
Abstract
Vascular calcification has now been recognized as a major problem in dialysis patients because of its strong influence on the prognosis. Along with the regulatory failure of calcification-inhibitory system, active phenotypic change of vascular smooth muscle cells (VSMCs) to osteoblast-like cells is also involved in the progression of vascular calcification.
Delaying or improving the vascular calcification is thought to be very important to improve the cardiovascular mortality in dialysis patients. Several interventional trials against vascular calcification using non-calcium-containing phosphate binders, low-dose active vitamin D plus cinacalcet, modification of dialysate calcium concentration, and sodium thiosulfate have been done, and some trials including non-calcium-containing phosphate binders showed beneficial effect on delaying vascular calcification in dialysis patients. However, delaying or improving vascular calcification has not been clearly proved to result in improved cardiovascular event and/or mortality rate by prospective interventional randomized controlled trials in dialysis patients. Whether the improvement of vascular calcification could directly lead to the improvement of survival is an urgent issue of clinical trials in dialysis patients.
Similar content being viewed by others


Background
As mentioned in a recent review [1], active atherosclerotic process has already begun in the early stages of chronic kidney disease (CKD), and atherosclerotic organ damages deteriorate along with the decreasing renal function. By the time of the initiation of renal replacement therapy (RRT), major atherosclerotic vascular damages have already been completed in many patients. Coronary artery stenosis (CAS) has been shown in almost 50% in patients with end-stage renal failure (ESRD) [2, 3], and almost 80% of diabetic ESRD patients have significant occult CAS at the initiation of RRT in spite of no chest symptom or no previous history of ischemic heart disease [2]. However, as Lindner et al. rung an alarm 42 years ago [4], atherosclerotic process intensively accelerates after the initiation of RRT.
One of the most characteristic features of atherosclerosis seen in dialysis patients is vascular calcification, especially “medial calcification.” Medial calcification was initially described in 1903, a hundred years ago by Johann Georg Mönckeberg, a German pathologist. Therefore, medial calcification may be called as “Mönckeberg’s mediasclerosis” or “Mönckeberg’s mediacalcinosis” [5]. Vascular calcification crossly associates with several target organ damages (TODs) including stroke, ischemic heart disease, and peripheral arterial disease. Vascular calcification causes TODs via the disturbance of vascular function, i.e., “vascular failure.”
Vascular calcification affects on the future cardiovascular events and/or mortality in dialysis patients [6,7,8,9,10,11,12,13,14,15,16,17,18,19,20]. Several clinical trials to aim to improve cardiovascular events and/or mortality in dialysis patients have been planned or reported. Among these trials, the largest randomized controlled trial (RCT), the LANDMARK study, which compares the effect of non-calcium-containing phosphate binder, lanthanum carbonate, with calcium-containing phosphate binder, is now ongoing in Japan [21]. The result of the LANDMARK study will soon be open.
Here, we want to summarize in the present situation about the clinical significance, mechanisms, and the behavior of vascular calcification by interventional trials and provide update information about clinical trials against vascular calcification in dialysis patients.
Clinical impact of vascular calcification
Several reports have shown the strong relationship between vascular calcification and clinical outcomes including cardiovascular events, and cardiovascular and all-cause mortality.
The number of calcified sites including carotid artery, abdominal aorta, ilio-femoral axis, and legs was a strong predictor of cardiovascular and all-cause mortality in an early study [6]. As to the relationship between coronary artery calcification score (CACS) and clinical outcomes, dialysis patients with higher CACS showed significantly higher rate of cardiovascular and all-cause mortality compared with those with mild or no CACS (Fig. 1) [7,8,9,10,11,12,13]. In these reports, cardiovascular events were also significantly correlated with high CACS. Aortic calcification also significantly correlated with cardiovascular and all-cause mortality and was an independent predictor of cardiovascular and all-cause mortality [14,15,16,17,18,19]. These associations between vascular calcification and clinical outcomes were independent even after adjusting traditional risk factors such as age, hemodialysis duration, hypertension, diabetes, smoking, and dyslipidemia. Clinical significance in other vascular sites has also been reported. Carotid artery calcification at the initiation of hemodialysis was an independent associating factor for cardiovascular events in incident hemodialysis patients [20]. Furthermore, lower limbs’ arterial calcification was crossly associated with the presence and severity of peripheral arterial disease (PAD) in hemodialysis patients [22]. Critical limb ischemia (CLI) strongly impacts on the prognosis of hemodialysis patients, and lower limbs’ arterial calcification in CLI patients was extremely high compared with non-PAD or non-CLI patients on hemodialysis (Fig. 2).

Impact of CACS on cardiovascular events, cardiovascular mortality, and all-cause mortality [10]. Patients with CACS >750 (N = 37) showed significantly higher cardiovascular event rate, cardiovascular mortality rate, and all-cause mortality rate compared with patients with CACS <750 (N = 37), according to the median value of CACS (748.2) of all 74 patients. a Cardiovascular events, b cardiovascular mortality, c all-cause mortality. Abbreviation: CACS coronary artery calcification score

a, b Lower limbs’ arterial calcification score and severity of peripheral arterial disease in hemodialysis patients [22]. Lower limbs’ arterial calcification score (above knee, below knee). *p < 0.01 vs. PAD(−) group, # p < 0.01 vs. PAD Fontaine 1 group. Abbreviations: SFA superficial femoral artery, BKA below-knee arteries, PAD peripheral arterial disease
Vascular calcification positively correlates with arterial stiffness. Aortic stiffness represented by aortic pulse wave velocity (PWV) was significantly associated with abdominal aortic calcification [23] and CACS [24]. Increased aortic stiffness (increased cardiac afterload) consequently leads to left ventricular hypertrophy (LVH). LVH could be also induced by elevated fibroblast growth factor 23 (FGF23) as proven in an animal experiment [25]. Both calcification-related cardiac ischemia and LVH concomitantly increase the risk of cardiovascular mortality (Fig. 3). Hemodynamic and functional changes associated with vascular calcification have strong clinical impact on morbidity and mortality in dialysis patients. Therefore, clarifying the mechanisms and accelerating factors for vascular calcification in dialysis patients is very important.

Clinical aspect of vascular calcification leading to cardiovascular complication. The critical key for vascular calcification is hyperphosphatemia due to decreased renal function. Hyperphosphatemia causes vascular calcification through several mechanisms. Vascular calcification, concomitantly with left ventricular hypertrophy and cardiac fibrosis, causes cardiovascular complication. Abbreviations: FGF23 fibroblast growth factor 23, VSMC vascular smooth muscle cell, iPTH intact parathyroid hormone, PWV pulse wave velocity
Mechanisms and associating factors for vascular calcification
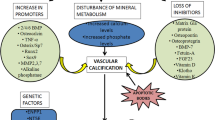
Serum levels of both calcium and phosphate in normal physiological condition are tightly regulated in narrow ranges. Formation of hydroxyapatite is limited in bones, and vascular calcification does not occur. Vascular smooth muscle cells (VSMCs) prevent ectopic calcification via inhibitory mechanisms in normal physiological condition. However, extensive and accelerating medial calcification ensues in CKD patients, especially in dialysis patients. Important mechanisms of vascular calcification in dialysis patients are (1) failure of inhibitory systems for vascular calcification and (2) differentiation of VSMCs to osteoblast-like cells (Fig. 4).

