
Abstract
The current treatment paradigm in head and neck cancer does not adequately address its clinical and biological heterogeneity. Data from genomic profiling studies in head and neck squamous cell carcinoma (HNSCC) have revealed the molecular features that are unique to HNSCC subgroups. This progress in the understanding of HNSCC biology provides an opportunity to develop personalized therapies for patients with distinct molecular subtypes to achieve better clinical outcomes including survival. However there are several well-recognized challenges that need to be overcome before genotype-matched therapies make precision medicine a reality for patients with HNSCC. Selection of appropriate patients for biomarker directed clinical trials based on sound scientific rationale will be critical in making cancer genomics more applicable in this malignancy.
Similar content being viewed by others


Background
Head and neck cancer is a heterogeneous disease comprising epithelial tumors of the upper aerodigestive tract. The majority (~90 %) are squamous cell carcinomas arising from the oral cavity, oropharynx, hypopharynx and larynx classed together as head and neck squamous cell carcinomas (HNSCC) [1]. The main risk factors for developing HNSCC are tobacco smoking, alcohol drinking [2] and human papillomavirus (HPV) infection [3]. HPV-positive HNSCC, which is usually found in oropharynx, is a sexually transmitted disease with a rising incidence in many developed countries [4]. It is biologically and clinically distinct from HPV-negative HNSCC that is classically associated with tobacco and alcohol exposure. Patients with HPV-positive disease have a better prognosis compared to those with HPV-negative disease [5].
The standard treatment paradigm for HNSCC is based on anatomical location and stage of the disease. The biological heterogeneity of HNSCC, however, is not routinely incorporated in the current clinical management algorithms. In general, surgery and/or radiotherapy represent the treatment of choice for early stage disease. Surgery is usually preferred for oral cavity tumors, with the need for post-operative adjuvant radiotherapy typically based on recurrence risks that are determined by stage and tumor pathology. Radiotherapy is generally given as primary treatment in oropharyngeal, hypopharyngeal and laryngeal tumors due to the interest in organ preservation, with salvage surgery considered for local recurrences after primary radiotherapy. Concurrent cisplatin-based chemo-radiotherapy is the standard of care for locoregionally advanced disease. For those patients who are deemed inappropriate candidates for platinum-based chemoradiotherapy, radiotherapy combined with the anti-epidermal growth factor receptor (EGFR) monoclonal antibody cetuximab, may be an alternative. The ideal risk-adapted therapeutic strategies aiming to optimize disease control with minimal long-term toxicities in favorable risk early stage disease, and to maximize survival outcomes with acceptable toxicities in poor risk disease, are still evolving.
For recurrent and metastatic HNSCC, platinum-based chemotherapy remains the mainstay of treatment, and addition of cetuximab to first line chemotherapy (cisplatin or carboplatin plus 5-fluorouracil) offers modest survival benefit [6]. Currently, there is no universally agreed second line therapy.
So far, the search for personalized therapy in patients with HNSCC remains elusive. Cetuximab is the only targeted biological agent approved for use in HNSCC and neither EGFR copy number nor level of EGFR expression was shown to predict its response [7, 8]. The hope, however, has been that as we sequence broader and deeper into HNSCC genomes, biological drivers in individual HNSCC will be identified with a high precision allowing development of genotype-matching therapy. The emerging data from HNSCC genome sequencing studies [9–13], including recent results from The Cancer Genome Atlas (TCGA) initiative [14], now provide an opportunity to develop genomically personalized therapy for patients with HNSCC.
Genomic landscape of HNSCC
Structural alterations
Genomic structural alterations are commonly seen in HNSCC regardless of HPV status. Both HPV-positive and HPV-negative tumors harbor amplifications of 1q, 3q, 5p and 8q and deletions of 3p, 5q, and 11q [9, 10, 12–14]. The amplification of 3q26/28 region containing squamous lineage transcription factors, TP63 and SOX2, and PIK3CA oncogene is seen in both, but more frequently in HPV-positive subtype [9, 10, 12–14]. In HPV-positive tumors, recurrent deletions in TRAF3 and 11q including ATM1 and focal amplification of E2F1 are also seen but 9p21.3 containing CDKN2A is usually intact [14]. In contrast, in HPV-negative tumors, 9p21.3 is commonly deleted while 11q13 containing CCND1, FADD and CTTN, and 11q22 containing BIRC2 and YAP1 are amplified [14]. It is noteworthy that 7p region that includes EGFR is less amplified in HPV-positive tumors [14].
From a biological perspective, recurrent CDKN2A deletions and CCND1 amplification seen in HPV-negative tumors and E2F1 amplifications in HPV-positive tumors indicate that loss of cell cycle regulation is the fundamental event in HNSCC carcinogenesis. The importance of mitogen activated protein kinase (MAPK) pathway in HPV-negative HNSCC is highlighted by EGFR amplification and PI3K-PTEN-AKT-mTOR pathway in both HPV-positive and HPV-negative tumors by PI3KCA amplification. Recurrent deletions in TRAF3 in HPV-positive tumors and amplification of FADD and BIRC2 in HPV-negative tumors showed that NF-kB pathway activation is an important biological driver in HNSCC. TRAF3 deletions also indicate defective innate immunity response in HPV-positive HNSCC.
Somatic mutations
Genes in cell cycle regulation
Alterations in genes that regulate cell cycle are commonly seen in HNSCC. In HPV-negative tumors, TP53 is mutated in 80–87 % and CDKN2A gene alterations are seen in 32–57 % [11, 12, 14]. CDKN2A can also be silenced by promoter hypermethylation in HPV-negative HNSCC [15] and it is noteworthy that CDKN2A expression is lost in almost all HPV-negative HNSCC [16]. On the other hand, TP53 and CDKN2A gene alterations are infrequent in HPV-positive tumors. A small subset of HPV-negative oral cavity squamous cell carcinoma do not have TP53 mutations but harbor activating HRAS mutations and inactivating CASP8 mutations constituting a distinct subset with a favorable prognosis [14]. RB1 mutations, although rare at <10 %, are seen predominantly in HPV-positive tumors [11]. MYC, in contrast, is amplified in 5–15 % of HPV-negative tumors [11, 14].
