TD, Vosslamber S, de Jager W, Raterman H, Voskuyl AE, Gelderman KA, et al. A1.7 Serum cytokine changes in rituximab-treated rheumatoid arthritis patients. Ann Rheum Dis. 2014;73(Suppl 1):A3 A.
El-Sherbiny YM, Churchman S, Emery P, Vital EM, Ponchel F. A5.18 Impact of Rituximab on Synovial Gene Expression. Ann Rheum Dis. 2013;72(Suppl 1):A37 A.
Taylor PC, Galien R, Van der Aa A, Jamoul C, Harrison P, Tasset C, Pan Y, Goyal L, Li W, Tarrant J. Monotherapy with Filgotinib, a JAK1-Selective Inhibitor, Reduces Disease-Related Biomarkers in Rheumatoid Arthritis Patients [abstract]. Arthritis Rheumatol. 2017;69(suppl 10). https://acrabstracts.org/abstract/monotherapy-with-filgotinib-a-jak1-selective-inhibitor-reduces-disease-related-biomarkers-in-rheumatoid-arthritis-patients/. Accessed Aug 20 2020.
Gabay CMJ, Daskalakis N, Barbot A, Zilberstein M, Boyapati A. Effect of Sarilumab on circulating biomarkers of bone and joint destruction in patients with rheumatoid arthritis with previous inadequate response to tumor necrosis factor inhibitors [abstract]. Arthritis Rheum. 2016;68(suppl 10):2016.
Boyle DL, Soma K, Hodge J, Kavanaugh A, Mandel D, Mease P, et al. The JAK inhibitor tofacitinib suppresses synovial JAK1-STAT signalling in rheumatoid arthritis. Ann Rheum Dis. 2015;74(6):1311–6.
Dennis G Jr, Holweg CT, Kummerfeld SK, Choy DF, Setiadi AF, Hackney JA, et al. Synovial phenotypes in rheumatoid arthritis correlate with response to biologic therapeutics. Arthritis Res Ther. 2014;16(2):R90.
Kennedy WP, Simon JA, Offutt C, Horn P, Herman A, Townsend MJ, et al. Efficacy and safety of pateclizumab (anti-lymphotoxin-alpha) compared to adalimumab in rheumatoid arthritis: a head-to-head phase 2 randomized controlled study (the ALTARA study). Arthritis Res Ther. 2014;16(5):467.
Herman A, Musselman D, Fischer S, Setiadi A, Gabay C, Kavanaugh A, et al. THU0130 Distinct Biomarkers Enrich for Clinical Response to Tocilizumab (TCZ) and Adalimumab (ADA) in Adacta: A Head-to-Head Monotherapy Study in Patients (PTS) with Methotrexate (MTX)-IR Rheumatoid Arthritis (RA). Ann Rheum Dis. 2013;72(Suppl 3):A206 A.
Emu B, Luca D, Offutt C, Grogan JL, Rojkovich B, Williams MB, et al. Safety, pharmacokinetics, and biologic activity of pateclizumab, a novel monoclonal antibody targeting lymphotoxin alpha: results of a phase I randomized, placebo-controlled trial. Arthritis Res Ther. 2012;14(1):R6.
Rosengren S, Wei N, Kalunian KC, Kavanaugh A, Boyle DL. CXCL13: a novel biomarker of B-cell return following rituximab treatment and synovitis in patients with rheumatoid arthritis. Rheumatology (Oxford). 2011;50(3):603–10.
van Baarsen LGM, Bos CL, van der Pouw KTCTM, Verweij CL. Transcription profiling of rheumatic diseases. Arthritis Res Ther. 2009;11(1):207.
Acknowledgements
Not applicable.
Funding
No specific funding was received from any bodies in the public, commercial or not-for-profit sectors to carry out the work described in this manuscript. K.B. is funded by Medical Research Council as a Clinical Training Research Fellowship (CTRF- MR/R001332/1 to K Bechman).
Author information
Authors and Affiliations
Contributions
All authors have approved the submitted version and agree to be personally accountable for the author’s own contributions. Involvement; KB, AD, CSB: design, analysis, interpretation of data and drafting manuscript. AC, JG: design, interpretation of data and drafting manuscript. Disclosures: J.B.G. has received honoraria for speaking or attending conferences from Pfizer, Bristol-Myers Squibb, UCB and Celgene. A.P.C. is a Trustee of the Kennedy Trust for Rheumatology Research, which has received royalty income linked to anti-TNF therapy when used in combination with methotrexate.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bechman, K., Dalrymple, A., Southey-Bassols, C. et al. A systematic review of CXCL13 as a biomarker of disease and treatment response in rheumatoid arthritis. BMC Rheumatol 4, 70 (2020). https://doi.org/10.1186/s41927-020-00154-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41927-020-00154-3