Cellular aspect of VSMCs leading to medial calcification. Elevated P induces Runx2 upregulation and promoted osteochondrocytic differentiation of VSMCs. At the same time, uremic milieu decreases endogenous inhibitors. Hyperphosphatemia stimulated excretion of extracellular matrix with high affinity for calcium from VSMCs. Concomitant calcium overload (or elevated calcium) enhances calcium deposition around VSMCs. Abbreviations: VSMC vascular smooth muscle cell, P phosphate, Ca calcium, MGP matrix Gla protein, ECM extracellular matrix
Inhibitory factors for vascular calcification include matrix Gla protein (MGP), pyrophosphate (produced in VSMCs), and circulating inhibitor fetuin A. On the other hand, activation of transcription factors “Runx2” and mineralization regulating protein “alkaline phosphatase (ALP)” are important key factors for osteochondrocytic differentiation of VSMCs. Uremic milieu concomitantly inactivates the production of inhibitors and promotes phenotypic changes and/or apoptosis of VSMCs resulting in medial calcification (Fig. 4) [26].
Inhibitory factors (with comment about the risk of warfarin use)
MGP is expressed in VSMCs and loaded in matrix vesicles (scaffold of calcification) around VSMCs, consequently inhibiting their calcification. MGP binds to calcium and pro-osteogenic factor bone morphologic protein 2 (BMP2) and inactivates it in normal condition [27]. However, loading of MGP is decreased in high calcium circumstances, and matrix calcification is promoted [28, 29]. Vitamin K is essential for MGP activation. Therefore, deficiency of vitamin K inhibits MGP activity, thus leading to vascular calcification enhancement. Warfarin, an antagonist to vitamin K, is a strong promoter of arterial calcification via blocking the activation of vitamin K-dependent MGP [30]. Many dialysis patients have cardiovascular complications, including artificial valve replacement, atrial fibrillation, and cardiogenic cerebral embolism, which might necessitate warfarin prescription. However, many cardiologists who prescribe warfarin do not know the profound risk of warfarin as a strong promoter of vascular calcification in dialysis patients. Not only nephrologists but cardiologists should be aware of the profound risk of warfarin use in dialysis patients.
Pyrophosphate binds to hydroxyapatite crystals and inhibits their further growth. ALP, which is upregulated in VSMCs in the early stages of vascular calcification and a key factor for mineralization, degrades pyrophosphate, thereby promoting calcification [31,32,33]. Osteogenic transcription factor Runx2 is thought to regulate the expression of ALP.
Fetuin A is a circulating inhibitor and forms a complex with calcium and phosphate, forming a calciprotein particle (CPP), thus preventing mineral deposition in vascular walls [34, 35]. Furthermore, fetuin A is taken up by synthetic VSMCs and secreted in a matrix around VSMCs, where it protects from calcification [36]. Therefore, fetuin A has dual inhibitory actions, one in circulation and the other in vascular walls. The levels of fetuin A are reduced in dialysis patients, and it might reflect the excessive CPP formation in dialysis patients [37,38,39,40].
Osteoblastic differentiation
Runx2 upregulation and ALP expression in VSMCs is the most important process in the early phase of osteoblast-like cell differentiation of VSMCs [41]. Expression of Runx2 is normally restricted in the bone and cartilage. However, VSMCs express Runx2 via the stimulation of several uremia-related factors including phosphate, oxidative stress [42], and aldosterone [43, 44], among which phosphate is the strongest stimulator of Runx2 upregulation.
Phosphate elevation is the most important and strong key factor for vascular calcification (Fig. 4). An early study by Shigematsu et al. provided that the primary culture of radial artery VSMCs from a dialysis patient showed increased excretion of extracellular matrix with high affinity for calcium when incubated with a high phosphate medium (Pi = 5.4 mg/dl) [45]. They provided the evidence that phosphate overload accelerates vascular calcium deposition in vitro. High phosphate signal around VSMCs was recognized by increased uptake of phosphate by VSMCs via Pit 1 and Pit 2, sodium-dependent phosphate transporters.
As to the implication of calcium, vessel rings from a dialysis patient showed calcium-induced calcification more potently than phosphate (at equivalent calcium-phosphate product) [46]. Higher calcium concentration accelerated calcium deposition more severely than high phosphate concentration in vessel rings. This result suggested calcium has stronger influence on vascular calcification than phosphate. Sustained hyperphosphatemia with episodic increase of calcium or calcium overload is thought to strongly influence the cellular defense mechanism against calcification.
Basic researches and epidemiological observational studies provided several clinical factors that are significantly associated with vascular calcification (Table 1) [10,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,, 22, 45–73]. Among these factors, mineral abnormalities including hyperphosphatemia, hypercalcemia, and elevated Ca × Pi product are the most important key factors for vascular calcification. Our previous study showed micro-inflammation, represented by elevated highly sensitive C-reactive protein, was another strong and independent predictor for CAC progression (Fig. 5) [10]. Other studies also provided the link between micro-inflammation and progression of CAC [24, 74, 75]. It has now been known that hyperphosphatemia itself is an important source of inflammation [76]. Inflammatory cytokine TNF-α upregulates Pit-1 expression and Na-Pi co-transporter and increases phosphate uptake into VSMCs [77]. Both phosphate overload and accompanying inflammation is thought to concomitantly enhance the vascular calcification.

Stratified hsCRP and the progression of CAC [10]. CACS of 56 patients on maintenance hemodialysis were evaluated repeatedly with 15 months interval, and delta CACS (changes of CACS) were shown according to stratified hsCRP. Progression of CACS was significantly correlated with baseline hsCRP values. Abbreviations: hsCRP high sensitive C-reactive protein, CACS coronary artery calcification score
FGF23/klotho axis and vascular calcification
Both fibroblast growth factor 23 (FGF23) and klotho, the key players in CKD-MBD, have recently attracted great concerns in relation to cardiovascular events and vascular calcification in CKD patients. FGF23 was found as a bone-derived (synthesized and excreted by osteoblast) hormone that regulates phosphate and 1,25-hydroxyvitamin D metabolism [78]. FGF23 binds to the FGF receptor with its co-receptor klotho and acts to increase renal phosphate excretion. In addition, FGF23 reduces the synthesis of 1,25-hydroxyvitamin D. Furthermore, FGF23 decreases parathyroid hormone synthesis and secretion. FGF23 induces left ventricular hypertrophy [25], and elevated FGF23 is known to associate with vascular calcification in HD patients [60,61,62].