PIK3CA & PTEN
PIK3CA is the most commonly altered oncogene in HNSCC [11, 12, 14]. The presence of hotspot mutations in PIK3CA helical domain is a unique feature of HPV-positive tumors, whereas, in HPV-negative tumors, mutations occur throughout the gene despite helical and kinase domain mutations are still common [11, 12, 14]. Twenty one percent of patients in the TCGA cohort had a PIK3CA mutation and of those, 25 % also had concurrent PIK3CA amplification [14]. An additional 20 % of tumors had PIK3CA amplification without mutations [14]. In addition to PIK3CA alterations, PTEN mutations or deletions are seen in ~11 % of HPV-positive HNSCC and 5 % of HPV-negative HNSCC [11, 17, 18].
Genes encoding receptor tyrosine kinase (RTK) and MAPK pathways
HPV-positive and HPV-negative HNSCC have alterations in genes encoding RTK and MAPK pathways at differing frequencies. While alterations in EGFR, FGFR1, and IGF1R are predominantly seen in HPV-negative tumors, FGFR2 and FGFR3 alterations including FGFR3 fusions are more frequent in HPV-positive tumors [12, 14]. MET amplification occurs in 2–13 % of HNSCC predominantly in the HPV-negative subtype [19]. ERBB2 alterations are seen in both subtypes at a low frequency (3–4 %) [14]. Mutations in MAPK pathway, mainly HRAS mutations in HPV-negative tumors and KRAS mutation in HPV-positive tumors, are seen in ~6 % of HNSCC [12, 14].
DNA damage response genes
Mutations in BRCA1, BRCA2 and ATR are seen in 6 %, 7 % and 4–10 % of HNSCC respectively [11]. ATM mutations are also seen in 1–16 % of HPV-positive HNSCC across studies [11, 12, 14].
NOTCH1
NOTCH1 is one of the most commonly mutated genes in HNSCC (11- 19 %) [9, 10, 14]. Inactivating mutations are predominantly found in HNSCC cases of the TCGA cohort whereas activating gain of function mutations with overexpression of downstream effectors are found predominantly in Chinese HNSCC cases [20–22]. It is well recognized that the biological role of NOTCH could be contextual [23] and NOTCH undoubtedly plays a complex biological role in HNSCC making it challenging to target without further biological insight.
Patterns of gene expression
In 2002, Belbin et al. first reported that HNSCC could be classified by gene expression patterns (N = 17) [24]. Four main subtypes of HNSCC (Group 1–4) based on expression pattern of 12814 genes were subsequently described by Chung et al. (N = 60) [25]. These four subtypes were validated independently in the University of North Carolina cohort (N = 138) and subtypes 1–4 were named as basal, mesenchymal, atypical and classical respectively [26]. The basal subtype is characterized by activation of EGFR pathway. The mesenchymal subtype has epithelial to mesenchymal transition (EMT) gene expression signature. The atypical subtype mainly consisted of HPV-positive tumors with high expression of CDKN2A, LIG1 and RPA2. The classical subtype showed tobacco-induced gene expression signature.
TCGA also independently validated these four signatures (N = 278) [14]. Integrated analysis of DNA alterations and RNA expression patterns in TCGA cohort showed that basal subtype tumors had NOTCH1 inactivation with intact oxidative stress signaling. This subtype also included tumors with CASP8 and HRAS mutations. In contrast, high expression of CD56, a natural killer cell marker, and HLA class I mutations were seen in the mesenchymal subtype. The atypical subtype again consisted mainly of HPV-positive tumors characterized by activating mutations in PIK3CA helical domain and a lack of chromosome 7 amplifications indicating a low level of EGFR amplification. In contrast, the classical subtype was seen in heavy smokers and at laryngeal site, characterized by TP53 mutation, CDKN2A loss, chromosome 3q amplification and alteration of oxidative stress genes.
These studies, however, suffer from underrepresentation of HPV-positive tumors. Gene expression analysis of a cohort enriched with HPV-positive HNSCC by et al. demonstrated that such tumors could be classified into classical-HPV and mesenchymal-HPV [27]. While both subtypes were characterized by enriched cell cycle genes, classical-HPV also had activation of putrescine (polyamine) degradation pathway possibly related to detoxification of tobacco use and mesenchymal-HPV had enrichment of immune response genes related to the tumoral CD8+ T lymphocytes infiltration [27]. HPV-negative mesenchymal tumors in this study also had enrichment of similar immune related genes [27] inferring mesenchymal tumors might respond better to immunotherapy.
Genomically driven dysregulated pathways and personalized therapies
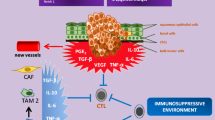
The gene alterations found in HNSCC across studies [9–14] perturb multiple biological pathways. Herein, these pathways are discussed in the context of rationally matched therapies (summarized in Fig. 1).

Genomically driven dysregulated pathways and potential matched therapies in HNSCC. Combination strategies currently being tested in ongoing clinical trials are also shown. Abbreviations: MAB, monoclonal antibody; TKI, tyrosine kinase inhibitor
TP53 and cell cycle regulation
TP53 is the most commonly mutated tumor suppressor in HNSCC and almost all HNSCC displayed a dysregulated cell cycle [14]. Loss of cell cycle regulation at G1 to S phase checkpoint is the dominant biological feature in HNSCC. TP53 and CDKN2A loss of function alterations and CCND1 amplification are the main driving mechanisms of this dysregulation in HPV-negative tumors [11, 12, 14]. In HPV-positive tumors, degradation of TP53 and RB1 by E6 and E7 viral proteins and focal amplification of E2F1 are the main culprits [12, 14].
To exploit cell cycle dysregulation as a drug target in HNSCC, cyclin dependent kinase (CDK) inhibitors are of particular interest for patients with CCND1 amplified tumors. CDK4/6 inhibitors, ribociclib (LEE011) and palbociclib, which prolong progression free survival of patients with hormone receptor positive, HER2-negative advanced breast cancer [28, 29], are currently in early phase development in HNSCC (Tables 1 and 2).