On the other hand, klotho, discovered by Kuro-o et al. in 1997 [79], is expressed in human vascular tissue in addition to its major expression in the kidney and parathyroid [80]. Vascular calcification is a prominent finding in mice with a klotho gene deletion, the same as in CKD patients, and klotho overexpression by adenoviral delivery to klotho−/− mice reverse the vascular calcification [81]. Suppressive mechanism of klotho on vascular calcification is multifactorial. Klotho prevents apoptosis of vascular smooth muscle cells [82] and acts as an anti-inflammatory modulator and restricts inflammatory process, thus protecting the vasculature [83].
Elevated FGF23 and hyperphosphatemia (along with klotho deficiency) are associated with vascular calcification in many observational studies. However, it should be noted that there is a controversy whether FGF23 is a direct contributor to vascular calcification. Scialla et al. recently reported that the baseline plasma FGF23 level was not associated with the prevalence or severity of coronary artery calcium content in patients with mild to moderate CKD (eGFR 20–70 ml/min/1.73 m2), suggesting that FGF23 is not associated with arterial calcification [84]. Furthermore, there is no in vitro study that proved direct action of FGF23 on vascular calcification. A major question that remains unresolved is whether FGF23 can directly act on vascular cells to promote or inhibit matrix. Further studies are necessary to investigate the role of FGF23/klotho axis on vascular calcification.
Potential tools against vascular calcification
Phosphate binders
The phosphate signal is thought to be an entry gate for the progression of CAC and future cardiovascular complications (Fig. 3). Another important key factor leading to the progression of CAC is calcium overload related to the use of calcium-based phosphate binders [85,86,87,88]. Even if levels of serum calcium are within normal ranges, total calcium intake (ingested as phosphate binder) significantly correlated with CAC progression [57]. Therefore, many interventional studies using non-calcium-containing phosphate binders have been performed.
Sevelamer hydrochloride, an available drug as the first non-calcium-containing phosphate binder other than aluminum-containing phosphate binder, has been proved to delay the progression of CAC compared with calcium carbonate [86,90,91,, 89–92]. Other than binding to phosphate, sevelamer hydrochloride has several pleiotropic effects to lower cholesterol, FGF23, advanced glycation end product, inflammatory markers, and C-reactive protein [86, 90, 93, 94]. Several clinical trials expected the beneficial inhibitory effect of sevelamer hydrochloride on the progression of vascular calcification. However, one meta-analysis study that analyzed 14 researches containing 3271 patients in total could not provide the effectiveness of sevelamer hydrochloride compared with calcium-based phosphate binders for delaying the progression of CAC [95]. Furthermore, the phosphate-binding capacity of sevelamer hydrochloride is rather weak compared with calcium carbonate and/or lanthanum carbonate [96, 97]. Phosphate-binding capacity is estimated as lanthanum carbonate > calcium carbonate > sevelamer hydrochloride, and the proportion of phosphate-binding capacity is approximately 3:1.5:1 [96, 97]. These might weaken the use of sevelamer hydrochloride in the clinical setting. The important matters to be required as a phosphate binder are good phosphate-binding capacity, good drug adherence, good phosphate control, and drug safety. In this respect, pleiotropic effects might be the next issue to these things.
Compared with sevelamer hydrochloride, lanthanum carbonate, which contains the rare earth element lanthanum, has stronger phosphate-binding capacity and enables good control of serum phosphate [96, 97]. There are several studies that compared the effect of lanthanum carbonate on vascular calcification with calcium-based phosphate binders [98,99,100,101]. We performed a prospective randomized interventional study that compared the effect of lanthanum carbonate and calcium carbonate on the progression of CAC (Fig. 6) [98]. Treatment with lanthanum carbonate was more effective compared to calcium carbonate in preventing the progression of CAC in patients on hemodialysis; regression by 6.4% was shown in the lanthanum-treated group vs. 41.2% progression in those receiving calcium carbonate. The serum levels of phosphate and calcium were not different between the two groups. In this study, even if low calcium dialysate (2.5 mEq/l) was used, CACS progressed in hemodialysis patients who are prescribed calcium-based phosphate binders. Meta-analysis, which compared calcium-based and non-calcium-based phosphate binders on survival and vascular calcification in dialysis patients, revealed beneficial effect in non-calcium-based phosphate binders on the progression of CAC [102].

Percent changes in CACS in the CC group and LC group [90]. Gray bar displays percent change in CACS in the 6-month lead-in period in a total of 42 patients. Open and closed bars display percent change during the 6-month intervention period in the calcium carbonate (CC) group (N = 23) and lanthanum carbonate (LC) group (N = 19). CACS of 42 hemodialysis patients using CC as phosphate binder increased 36.8% during the 6-month lead-in period. In interventional period, they were divided into the CC group and LC group. Mean CACS increased 41.2% in the CC group, while mean CACS decreased 6.4% in the LC group (p = 0.024). Abbreviation: CACS coronary artery calcification score
Vascular calcification might be merely one surrogate. The true target is to prevent a cardiovascular event and improve patients’ survival. In this regard, recent observational studies provided beneficial effect of lanthanum carbonate on hemodialysis patients’ survival [103, 104]. A large RCT, the LANDMARK study, is now ongoing in Japan to evaluate the cardiovascular event and patient survival in the lanthanum carbonate group and calcium carbonate group [21]. As to the safety of lanthanum, side effects and bone toxicity have been evaluated [105,106,107] and at present, severe side effect or bone toxicity have not been shown. However, because the nature of lanthanum is metal, careful observation for a long period is necessary to conclude the safety of lanthanum carbonate.
Calcimimetic and active vitamin D
Both high and low turnover bone condition could associate with CAC progression [51,57,58,, 56–59, 70, 85, 89]. Increased release of calcium and phosphate from the bone in patients with high turnover bone and decreased uptake of calcium and phosphate into the bone (disturbed buffer function of the bone) in patients with low turnover bone might cause advanced vascular calcification in such patients. Dialysis patients with intact parathyroid hormone (iPTH) levels >400 pg/ml have, in general, high turnover bone, and those with iPTH levels <150 pg/ml often present adynamic or low turnover bone. Therefore, a control target of iPTH of 150–400 pg/ml might be reasonable from the point of view of preventing vascular calcification.
A randomized interventional study (ADVANCE study) provided that the rate of progression of CAC and aortic valve calcification was reduced when cinacalcet was added to low-dose active vitamin D compared to larger doses of active vitamin D therapy alone [108, 109]. However, significant benefits in overall survival or cardiovascular events by cinacalcet were not observed in a large RCT (EVOLVE trial) in 3883 hemodialysis patients after 5 years’ follow-up [110]. Recent manuscripts partly analyzed the data of patients in the EVOLVE trial whose FGF23 decreased more than 30% by cinacalcet within 20 weeks. One showed significant improvement in time to primary endpoint (death or first nonfatal cardiovascular event) compared with placebo [111], and another provided the improved primary composite outcome (death and major cardiovascular event) by cinacalcet in patients more than 65 years old [112]. The results of the EVOLVE trial are somewhat inconclusive and should be carefully interpreted.
Increased calcium and phosphate absorption by active vitamin D might influence vascular calcification. However, active vitamin D increased klotho and osteopontin expression in arterial walls while decreasing aortic calcification in CKD mice fed a high phosphate diet [113]. Therefore, low-dose active vitamin D, in the dose that does not increase calcium and phosphate load, might be useful for preventing vascular calcification [80, 114].
Vitamin K
Vitamin K is required as a cofactor in the process of gamma-carboxylation of extracellular matrix protein. Green leaf vegetables contain vitamin K1, and cheese, natto, and animals contain vitamin K2. Coagulation factors require vitamin K1 for their carboxylation process, and MGP requires vitamin K2 for its carboxylation, converting to active form [115]. MGP (in active form) inhibits extracellular matrix calcification and prevents arterial calcification. On the other hand, warfarin, an antagonist to vitamin K, promotes arterial calcification via blocking the activation of MGP as previously described [30]. Furthermore, many hemodialysis patients have vitamin K deficiency. On the basis of these findings, clinical trials intending to prevent vascular calcification by vitamin K1 or K2 supplementation in dialysis patients are now ongoing [116, 117].
Dialysate modification
Calcium overload could occur not only by the use of calcium-containing phosphate binders or high-dose active vitamin D but also by high calcium concentration dialysate. High calcium concentration dialysate (1.75 mmol/l) yields net calcium influx of 978 mg into the body during one hemodialysis session in a patient with serum calcium 9.1 mg/dl [118]. Positive calcium balance excessively inhibits parathyroid function and may cause low turnover bone, which is a known risk factor for vascular calcification.
In a recent randomized controlled study, effect of lowering the dialysate calcium level on the progression of CAC and the histologic bone abnormalities in 425 hemodialysis patients was examined [119]. As a result, progression rate of CAC was significantly lower in the low calcium dialysate (1.25 mmol/l) group than in the high calcium dialysate (1.75 mmol/l) group. Furthermore, the prevalence of histologically diagnosed low turnover bone significantly decreased in the low calcium group (from 85 to 41.8%, p = 0.001). Lowering dialysate calcium levels delayed the progression of CAC and improved bone turnover in patients on hemodialysis with baseline iPTH levels ≤300 pg/ml.
As to other changeable components of dialysate, one recent cross-sectional observational study in our dialysis center provided that pre-dialysis serum bicarbonate levels were significantly associated with coronary CAC score [64]. Acid-base status in dialysis patients associates with vascular calcification, and dialysate bicarbonate could be modifiable. Ultrapure dialysate might be efficacious to delay vascular calcification because micro-inflammation is a strong promoter for vascular calcification [10, 24, 74, 75]. Furthermore, because uremic milieu promotes atherosclerosis in dialysis patients, change of dialysis modality might also affect the progression of vascular calcification. Further interventional study to change dialysate fluid or dialysis modality might bring the benefit for delaying the progression of vascular calcification.
Bisphosphonate
Bisphosphonates are synthetic analogs of inorganic pyrophosphate and suppress bone resorption. Elution of calcium and phosphate (substrate of calcification) from the bone could be decreased or stopped by bisphosphonate. Therefore, bisphosphonates might be beneficial to prevent the progression of vascular calcification. Earlier studies provided the evidence that oral and parenteral etidronate delayed the progression of CAC and aortic calcification [120, 121]. However, this effect was not proven in alendronate [122].
Bisphosphonate use in osteoporotic post-menopausal women increased the risk of calcification in coronary artery and cardiac valves [123]. Furthermore, long-term safety (and efficacy) of bisphosphonate in CKD patients has not been confirmed. Thus, the recent Kidney Disease Improving Global Outcomes (KDIGO) recommendation suggested not to prescribe bisphosphonates in patients with an eGFR <30 ml/min/1.73 m2 [124], and the Japanese Clinical Practice Guideline for the Management of Chronic Kidney Disease-Mineral and Bone Disorder suggested bisphosphonate use should not be recommended for osteoporosis in dialysis patients [125].
Sodium thiosulfate
Sodium thiosulfate (STS) is a chelating agent and has been applied for calcific uremic arteriolopathy [126, 127]. Calcium-thiosulfate complex is more soluble than calcium oxalate and calcium phosphate, and STS has antioxidant activity [128, 129]. Intravenous infusion of 25% STS solution immediately after hemodialysis for 60 times during 5 months was well tolerated in most patients [126], and calcific uremic arteriolopathy improved in a large observational study [127]. In one study which evaluated the effect of STS on CAC, twice weekly STS infusion post-hemodialysis for 4 months delayed the progression of CAC compared with non-treated control (p = 0.03) [130]. CACS was unchanged in the STS-treated group but increased significantly in the non-treated control group. Considering the chelating and removing nature of STS for precipitated calcium from the vascular walls, long-term use of STS might not only delay the progression of calcification but also decrease the calcification score. Gastrointestinal side effects and subsequent malnutrition and the potential risk of STS to decrease bone mineral density of normal bones should be considered when used for a long period.
Conclusions
Vascular calcification is an independent and important risk factor for cardiovascular events and all-cause mortality in patients on hemodialysis. The mechanism of vascular calcification is multifactorial, and the active process of calcification advances along with dialysis duration. The decrease of three major inhibitory factors of calcification including fetuin A, MGP, and pyrophosphate in the vascular walls and the active osteoblast-like cell differentiation of VSMCs due to Runx2 cascade by phosphate and calcium metabolism abnormality, inflammation, and oxidative stress are involved in vascular calcification.
Several prospective interventional trials against vascular calcification have been performed and are now ongoing. However, no prospective trial has yet proved to improve cardiovascular events and survival in hemodialysis patients. Some observational retrospective studies provided the efficacy of non-calcium-containing phosphate binder. Because vascular calcification strongly influences the outcome of dialysis patients, we wish future interventional RCTs against vascular calcification would clearly provide the beneficial effect on the prognosis of hemodialysis patients.
References
Kobayashi S. Cardiovascular events in chronic kidney disease (CKD)—an importance of vascular calcification and microcirculatory impairment. Renal Replacement Therapy. 2016;2:55. doi:10.1186/s41100-016-0062-y.
Ohtake T, Kobayashi S, Moriya H, Negishi K, Okamoto K, Maesato K, et al. High prevalence of occult coronary artery stenosis in patients with chronic kidney disease at the initiation of renal replacement therapy: an angiographic examination. J Am Soc Nephrol. 2005;16:1141–8.
Joki N, Hase H, Nakamura R, Yamaguchi T. Onset of coronary artery disease prior to initiation of haemodialysis in patients with end stage renal disease. Nephrol Dial Transplant. 1997;12:718–23.