In vitro inhibition of G2 to M phase checkpoint enhances apoptosis induced by DNA damaging agents in TP53 mutant HNSCC [30]. A WEE-1 kinase inhibitor, AZD1775, which disrupts G2 checkpoint by inhibiting CDK1 phosphorylation, is currently being tested in HNSCC in the neo-adjuvant setting in combination with chemotherapy (NCT02508246) and in the locoregionally advanced setting combined with chemoradiotherapy (NCT02585973). Inhibition of checkpoint kinase 1/2 (CHK1/2) also induces mitotic catastrophe followed by cell death in TP53 mutant HNSCC in vitro [31]. As such combining CHK1/2 inhibitors with radiotherapy or DNA damaging agents seems a rational strategy in HNSCC with TP53 loss of function mutations. On the other hand, in breast, ovarian, colon and prostate cancer cells, WEE-1 was found to be a synthetic lethal partner of CHK1 and combined inhibition of WEE-1 and CHK-1 results in tumor growth inhibition in vitro and in vivo regardless of TP53 status [32]. Interestingly, in HPV-positive HNSCC, combining radiotherapy with WEE-1 kinase inhibition increases apoptosis in vitro that is caspase mediated but independent of TP53 [33]. Currently, the role of TP53 mutation status in predicting response to WEE-1 and CHK1/2 inhibition remains unclear in HNSCC.
PI3K-PTEN-AKT-mTOR pathway
PIK3CA is the most commonly mutated oncogene in HNSCC and, combined with other gene alterations, PI3K-PTEN-AKT-mTOR pathway is dysregulated in ~30 % of HNSCC [11, 14, 34]. One of the most “actionable” target in this pathway is the activating gain of function PIK3CA mutations, as PI3K inhibitors of different classes, either alone or in combination with other targeted agents, are in active development in HNSCC (Tables 1 and 2). On the other hand, loss of function gene alterations in PTEN is a challenge to target directly, since the restoration of its tumor suppressive function is not straightforward.
In in vitro studies and patient derived xenografts, PIK3CA mutations sensitized response to PI3K and mTOR inhibitors [34, 35]. However, no significant improvement in clinical outcomes was seen with PX-866, an irreversible PI3K inhibitor, in unselected population of HNSCC when combined with doxcetaxel or cetuximab [36, 37]. In particular, 8 patients with PIK3CA mutant tumors did not respond to the PX-866 and cetuximab combination raising doubts over the role of PIK3CA mutations in predicting response to PI3K pathway inhibition. On the other hand, promising tumor activity was seen with BYL719, a PI3K class I α isoform inhibitor, when combined with cetuximab in a phase Ib study [38] suggesting that efficacy of isoform specific PI3K inhibitors may be different from that of pan-PI3K inhibitors in HNSCC. It is plausible that mutations causing different amino acid substitutions at different positions might have different biological functions with variable sensitivities to PI3K inhibitors [39]. BYL719 is also currently being evaluated with cisplatin-based chemoradiotherapy in locoregionally advanced setting in a phase I study (NCT02537223).
Previous studies demonstrated that mTOR inhibitors, temsirolimus and everolimus, have limited antitumor activity in platinum-refractory recurrent or metastatic HNSCC [40, 41]. Although response rates were higher in platinum naïve setting [42, 43], these studies were performed in non-selected populations and as such the role of PIK3CA or PTEN alterations in predicting response to mTOR inhibitors in HNSCC is not clearly known.
Receptor tyrosine kinase pathways
EGFR
Overexpression of EGFR was seen in >90 % of HNSCC and associated with a poor prognosis [44–48]. Cetuximab improves survival of patients in both locoregionally advanced setting and recurrent or metastatic setting when combined with radiotherapy and chemotherapy respectively [6, 49]. However, single agent response to cetuximab in recurrent or metastatic HNSCC is 13 % at best indicating there is primary resistance to EGFR inhibition [50].
EGFR alterations are found in ~15 % of patients with HNSCC predominantly in HPV-negative tumors [12, 14]. Neither EGFR copy number nor level of EGFR expression was shown to predict cetuximab response [7, 8]. EGFR mutations including EGFRvIII are also extremely rare in HNSCC and unlikely to be useful as predictive biomarkers for EGFR targeted therapy [14, 47, 51–53]. However, alterations in ERBB2, MET, PIK3CA, PTEN and HRAS can co-occur with EGFR alterations and may explain mechanisms of EGFR inhibitor resistance in individual HNSCC [14]. Combined inhibition of EGFR and other RTK and/or downstream pathways based on genotype of tumors might overcome primary and secondary resistance to EGFR inhibition leading to improved clinical outcomes. Currently, the safety and efficacy of cetuximab combined with multiple molecular targeted therapies are being tested in various phase 1 and 2 clinical trials (Table 2).
ERBB
ERBB2 alteration (amplification plus mutation) is seen in ~4 % of HPV-negative HNSCC and ~3 % of HPV-positive HNSCC [14]. Afatinib, an irreversible pan-ERBB inhibitor, was recently shown to improve progression-free-survival in a non-selected population of recurrent or metastatic HNSCC when compared to methotrexate in the second line setting, indicating that targeting ERBB pathway is a valid therapeutic strategy in HNSCC [54]. Dacomitinib, another irreversible pan-ERBB inhibitor, also demonstrated a single agent response rate of ~13 % in a phase 2 study [55]. Presence of mutations in PIK3CA or PTEN seems to predict poor PFS in patients treated with dacomitinib in a separate study [56] indicating dual inhibition of ERBB and PI3K pathways might produce better clinical benefit in patients with PIK3CA or PTEN alterations. A recent phase Ib study has already established maximum tolerated dose of the combination of cetuximab and afitinib [57]. From a genomic perspective, patients with EGFR and ERBB aberrations are likely to derive better clinical benefit from this novel combination. ERBB targeted agents are currently in active clinical development in HNSCC (Tables 1 and 2).
FGFR
FGFR1 alterations (amplification or mutations) are seen in ~12 % of HPV-negative HNSCC and FGFR3 alterations (mutations or fusions) in ~11 % of HPV-positive HNSCC [14]. In a preclinical study FGFR1 mRNA and protein expression level but not FGFR1 copy number was found to be associated with response to a pan-FGFR inhibitor BGJ398 [58]. Data from squamous cell lung cancer suggest that FGFR1-amplified tumor cells with co-expression of MYC are more sensitive to FGFR inhibition [59]. Currently there is no clinical data to indicate the efficacy of FGFR inhibitors in HNSCC as they are still in early stages of drug development.
MET
MET amplification is found in 2–13 % of HNSCC and it is mutated in ~6 % [19]. Acquired MET amplification is a well-recognized biological mechanism of resistance to EGFR inhibition [60–62] and MET overexpression is frequently seen in HNSCC [19]. However, in a phase II study of foretinib, an oral multikinase inhibitor of MET and VEGFR2, in an unselected HNSCC population, no partial or complete response was seen [63]. Despite this disappointing result, combination treatment of MET inhibitors with cetuximab or other RTK inhibitors in selected population is biologically rational and still worth investigating.
Immune related pathways and genomic predictors for immunotherapy
HNSCC is an immunosuppressive disease and immune checkpoint inhibitors (ICI) are emerging as a promising therapy for patients with HNSCC. Low frequency mutations of HLA-A, HLA-B and B2M are seen in both HPV-positive and HPV-negative tumors implicating that tumor antigen presentation may be disrupted in tumors harboring these genetic alterations [14]. Early data indicate that single agent response to PD-1/PD-L1 pathway inhibition in HNSCC is ~12–20 % [64, 65]. Although objective response rates are higher in patients with PD-L1 positive tumors, PD-L1 expression was not a binary predictive biomarker as PD-L1 negative tumors can demonstrate meaningful tumor shrinkage or clinical benefit [64, 65]. No definite difference in response rate to ICI between HPV-positive and HPV-negative subtypes has been observed [64, 65]. CheckMate 141 (NCT02105636), a phase III study that is comparing the efficacy of nivolumab, an anti-PD-1 inhibitor, in the platinum-resistant recurrent or metastatic setting with investigator’s choice of therapy has been stopped early because it met the primary overall survival endpoint at a preplanned interim analysis [66].
In squamous cell lung cancer, response to nivolumab was shown to associate with tumor mutation burden, neo-antigen load and smoking mutation signature [67]. Although hypermutated phenotype is rare in HNSCC, neo-antigen landscape and smoking signature may be relevant in predicting immunotherapy response in HNSCC. Seventy five percent of HPV-positive HNSCC and 23 % of HPV-negative HNSCC in the combined University of Chicago and TCGA cohort (N = 134 + 424 = 558) have an inflamed tumor phenotype [62]. This phenotype is characterized by enrichment of immune response genes related to intra-tumoral CD8+ T lymphocyte infiltration, PD-L1 expression and expression of CTLA-4, LAG3, PD-L2 and IDO [68]. There is also a strong correlation between inflamed tumor phenotype and mesenchymal subtype [68]. Considering these emerging data, it is tempting to speculate that HNSCC with this gene expression signature may respond better to ICI. This hypothesis, however, needs further prospective clinical validation.
DNA damage response and homologous recombination (HR) deficiency
Interestingly somatic mutations in genes involved in DNA damage response including HR are seen in HNSCC at various frequencies [11]. Somatic mutations in BRCA1 (~6 %), BRCA2 (~7 %), ATR (4–10 %) and ATM (1-16 %) are all reported providing the rationale for targeting DNA repair pathway in these tumors [12]. Inactivation of Fanconi anemia/BRCA pathway via promoter hypermethylation of FANCF was also seen in 15 % of patients with HNSCC in a previous study [69]. PARP inhibitors and ATR inhibitors, in combination with radiotherapy or cisplatin, are of particular interest for patients with HR deficient genotype. Currently ATR inhibitors, AZD6738 and VX-970, are in early phase of development in HNSCC (NCT02264678; NCT02567422). Furthermore, in a phase I study of AZD1775, a wee-1 kinase inhibitor, a HNSCC patient with a BRCA mutation achieved a partial response [70] supporting that DNA repair pathway might be a valid therapeutic target in selected HNSCC population.
Notch pathway
NOTCH1 is one of the most commonly mutated genes in HNSCC (~10–15 %) [9, 10, 14] and associated with a poor prognosis [21]. NOTCH pathway plays complex biological roles including cell differentiation, proliferation, angiogenesis and survival [71–73]. As such NOTCH pathway inhibition could be a valid therapeutic strategy in HNSCC. A recent phase I study reported that 2 out of 15 HNSCC patients (13 %) treated with combination of NOTCH inhibitor MK-0752 and mTOR inhibitor ridaforolimus achieved partial response [74]. However, considering the fact that both activating and inactivating NOTCH mutations are seen in HNSCC and the increased risk of squamous cell carcinoma of skin observed with gamma-secretase inhibitors in patients with Alzheimer’s disease [75, 76], targeting NOTCH pathway in HNSCC in the right biological context with minimal risk will be challenging. Further biological insight will be necessary before NOTCH pathway inhibition can be optimally exploited for treatment of patients with HNSCC.
Challenges & future directions
The recent genomic profiling studies identified biologically distinct HNSCC subgroups providing rationale for developing genomically directed personalized therapies. However, there are well-recognized challenges. Currently available genomic data in HNSCC is limiting as they were mostly derived from early stage resectable and locally advanced tumors with gross underrepresentation of recurrent/metastatic disease (Table 3) and do not truly inform the biological drivers of recurrent and metastatic HNSCC in which most novel targeted agents are currently being tested. Most studies also included only small number of HPV-positive cases (Table 3). Moreover, they were conducted in heterogeneous patient populations without detailed clinical annotation and as such lack power to determine prognostic and predictive value of genetic alterations identified.