Lindner A, Charra B, Sherrard DJ, Scribner BH. Accelerated atherosclerosis in prolonged maintenance hemodialysis. N Engl J Med. 1974;290:697–701.
Möncheberg JG. Uber die reine Mediaverkalkung der Extremitätenarterien und ihr verhalten zur Arterosklerose. Virchow Arch Pathol Anat. 1903;171:141–67.
Blacher J, Guerin AP, Pannier B, Marchais SJ, London GM. Arterial calcifications, arterial stiffness, and cardiovascular risk in ESRD. Hypertension. 2001;38:938–42.
London GM. Cardiovascular calcifications in uremic patients: clinical impact on cardiovascular function. J Am Soc Nephrol. 2003;14:S305–9.
Block GA, Raggi P, Bellasi A, Kooienga L, Spiegel DM. Mortality effect of coronary calcification and phosphate binder choice in incident hemodialysis patients. Kidney Int. 2007;71:438–41.
Matsuoka M, Iseki K, Tamashiro M, Fujimoto N, Higa N, Touma T, et al. Impact of high coronary artery calcification score (CACS) on survival in patients on chronic hemodialysis. Clin Exp Nephrol. 2004;8:54–8.
Ohtake T, Ishioka K, Honda K, Oka M, Maesato K, Mano T, et al. Impact of coronary artery calcification in hemodialysis patients: risk factors and associations with prognosis. Hemodial Int. 2010;14:218–25.
Shantouf RS, Budoff MJ, Ahmadi N, Ghaffari A, Flores F, Gopal A, et al. Total and individual coronary artery calcium scores as independent predictors of mortality in hemodialysis patients. Am J Nephrol. 2010;31:419–25.
Shimoyama Y, Tsuruta Y, Niwa T. Coronary artery calcification score is associated with mortality in Japanese hemodialysis patients. J Ren Nutr. 2012;22:139–42.
Wilkieson T, Rahman MO, Gangii AS, Voss M, Ingram AJ, Ranganath N, et al. Coronary artery calcification, cardiovascular events, and death: a prospective cohort study of incident patients on hemodialysis. Canadian J Kidney Disease. 2015;2:29.
Okuno S, Ishimura E, Kitatani K, Fujino Y, Kohno K, Maeno Y, et al. Presence of abdominal aortic calcification is significantly associated with all-cause and cardiovascular mortality in maintenance hemodialysis patients. Am J Kidney Dis. 2007;49:417–25.
Verbeke F, Van Biesen W, Honkanen E, Wikstrom B, Jensen PB, Krzesinski JM, et al. CORD study investigators: prognostic value of aortic stiffness and calcification for cardiovascular events and mortality in dialysis patients: outcome of the Calcification Outcome in Renal Disease (CORD) study. Clin J Am Soc Nephrol. 2011;6:153–9.
Noordzij M, Cranenburg EM, Engelsman LF, Hermans MM, Boeschoten EW, Brandenburg VM, Bos WJ, et al. NECOSAD study group: progression of aortic calcification is associated with disorders of mineral metabolism and mortality in chronic dialysis patients. Nephrol Dial Transplant. 2011;26:1662–9.
Inoue T, Ogawa T, Ishida H, Ando Y, Nitta K. Aortic arch calcification evaluated on chest X-ray is a strong independent predictor of cardiovascular events in chronic hemodialysis patients. Heart Vessels. 2012;27:135–42.
Ohya M, Otani H, Kimura K, Saika Y, Fujii R, Yukawa S, et al. Vascular calcification estimated by aortic calcification area index is a significant predictive parameter of cardiovascular mortality in hemodialysis patients. Clin Exp Nephrol. 2011;15:877–83.
Komatsu M, Okazaki M, Tsuchiya K, Kawaguchi H, Nitta K. Aortic arch calcification predicts cardiovascular and all-cause mortality in maintenance hemodialysis patients. Kidney Blood Press Res. 2014;39:658–67.
Nakayama M, Ura Y, Nagata M, Okada Y, Sumida Y, Nishida K, et al. Carotid artery calcification at the initiation of hemodialysis is a risk factor for cardiovascular events in patients with end-stage renal disease: a cohort study. BMC Nephrol. 2011;12:56.
Ogata H, Fukagawa M, Hirakata H, Kaneda H, Kagimura T, Akizawa T, LANDMARK study group: design and baseline characteristics of the LANDMARK study. Clin Exp Nephrol. 2016. in press.
Ohtake T, Oka M, Ikee R, Mochida Y, Ishioka K, Moriya H, et al. Impact of lower limbs’ arterial calcification on the prevalence and severity of PAD in patients on hemodialysis. J Vasc Surg. 2011;53:676–83.
Raggi P, Bellasi A, Ferramosca E, Islam T, Muntner P, Block GA. Association of pulse wave velocity with vascular and valvular calcification in hemodialysis patients. Kidney Int. 2007;71:802–7.
Haydar AA, Covic A, Colhoun H, Rubens M, Goldsmith DJ. Coronary artery calcification and aortic pulse wave velocity in chronic kidney disease patients. Kidney Int. 2004;65:1790–4.
Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011. doi:10.1172/JCI46122.
Shanahan CM. Mechanisms of vascular calcification in CKD-evidence for premature ageing? Nat Rev Nephrol. 2013;9:661–70.
Yao Y, Bennett BJ, Wang X, Rosenfeld ME, Giachelli C, Lusis AJ, et al. Inhibition of bone morphogenic protein protects against atherosclerosis and vascular calcification. Circ Res. 2010;107:485–94.
Reynolds JL, Joannides AJ, Skepper JN, McNair R, Schurgers LJ, Proudfoot D, et al. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol. 2004;15:2857–67.
Kapustin AN, Davies JD, Reynolds JL, McNair R, Jones GT, Sidibe A, et al. Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circ Res. 2011;109:e1–e212.
Palaniswamy C, Sekhri A, Aronow WS, Kalra A, Peterson SJ. Association of warfarin use with valvular and vascular calcification: a review. Clin Cardiol. 2011;34:74–81.
Lomashvili KA, Garg P, Narisawa S, Millan JL, O’Neill WC. Upregulation of alkaline phosphatase and pyrophosphate hydrolysis: potential mechanism for uremic vascular calcifications. Kidney Int. 2008;73:1024–30.
Narisawa S, Harmey D, Yadav MC, O’Neill WC, Hoylaerts MF, Millan JL. Novel inhibitors of alkaline phosphatase suppress vascular smooth muscle cell calcification. J Bone Miner Res. 2007;22:1700–10.
O’Neill WC. Pyrophosphate, alkaline phosphatase, and vascular calcification. Circ Res. 2006;99, e2.
Kuro-o M. A phosphate-centric paradigm for pathophysiology and therapy of chronic kidney disease. Kidney Int Suppl. 2013;3:420–6.