From a biological point of view, the main challenges are those posed by spatial and temporal tumor heterogeneity. Considering profound intra-tumor genetic heterogeneity found in multiple solid tumor types [77–79] including HNSCC [80, 81], it will be difficult to make accurate assessment of tumor genetic characteristics from a single tumor biopsy. To address this in patients with multiple metastatic sites, ideally multiple tumor biopsies from all disease sites will be necessary to assess tumor genomic profile accurately. Cleary, this is not logistically feasible in current oncology practice and innovative tumor sampling methods will be necessary to overcome this challenge. One potential method is mutation profiling of plasma-derived circulating tumor DNA (ctDNA) using next generation sequencing (NGS). As tumor DNA from different metastatic sites are shed into circulation, it could be argued that ctDNA contains mutations or genetic alterations derived from all sub-clones of tumors. With rapid advances in NGS technologies, several groups have demonstrated that molecular characterization of ctDNA is feasible [82–87]. Analysis of serial ctDNA samples could reveal clonal evolution of tumors highlighting the future potential of ctDNA mutation profiling in addressing both spatial and temporal intra-tumor genetic heterogeneity. In appropriate HNSCC cases, it might also be possible to study tumor mutations from DNA isolated from saliva. The feasibility of tumor specific mutation testing from ctDNA and saliva in patients with HNSCC has been explored in a recent study [88].
The obvious drug targets in cancer genomes are activating mutations in driver oncogenes. In HNSCC PIK3CA is the most frequently mutated oncogene (~20 %) and PI3K inhibitors are in active development. However, despite the preclinical evidence showing PIK3CA mutations sensitize response to PIK3A inhibitors, clinical results so far have been disappointing except possibly for BYL719, a PI3K class I α isoform inhibitor, in combination with cetuximab where some signals of activity have been observed. Further biological insight will be needed to advance development of PI3K inhibitors in HNSCC. Beyond PIK3CA, actionable mutations in other oncogenic driver genes such as ERBB, FGFR, and MET are relatively rare making it challenging to conduct biomarker directed clinical trials. Future trials with innovative designs will be needed to address this issue.
Although genetic alterations in tumor suppressor genes TP53 and CDKN2A are common in HNSCC, these alterations are notoriously difficult to exploit for targeted therapeutics at present. Again, mutations in other tumor suppressor genes are seen at low frequencies. Further studies will be needed to elucidate synthetic lethal interactions between these genetic events using computational algorithms such as DAISY (data mining synthetic lethality identification pipeline) [89] or using si-RNA, sh-RNA or CRISPR (clustered regularly interspaced short palindromic repeats) screens so that rational treatment strategies could be developed based on tumor genomic profiles [90]. Currently, one of the most exciting synthetic lethality opportunities involves the use of DNA damaging agents such as PARP inhibitors or ATR inhibitors in patients with HR deficient tumors.
From a genomic perspective, it will be necessary to understand the clonal composition and progression of tumors to develop effective genotype-matched therapy. As clonal selection and progression would have occurred that led to the development of clinically detectable recurrent or metastatic disease, actual clonal composition of advanced HNSCC could be different from that of primary tumors and arguably might contain more genetic driver events that are actionable. Accurate charting of clonal and sub-clonal genetic events in recurrent and metastatic HNSCC based on absolute quantification of somatic mutation events will be critical in finding the relevant therapeutic targets in individual tumors [91, 92]. It is plausible that some genetic alterations are present only in minor sub-clones and targeting those might not produce meaningful clinical benefits.
Considering different biological pathways are active in different subtypes of HNSCC, single agent activity of targeted agents in non-selected population is likely to be modest. To significantly improve clinical outcomes, rational combination treatment strategies should be tested prospectively in selected populations enriched by unique tumor molecular features present in the recurrent or metastatic tumors. Currently, based on available genomic data mainly derived from early stage tumors, the proportion of HNSCC patients who could benefit from personalized therapy remains relatively small considering the most common genetic events in HNSCC occur in tumor suppressor genes. However, concerted large scale genomic profiling programs such as SPECTA (Screening Patients for Efficient Clinical Trial Access) of EORTC (European Organization for Research and Treatment of Cancer) that aims to profile advanced solid tumors to offer genotype-matched therapies could more accurately inform targetable genetic events in recurrent/metastatic HNSCC. These initiatives may also shed insight on the proportion of patients who truly benefit from personalized treatment strategies with acceptable toxicity profiles.
Beyond targeted therapy, ICI are now emerging as a promising new therapy in HNSCC. The genomic predictors of response to ICI clearly exist in other cancer types [67, 93] and it would be prudent to identify genomic and immune mechanisms that underlie tumor immune escape and ICI resistance in HNSCC. As future therapeutic opportunities arise with advances in our understanding of HNSCC biology, there will also be new challenges of translating these advances to personalized therapies. The vision for precision medicine in HNSCC requires concerted interest and continuous effort in the conduct of innovative but complex biomarker directed multidisciplinary trials.
Conclusion
New treatment paradigms for patients with HNSCC are currently evolving. At present the two main subtypes of HNSCC classified by HPV status remains the most clinically relevant. Development of personalized therapy in HNSCC is still in early stage and results from ongoing preclinical and clinical studies will provide further insight into future novel therapeutic strategies. It is, however, critical that future studies select appropriate patients for potential matched therapies based on sound biological rationale to realize precision medicine in head and neck cancer.
Abbreviations
CDK, Cyclin Dependent Kinase; CRISPR, Clustered Regularly Interspaced Short Palindromic Repeats; ctDNA, circulating tumor DNA; DAISY, Data Mining Synthetic Lethality Identification Pipeline; EGFR, Epidermal Growth Factor Receptor; EMT, Epithelial Mesenchymal Transition; EORTC, European Organization for Research and Treatment of Cancer; HNSCC, Head and Neck Squamous Cell Carcinoma; HPV, Human Papillomavirus; HR, Homologous Recombination; ICI, Immune Checkpoint Inhibitors; MAPK, Mitogen Activated Protein Kinase; NGS, Next Generation DNA Sequencing; RTK, Receptor Tyrosine Kinase; SPECTA, Screening Patients for Efficient Clinical Trial Access; TCGA, The Cancer Genome Atlas.
References
Pai SI, Westra WH. Molecular pathology of head and neck cancer: implications for diagnosis, prognosis, and treatment. Annu Rev Pathol. 2009;4:49–70.
Maier H, Dietz A, Gewelke U, Heller WD, Weidauer H. Tobacco and alcohol and the risk of head and neck cancer. Clin Investig. 1992;70:320–7.
Mork J, Lie AK, Glattre E, et al. Human papillomavirus infection as a risk factor for squamous-cell carcinoma of the head and neck. N Engl J Med. 2001;344:1125–31.
Chaturvedi AK, Engels EA, Pfeiffer RM, et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol. 2011;29:4294–301.
Ang KK, Harris J, Wheeler R, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med. 2010;363:24–35.
Vermorken JB, Mesia R, Rivera F, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. 2008;359:1116–27.
Licitra L, Mesia R, Rivera F, et al. Evaluation of EGFR gene copy number as a predictive biomarker for the efficacy of cetuximab in combination with chemotherapy in the first-line treatment of recurrent and/or metastatic squamous cell carcinoma of the head and neck: EXTREME study. Ann Oncol. 2011;22:1078–87.
Licitra L, Storkel S, Kerr KM, et al. Predictive value of epidermal growth factor receptor expression for first-line chemotherapy plus cetuximab in patients with head and neck and colorectal cancer: analysis of data from the EXTREME and CRYSTAL studies. Eur J Cancer. 2013;49:1161–8.
Stransky N, Egloff AM, Tward AD, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–60.
Agrawal N, Frederick MJ, Pickering CR, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333:1154–7.
Chung CH, Guthrie VB, Masica DL, et al. Genomic alterations in head and neck squamous cell carcinoma determined by cancer gene-targeted sequencing. Ann Oncol. 2015;26:1216–23.
Seiwert TY, Zuo Z, Keck MK, et al. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin Cancer Res. 2015;21:632–41.
Pickering CR, Zhang J, Yoo SY, et al. Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer Discov. 2013;3:770–81.
Cancer Genome Atlas N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576–82.
Guerrero-Preston R, Michailidi C, Marchionni L, et al. Key tumor suppressor genes inactivated by “greater promoter” methylation and somatic mutations in head and neck cancer. Epigenetics. 2014;9:1031–46.
Lim AM, Do H, Young RJ, et al. Differential mechanisms of CDKN2A (p16) alteration in oral tongue squamous cell carcinomas and correlation with patient outcome. Int J Cancer. 2014;135:887–95.
Shao X, Tandon R, Samara G, et al. Mutational analysis of the PTEN gene in head and neck squamous cell carcinoma. Int J Cancer. 1998;77:684–8.
Pedrero JM, Carracedo DG, Pinto CM, et al. Frequent genetic and biochemical alterations of the PI 3-K/AKT/PTEN pathway in head and neck squamous cell carcinoma. Int J Cancer. 2005;114:242–8.
Seiwert TY, Jagadeeswaran R, Faoro L, et al. The MET receptor tyrosine kinase is a potential novel therapeutic target for head and neck squamous cell carcinoma. Cancer Res. 2009;69:3021–31.
Izumchenko E, Sun K, Jones S, et al. Notch1 mutations are drivers of oral tumorigenesis. Cancer Prev Res (Phila). 2015;8:277–86.
Song X, Xia R, Li J, et al. Common and complex Notch1 mutations in Chinese oral squamous cell carcinoma. Clin Cancer Res. 2014;20:701–10.
Sun W, Gaykalova DA, Ochs MF, et al. Activation of the NOTCH pathway in head and neck cancer. Cancer Res. 2014;74:1091–104.
Bolos V, Grego-Bessa J, de la Pompa JL. Notch signaling in development and cancer. Endocr Rev. 2007;28:339–63.
Belbin TJ, Singh B, Barber I, et al. Molecular classification of head and neck squamous cell carcinoma using cDNA microarrays. Cancer Res. 2002;62:1184–90.
Chung CH, Parker JS, Karaca G, et al. Molecular classification of head and neck squamous cell carcinomas using patterns of gene expression. Cancer Cell. 2004;5:489–500.
Walter V, Yin X, Wilkerson MD, et al. Molecular subtypes in head and neck cancer exhibit distinct patterns of chromosomal gain and loss of canonical cancer genes. PLoS One. 2013;8, e56823.
Keck MK, Zuo Z, Khattri A, et al. Integrative analysis of head and neck cancer identifies two biologically distinct HPV and three non-HPV subtypes. Clin Cancer Res. 2015;21:870–81.
Finn RS, Crown JP, Lang I, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015;16:25–35.
Turner NC, Ro J, Andre F, et al. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. N Engl J Med. 2015;373:209–19.
Osman AA, Monroe MM, Ortega Alves MV, et al. Wee-1 kinase inhibition overcomes cisplatin resistance associated with high-risk TP53 mutations in head and neck cancer through mitotic arrest followed by senescence. Mol Cancer Ther. 2015;14:608–19.
Gadhikar MA, Sciuto MR, Alves MV, et al. Chk1/2 inhibition overcomes the cisplatin resistance of head and neck cancer cells secondary to the loss of functional p53. Mol Cancer Ther. 2013;12:1860–73.
Carrassa L, Chila R, Lupi M, et al. Combined inhibition of Chk1 and Wee1: in vitro synergistic effect translates to tumor growth inhibition in vivo. Cell Cycle. 2012;11:2507–17.
Tanaka N, Patel AA, Wang J, et al. Wee-1 Kinase Inhibition Sensitizes High-Risk HPV+ HNSCC to Apoptosis Accompanied by Downregulation of MCl-1 and XIAP Antiapoptotic Proteins. Clin Cancer Res. 2015;21:4831–44.
Lui VW, Hedberg ML, Li H, et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013;3:761–9.