Schinke T, Amendt C, Trindl A, Pöschke O, Müller-Esterl W, Jahnen-Dechent W. The serum protein alpha2-HS glycoprotein/fetuin inhibits apatite formation in vitro and in mineralizing calvaria cells. A possible role in mineralization and calcium homeostasis. J Biol Chem. 1996;271:20789–96.
Reynolds JL, Skepper JN, McNair R, Kasama T, Gupta K, Weissberg PL, et al. Multifunctional roles for serum protein fetuin-a in inhibition of human vascular smooth muscle cell calcification. J Am Soc Nephrol. 2005;16:2920–30.
Ketteler M, Bongartz P, Westenfeld R, Wildberger JE, Mahnken AH, Böhm R, et al. Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: a cross-sectional study. Lancet. 2003;361:827–33.
Smith ER, Cai MM, McMahon LP, Pedegogos E, Toussaint ND, Brumby C, et al. Serum fetuin-A concentration and fetuin-A-containing carciprotein particles in patients with chroic inflammatory disease and renal failure. Nephrology (Carlton). 2013;18:215–21.
Smith ER, Ford ML, Tomlinson LA, Rajkumar C, McMahon LP, Holt SG. Phosphorylated fetuin-A-containing carciprotein particles are associated with aortic stiffness and a procalcific milieu in patients with pre-dialysis CKD. Nephrol Dial Transplant. 2012;27:1957–66.
Hamano T, Matsui I, Mikami S, Tomida K, Fujii N, Imai E, et al. Fetuin-mineral complex reflects extraosseous calcification stress in CKD. J Am Soc Nephrol. 2010;21:1998–2007.
Lyemere VP, Proudfoot D, Weissberg PL, Shanahan CM. Vascular smooth muscle cell phenotypic plasticity and the regulation of vascular calcification. J Intern Med. 2006;260:192–210.
Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J Biol Chem. 2008;283:15319–27.
Li X, Giachelli CM. Sodium-dependent phosphate cotoransporters and vascular calcification. Curr Opin Nephrol Hypertens. 2007;16:325–8.
Voelkl J, Alesutan I, Leibrock CB, Quintanilla-Martinez L, Kuhn V, Feger M, et al. Spironolactone ameliorates PIT1-dependent vascular osteoinduction in klotho-hypomorphic mice. J Clin Invest. 2013;123:812–22.
Shigematsu T, Kono T, Satoh K, Yokoyama K, Yoshida T, Hosoya T, et al. Phosphate overload accelerates vascular calcium deposition in end-stage renal disease patients. Nephrol Dial Trasplant. 2003;18 Suppl 3:iii86–9.
Shroff RC, McNair R, Skepper JN, Figg N, Schurgers LJ, Deanfield J, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol. 2010;21:103–12.
Nitta K, Akiba T, Uchida K, Kawashima A, Yumura W, Kabaya T, et al. The progression of vascular calcification and serum osteoprotegerin levels in patients on long-term hemodialysis. Am J Kidney Dis. 2003;42:303–9.
Taki K, Takayama F, Tsuruta Y, Niwa T. Oxidative stress, advanced glycation end product, and coronary artery calcification in hemodialysis patients. Kidney Int. 2006;70:218–24.
McCullough PA, Soman S. Cardiovascular calcification in patients with chronic renal failure: are we on target with this risk factor? Kidney Int Suppl. 2004;90:S18–24.
McCullough PA. Effect of lipid modification on progression of coronary calcification. J Am Soc Nephrol. 2005;16 Suppl 2:S115–9.
Shantouf R, Kovesdy CP, Kim Y, Ahmadi N, Luna A, Luna C, et al. Association of serum alkaline phosphatase with coronary artery calcification in maintenance hemodialysis patients. Clin J Am Soc Nephrol. 2009;4:1106–14.
Barreto DV, Barreto Fde C, Carvalho AB, Cuppari L, Draibe SA, Dalboni MA, et al. Association of changes in bone remodeling and coronary artery calcification in hemodialysis patients: a prospective study. Am J Kidney Dis. 2008;52:1139–50.
Kirkpantur A, Altun B, Hazirolan T, Akata D, Arici M, Kirazli S, et al. Association among serum fetuin-A level, coronary artery calcification, and bone mineral densitometry in maintenance hemodialysis patients. Art Organs. 2009;33:844–54.
Moe SM, Reslerova M, Ketteler M, O’Neill K, Duan D, Koczman J, et al. Role of calcification inhibitors in the pathogenesis of vascular calcification in chronic kidney disease (CKD). Kidney Int. 2005;67:2295–304.
Mehrotra R, Westenfeld R, Christenson P, Budoff M, Ipp E, Takasu J, et al. Serum fetuin-A in nondialyzed patients with diabetic nephropathy: relationship with coronary artery calcification. Kidney Int. 2005;67:1070–7.
Kim SC, Kim HW, Oh SW, Tang HN, Kim MG, Jo SK, et al. Low iPTH can predict vascular and coronary calcifications in patients undergoing peritoneal dialysis. Nephron Clin Prac. 2011;117:c113–9.
London GM, Marty C, Marchais SJ, Guerin AP, Metivier F, de Vernejoul MC. Arterial calcifications and bone histomorphometry in end-stage renal disease. J Am Soc Nephrol. 2004;15:1943–51.
Adragao T, Herberth J, Monier-Faugere MC, Branscum AJ, Ferreira A, Frazao JM, et al. Low bone volume—a risk factor for coronary calcification in hemodialysis patients. Clin J Am Soc Nephrol. 2009;4:450–5.
Coen G, Ballanti P, Mantilla D, Manni M, Lippi B, Pierantozzi A, et al. Bone turnover, osteopenia and vascular calcifications in hemodialysis patients. Am J Nephrol. 2009;29:145–52.
Kahn AM, Chirinos JA, Litt H, Yang W, Rosas SE. FGF-23 and the progression of coronary arterial calcification in patients new to dialysis. Clin J Am Soc Nephrol. 2012;7:2017–22.
Ozkok A, Kekik C, Karahan GE, Sakaci T, Ozel A, Unsal A, et al. FGF-23 associated with the progression of coronary artery calcification in hemodialysis patients. BMC Nephrol. 2013;14:241.
Nasrallah MM, El-Shehaby AR, Salem MM, Osman NA, El Sheikh E, Sharaf El Din UA. Fibroblast growth factor-23 (FGF-23) is independently correlated to aortic calcification in haemodialysis patients. Nephrol Dial Transplant. 2010;25:2679–85.
Jean G, Bresson E, Terrat JC, Vanel T, Hurot JM, Lorriaux C, et al. Peripheral vascular calcification in long-hemodialysis patients: associated factors and survival consequences. Nephrol Dial Transplant. 2009;24:948–55.
Oka M, Ohtake T, Mochida Y, Ishioka K, Maesato K, Moriya H, et al. Correlation of coronary artery calcification with pre-hemodialysis bicarbonate levels in patients on hemodialysis. Ther Apher Dial. 2012;16:267–71.
Qunibi WY. Dyslipidemia and progression of cardiovascular calcification (CVC) in patients with end-stage renal disease (ESRD). Kidney Int Suppl. 2005;95:S43–50.
Greif M, Arnoldt T, von Ziegler F, Ruemmler J, Becker C, Wakili R, et al. Lipoprotein (a) is independently correlated with coronary artery calcification. Eur J Intern Med. 2013;24:75–9.
Ozkok A, Caliskan Y, Sakaci T, Erten G, Karahan G, Ozel A, et al. Osteoprotegerin/RANKL axis and progression of coronary artery calcification in hemodialysis patients. Clin J Am Soc Nephrol. 2012;7:965–73.
Barreto DV, Barreto FC, Carvalho AB, Cuppari L, Cendoroglo M, Draibe SA, et al. Coronary calcification in hemodialysis patients: the contribution of traditional and uremia-related risk factors. Kidney Int. 2005;67:1576–82.
Morena M, Dupuy AM, Jaussent I, Vernhet H, Gahide G, Klouche K, et al. A cut-off value of plasma osteoprotegerin level may predict the presence of coronary artery calcifications in chronic kidney disease patients. Nephrol Dial Transplant. 2009;24:3389–97.
Malluche HH, Blomquist G, Monier-Faugere MC, Cantor TL, Davenport DL. High parathyroid hormone level and osteoporosis predict progression of coronary artery calcification in patients on dialysis. J Am Soc Nephrol. 2015;26:2534–44.
Wang YN, Sun Y, Wang Y, Jia YL. Serum S100A12 and progression of coronary artery calcification over 4 years in hemodialysis patients. Am J Nephrol. 2015;42:4–13.
Zhang H, Wang LJ, Si DL, Wang C, Yang JC, Jiang P, et al. Correlation between osteocalcin-positive endothelial progenitor cells and spotty calcification in patients with coronary artery disease. Clin Exp Pharmacol Physiol. 2015;42:734–9.
Wang M, Li H, You L, Yu X, Zhang M, Zhu R, et al. Association of serum phosphate variability with coronary artery calcification among hemodialysis patients. PLoS One. 2014;9, e93360.
Jung HH, Kim SW, Han H. Inflammation, mineral metabolism and progressive coronary artery calcification in patients on haemodialysis. Nephrol Dial Transplant. 2006;21:1915–20.
Stompór T, Pasowicz M, Sullowicz W, Dembinska-Kiec A, Janda K, Wöjcik K, et al. An association between coronary artery calcification score, lipid profile, and selected markers of chronic inflammation in ESRD patients treated with peritoneal dialysis. Am J Kidney Dis. 2003;41:203–11.
Izumi M, Morita S, Nishian Y, Miyamoto T, Kasumoto H, Oue M, et al. Switching from calcium carbonate to sevelamer hydrochloride has suppressive effects on the progression of aortic calcification in hemodialysis patients: assessment using plain chest X-ray films. Ren Fail. 2008;30:952–8.
Koleganova N, Piecha G, Ritz E, Schirmacher P, Müller A, Meyer HP, et al. Arterial calcification in patients with chronic kidney disease. Nephrol Dial Transplant. 2009;24:2488–96.
Riminucci M, Collins MT, Fedarko NS, Cherman N, Corsi A, White KE, et al. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest. 2003;112:683–92.
Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51.
Lim K, Lu TS, Molostvov G, Lee C, Lam FT, Zehnder D, et al. Vascular klotho deficiency potentiates the development of human artery calcification and mediates resistance to fibroblast growth factor 23. Circulation. 2012;125:2243–55.
Shiraki-Iida T, Iida A, Nabeshima Y, Anazawa H, Nishikawa S, Noda M, et al. Improvement of multiple pathophysiological phenotypes of klotho (kl/kl) mice by adenovirus-mediated expression of the klotho gene. J Gene Med. 2000;2:233–42.
Nakano-Kurimoto R, Ikeda K, Uraoka M, Nakagawa Y, Yutaka K, Koide M, et al. Replicative senescence of vascular smooth muscle cells enhances the calcification through initiating the osteoblastic transition. Am J Physiol Heart Circ Physiol. 2009;297:H1673–84.
Zhao Y, Banerjee S, Dey N, LeJeune WS, Sarkar PS, Brobey R, et al. Klotho depletion contributes to increased inflammation in kidney of the db/db mouse model of diabetes via Re1A (Serine)536 phosphorylation. Diabetes. 2011;60:1907–16.
Scialla JJ, Lau WL, Reilly MP, Isakova T, Yang HY, Crouthamel MH, et al. Fibroblast growth factor 23 is not associated with and does not induce arterial calcification. Kidney Int. 2013;83:1159–68.
Chertow GM, Raggi P, Chasan-taber S, Bommer J, Holzer H, Burke SK. Determinants of progressive vascular calcification in hemodialysis patients. Nephrol Dial Transplant. 2004;19:1489–96.
Chertow GM, Burke SK, Raggi P. Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int. 2002;62:245–52.
Goodman WG, Goldin J, Kuizon BD, Yoon C, Gales B, Sider D, et al. Coronary artery calcification in young adults with end-stage renal disease who are undergoing dialysis. N Engl J Med. 2000;342:1478–83.
Guerin AP, London GM, Marchais SJ, Metivier F. Arterial stiffness and vascular calcifications in end-stage renal disease. Nephrol Dial Transplant. 2000;15:1014–21.
Block GA, Spiegel DM, Ehrlich J, Mehta R, Lindbergh J, Dreisbach A, et al. Effects of sevelamer and calcium on coronary artery calcification in patients new to hemodialysis. Kidney Int. 2005;68:1815–24.
Kakuta T, Tanaka R, Hyodo T, Suzuki H, Kanai G, Nagaoka M, et al. Effect of sevelamer and calcium-based phosphate binders on coronary artery calcification and accumulation of circulating advanced glycation end products in hemodialysis patients. Am J Kidney Dis. 2011;57:422–31.
Shantouf R, Ahmadi N, Flores F, Tiano J, Gopal A, Kalantar-Zadeh K, et al. Impact of phosphate binder type on coronary artery calcification in hemodialysis patients. Clin Nephrol. 2010;74:12–8.
Asmus HG, Braun J, Krause R, Brunkhorst R, Holzer H, Schulz W, et al. Two year comparison of sevelamer and calcium carbonate effects on cardiovascular calcification and bone density. Nephrol Dial Transplant. 2005;20:1653–61.
Vlassara H, Uribarri J, Cai W, Goodman S, Pyzik R, Post J, et al. Effects of sevelamer on HbA1c, inflammation, and advanced glycation end products in diabetic kidney disease. Clin J Am Soc Nephrol. 2012;7:934–42.
Guida B, Cataldi M, Riccio E, Grumetto L, Pota A, Borrelli S, et al. Plasma p-cresol lowering effect of sevelamer in peritoneal dialysis patients: evidence from a Cross-Sectional Observational Study. PLoS One. 2013;8, e73558.