Mazumdar T, Byers LA, Ng PK, et al. A comprehensive evaluation of biomarkers predictive of response to PI3K inhibitors and of resistance mechanisms in head and neck squamous cell carcinoma. Mol Cancer Ther. 2014;13:2738–50.
Jimeno A, Bauman JE, Weissman C, et al. A randomized, phase 2 trial of docetaxel with or without PX-866, an irreversible oral phosphatidylinositol 3-kinase inhibitor, in patients with relapsed or metastatic head and neck squamous cell cancer. Oral Oncol. 2015;51:383–8.
Jimeno A, Shirai K, Choi M, et al. A randomized, phase II trial of cetuximab with or without PX-866, an irreversible oral phosphatidylinositol 3-kinase inhibitor, in patients with relapsed or metastatic head and neck squamous cell cancer. Ann Oncol. 2015;26:556–61.
Gonzalez-Angulo AM, Juric D, Argiles G, et al. Safety, pharmacokinetics, and preliminary activity of the alpha specific PI3K inhibitor: Results from the first-in-human study. J Clin Oncol 31, 2013 (suppl; abstr 2531).
Barbareschi M, Buttitta F, Felicioni L, et al. Different prognostic roles of mutations in the helical and kinase domains of the PIK3CA gene in breast carcinomas. Clin Cancer Res. 2007;13:6064–9.
Grunwald V, Keilholz U, Boehm A, et al. TEMHEAD: a single-arm multicentre phase II study of temsirolimus in platin- and cetuximab refractory recurrent and/or metastatic squamous cell carcinoma of the head and neck (SCCHN) of the German SCCHN Group (AIO). Ann Oncol. 2015;26:561–7.
Massarelli E, Lin H, Ginsberg LE, et al. Phase II trial of everolimus and erlotinib in patients with platinum-resistant recurrent and/or metastatic head and neck squamous cell carcinoma. Ann Oncol. 2015;26:1476–80.
Fury MG, Sherman E, Ho A, et al. A phase I study of temsirolimus plus carboplatin plus paclitaxel for patients with recurrent or metastatic (R/M) head and neck squamous cell cancer (HNSCC). Cancer Chemother Pharmacol. 2012;70:121–8.
Saba NF, Hurwitz SJ, Magliocca K, et al. Phase 1 and pharmacokinetic study of everolimus in combination with cetuximab and carboplatin for recurrent/metastatic squamous cell carcinoma of the head and neck. Cancer. 2014;120:3940–51.
Ozanne B, Richards CS, Hendler F, Burns D, Gusterson B. Over-expression of the EGF receptor is a hallmark of squamous cell carcinomas. J Pathol. 1986;149:9–14.
Grandis JR, Tweardy DJ. Elevated levels of transforming growth factor alpha and epidermal growth factor receptor messenger RNA are early markers of carcinogenesis in head and neck cancer. Cancer Res. 1993;53:3579–84.
Ang KK, Berkey BA, Tu X, et al. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res. 2002;62:7350–6.
Hama T, Yuza Y, Saito Y, et al. Prognostic significance of epidermal growth factor receptor phosphorylation and mutation in head and neck squamous cell carcinoma. Oncologist. 2009;14:900–8.
Keren S, Shoude Z, Lu Z, Beibei Y. Role of EGFR as a prognostic factor for survival in head and neck cancer: a meta-analysis. Tumour Biol. 2014;35:2285–95.
Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354:567–78.
Vermorken JB, Trigo J, Hitt R, et al. Open-label, uncontrolled, multicenter phase II study to evaluate the efficacy and toxicity of cetuximab as a single agent in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck who failed to respond to platinum-based therapy. J Clin Oncol. 2007;25:2171–7.
Lee JW, Soung YH, Kim SY, et al. Somatic mutations of EGFR gene in squamous cell carcinoma of the head and neck. Clin Cancer Res. 2005;11:2879–82.
Loeffler-Ragg J, Witsch-Baumgartner M, Tzankov A, et al. Low incidence of mutations in EGFR kinase domain in Caucasian patients with head and neck squamous cell carcinoma. Eur J Cancer. 2006;42:109–11.
Khattri A, Zuo Z, Bragelmann J, et al. Rare occurrence of EGFRvIII deletion in head and neck squamous cell carcinoma. Oral Oncol. 2015;51:53–8.
Machiels JP, Haddad RI, Fayette J, et al. Afatinib versus methotrexate as second-line treatment in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck progressing on or after platinum-based therapy (LUX-Head & Neck 1): an open-label, randomised phase 3 trial. Lancet Oncol. 2015;16:583–94.
Abdul Razak AR, Soulieres D, Laurie SA, et al. A phase II trial of dacomitinib, an oral pan-human EGF receptor (HER) inhibitor, as first-line treatment in recurrent and/or metastatic squamous-cell carcinoma of the head and neck. Ann Oncol. 2013;24:761–9.
Kim HS, Kwon HJ, Jung I, et al. Phase II clinical and exploratory biomarker study of dacomitinib in patients with recurrent and/or metastatic squamous cell carcinoma of head and neck. Clin Cancer Res. 2015;21:544–52.
Janjigian YY, Smit EF, Groen HJ, et al. Dual inhibition of EGFR with afatinib and cetuximab in kinase inhibitor-resistant EGFR-mutant lung cancer with and without T790M mutations. Cancer Discov. 2014;4:1036–45.
Goke F, Franzen A, Hinz TK, et al. FGFR1 Expression Levels Predict BGJ398 Sensitivity of FGFR1-Dependent Head and Neck Squamous Cell Cancers. Clin Cancer Res. 2015;21:4356–64.
Malchers F, Dietlein F, Schottle J, et al. Cell-autonomous and non-cell-autonomous mechanisms of transformation by amplified FGFR1 in lung cancer. Cancer Discov. 2014;4:246–57.
Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43.
Chau NG, Perez-Ordonez B, Zhang K, et al. The association between EGFR variant III, HPV, p16, c-MET, EGFR gene copy number and response to EGFR inhibitors in patients with recurrent or metastatic squamous cell carcinoma of the head and neck. Head Neck Oncol. 2011;3:11.
Rabinowits G, Haddad RI. Overcoming resistance to EGFR inhibitor in head and neck cancer: a review of the literature. Oral Oncol. 2012;48:1085–9.
Seiwert T, Sarantopoulos J, Kallender H, McCallum S, Keer HN, Blumenschein Jr G. Phase II trial of single-agent foretinib (GSK1363089) in patients with recurrent or metastatic squamous cell carcinoma of the head and neck. Invest New Drugs. 2013;31:417–24.
Seiwert TY, Haddad RI, Gupta S, et al. Antitumor activity and safety of pembrolizumab in patients with advanced squamous carcinoma of head and neck: Preliminary results from KEYNOTE-012 expansion cohort. J Clin Oncol 2015;33:suppl: abstr LBA6008.
Segal NH, Ou SI, Balmanoukian AS, et al. Safety and effecacy of MEDI4736, an anti-PD-L1 antibody, in patients from a squamous cell carcinoma of the head and neck (SCCHN) expansion cohort. J Clin Oncol. 2015;33.
Gillison ML, Blumenschein G, Fayette J, et al. Nivolumab Versus Investigator’s Choice (IC) for Recurrent or Metastatic (R/M) Head and Neck Squamous Cell Carcinoma (SCCHN): CheckMate-141. Presented at: AACR 2016 Annual Meeting, New Orleans; April 16–20, 2016. Abstract CT099.
Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–8.
Saloura V, Zuo Z, Koeppen H, et al. Correlation of T-cell inflamed phenotype with mesenchymal sutype, PD-L1 and other immune checkpoints in head and neck cancer. J Clin Oncol. 2014;32(suppl; abstr 6009):5s.
Marsit CJ, Liu M, Nelson HH, Posner M, Suzuki M, Kelsey KT. Inactivation of the Fanconi anemia/BRCA pathway in lung and oral cancers: implications for treatment and survival. Oncogene. 2004;23:1000–4.
Do K, Wilsker D, Ji J, et al. Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients With Refractory Solid Tumors. J Clin Oncol. 2015;33:3409–15.
Hori K, Sen A, Artavanis-Tsakonas S. Notch signaling at a glance. J Cell Sci. 2013;126:2135–40.
Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008;358:2039–49.
Struhl G, Adachi A. Nuclear access and action of notch in vivo. Cell. 1998;93:649–60.
Piha-Paul SA, Munster PN, Hollebecque A, et al. Results of a phase 1 trial combining ridaforolimus and MK-0752 in patients with advanced solid tumours. Eur J Cancer. 2015;51:1865–73.
Coric V, Salloway S, van Dyck CH, et al. Targeting Prodromal Alzheimer Disease With Avagacestat: A Randomized Clinical Trial. JAMA Neurol. 2015;72:1324–33.
Henley DB, Sundell KL, Sethuraman G, Dowsett SA, May PC. Safety profile of semagacestat, a gamma-secretase inhibitor: IDENTITY trial findings. Curr Med Res Opin. 2014;30:2021–32.
Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92.
Gerlinger M, Catto JW, Orntoft TF, Real FX, Zwarthoff EC, Swanton C. Intratumour heterogeneity in urologic cancers: from molecular evidence to clinical implications. Eur Urol. 2015;67:729–37.
de Bruin EC, McGranahan N, Mitter R, et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science. 2014;346:251–6.
Mroz EA, Tward AD, Pickering CR, Myers JN, Ferris RL, Rocco JW. High intratumor genetic heterogeneity is related to worse outcome in patients with head and neck squamous cell carcinoma. Cancer. 2013;119:3034–42.
Mroz EA, Tward AD, Hammon RJ, Ren Y, Rocco JW. Intra-tumor genetic heterogeneity and mortality in head and neck cancer: analysis of data from the Cancer Genome Atlas. PLoS Med. 2015;12, e1001786.
Forshew T, Murtaza M, Parkinson C, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med. 2012;4:136ra68.
Murtaza M, Dawson SJ, Tsui DW, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013;497:108–12.
Dawson SJ, Tsui DW, Murtaza M, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;368:1199–209.
Hong MK, Macintyre G, Wedge DC, et al. Tracking the origins and drivers of subclonal metastatic expansion in prostate cancer. Nat Commun. 2015;6:6605.
Murtaza M, Dawson SJ, Pogrebniak K, et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat Commun. 2015;6:8760.
Girotti MR, Gremel G, Lee R, et al. Application of Sequencing, Liquid Biopsies, and Patient-Derived Xenografts for Personalized Medicine in Melanoma. Cancer Discov. 2016;6:286–99.
Wang Y, Springer S, Mulvey CL, et al. Detection of somatic mutations and HPV in the saliva and plasma of patients with head and neck squamous cell carcinomas. Sci Transl Med. 2015;7:293ra104.
Jerby-Arnon L, Pfetzer N, Waldman YY, et al. Predicting cancer-specific vulnerability via data-driven detection of synthetic lethality. Cell. 2014;158:1199–209.
Thompson JM, Nguyen QH, Singh M, Razorenova OV. Approaches to identifying synthetic lethal interactions in cancer. Yale J Biol Med. 2015;88:145–55.
Carter SL, Cibulskis K, Helman E, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–21.
Van Loo P, Campbell PJ. ABSOLUTE cancer genomics. Nat Biotechnol. 2012;30:620–1.
Snyder A, Wolchok JD, Chan TA. Genetic basis for clinical response to CTLA-4 blockade. N Engl J Med. 2015;372:783.
Funding
Both authors are employees of the University Health Network, Toronto, Canada and there is no other source of funding directly related to this manuscript.
Availability of data and materials
This is a review article and there is no raw data related to this manuscript for data sharing.
Authors’ contributions
KA drafted the manuscript. LS corrected and finalized the manuscript. Both authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Aung, K.L., Siu, L.L. Genomically personalized therapy in head and neck cancer. Cancers Head Neck 1, 2 (2016). https://doi.org/10.1186/s41199-016-0004-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41199-016-0004-y