Zhang Q, Li M, Lu Y, Li H, Gu Y, Hao C, et al. Meta-analysis comparing sevelamer and calcium-based phosphate binders on cardiovascular calcification in hemodialysis patients. Nephron Clin Pract. 2010;115:c259–67.
Akizawa T. Importance of serum phosphate management and feature of phosphate binder in hemodialysis patients. Therapeutic Research. 2014;35:285–91 (in Japanese).
Daugirdas JT, Finn WF, Emmett M, Chertow GM. Frequent Hemodialysis Network Trial Group. The phosphate binder equivalent dose. Semin Dial. 2011;24:41–9.
Ohtake T, Kobayashi S, Oka M, Furuya R, Iwagami M, Tsutsumi D, et al. Lanthanum carbonate delays progression of coronary artery calcification compared with calcium-based phosphate binders in patients on hemodialysis: a pilot study. J Cardiovasc Pharm Ther. 2013;18:439–46.
Toussaint ND, Lau KK, Polkinghorne KR, Kerr PG. Attenuation of aortic calcification with lanthanum carbonate versus calcium-based phosphate binders in haemodialysis: a pilot randomized controlled trial. Nephrology. 2011;16:290–8.
Kalil RS, Flanigan M, Stanford W, Haynes WG. Dissociation between progression of coronary artery calcification in hemodialysis patients: a prospective pilot study. Clin Nephrol. 2012;78:1–9.
Wada K, Wada Y. Evaluation of aortic calcification with lanthanum carbonate vs. calcium-based phosphate binders in maintenance hemodialysis patients with type 2 diabetes mellitus: an open-label randomized controlled trial. Ther Apher Dial. 2014;18:353–60.
Jamal SA, Vandermeer B, Raggi P, Mendelssohn DC, Chatterley T, Dorgan M, et al. Effect of calcium-based versus non-calcium-based phosphate binders on mortality in patients with chronic kidney disease: an updated systematic review and meta-analysis. Lancet. 2013;382:1268–77.
Komaba H, Kakuta T, Suzuki H, Hida M, Suga T, Fukagawa M. Survival advantage of lanthanum carbonate for hemodialysis patients with uncontrolled hyperphosphatemia. Nephrol Dial Transplant. 2015;30:107–14.
Tsuchida K, Nagai K, Yokota N, Minakuchi J, Kawashima S. Impact of lanthanum carbonate on prognosis of chronic hemodialysis patients: a retrospective cohort study (Kawashima Study). Ther Apher Dial. 2016;20:142–8.
Shigematsu T. Lanthanum Carbonate Research Group: three-year extension study of lanthanum carbonate therapy in Japanese hemodialysis patients. Clin Exp Nephrol. 2010;14:589–97.
Shigematsu T, Nakashima Y, Ohya M, Tatsuta K, Koreeda D, Yoshimoto W, et al. The management of hyperphosphatemia by lanthanum carbonate in chronic kidney disease patients. Int J Nephrol Renovasc Dis. 2012;5:81–9.
Shigematsu T, Ohya M, Negi S, Matsumoto AR, Nakashima YM, Iwatani Y, et al. Safety and efficacy evaluation of lanthanum carbonate in end-stage renal disease patients. Contrib Nephrol. 2015;185:42–55.
Raggi P, Chertow GM, Torres PU, Csiky B, Naso A, Nossuli K, et al. The ADVANCE study: a randomized study to evaluate the effect of cinacalcet plus low-dose vitamin D on vascular calcification in patients on hemodialysis. Nephrol Dial Transplant. 2011;26:1327–39.
Urena-Torres P, Bridges I, Christiano C, Counoyer SH, Cooper K, Farouk M, et al. Efficiency of cinacalcet with low-dose vitamin D in incidental hemodialysis subjects with secondary hyperparathyroidism. Nephrol Dial Transplant. 2013;28:1241–54.
Chertow GM, Block GA, Correa-Rotter R, Drüeke TB, Floege J, Goodman WG, et al. Effect of cinacalcet on cardiovascular disease in patients undergoing dialysis. N Engl J Med. 2012;367:2482–94.
Moe SM, Chertow GM, Parfrey PS, Kubo Y, Block GA, Correa-Rotter R, et al. Cinacalcet, fibroblast growth factor 23, and cardiovascular disease in hemodialysis: the evaluation of cinacalcet HCL therapy to lower cardiovascular events (EVOLVE) trial. Circulation. 2015;132:27–39.
Parfrey P, Drüeke TB, Block GA, Correa-Rotter R, Floege J, Herzog CA, et al. The effects of cinacalcet in older and younger patients on hemodialysis: the evaluation of cinacalcet HCL therapy to lower cardiovascular events (EVOLVE) trial. Clin J Am Soc Nephrol. 2015;10:791–9.
Lau WL, Leaf EM, Hu MC, Takeno MM, Kuro-o M, Moe OW, et al. Vitamin D receptor agonists increase klotho and osteopontin while decreasing aortic calcification in mice with chronic kidney disease fed a high phosphate diet. Kidney Int. 2012;82:1261–70.
Mathew S, Lund RJ, Chaudhary LR, Geurs T, Hruska KA. Vitamin D receptor activators can protect against vascular calcification. J Am Soc Nephrol. 2008;19:1509–19.
Price PA. Gla-containing proteins of bone. Connect Tissue Res. 1989;21:51–7.
Holden RM, Booth SL, Day AG, Clase CM, Zimmerman D, Moist L, et al. Inhibiting the progression of arterial calcification with vitamin K in hemodialysis patients (iPACK-HD) trial: rationale and study design for a randomized trial of vitamin K in patients with end stage kidney disease. Can J Kidney Health Dis. 2015;2:17.
Caluwé R, Pyfferoen L, De Boeck K, De Vriese AS. The effects of vitamin K supplementation and vitamin K antagonists on progression of vascular calcification: ongoing randomized controlled trials. Clin Kideny J. 2016;9:273–9.
Hou SH, Zhao J, Ellman CF, Hu J, Griffin Z, Spiegel DM, et al. Calcium and phosphate fluxes during hemodialysis with low calcium dialysate. Am J Kidney Dis. 1991;18:217–24.
Ok E, Asci G, Bayraktaroglu S, Toz H, Ozkahya M, Yilmaz M, et al. Reduction of dialysate calcium level reduces progression of coronary artery calcification and improves low bone turnover in patients on hemodialysis. J Am Soc Nephrol. 2016;27:2475–86.
Nitta K, Akiba T, Suzuki K, Uchida K, Watanabe R, Majima K, et al. Effects of cyclic intermittent etidronate therapy on coronary artery calcification in patients receiving long-term hemodialysis. Am J Kideny Dis. 2004;44:680–8.
Hashiba H, Aizawa S, Tamura K, Shigematsu T, Kogo H. Inhibitory effects of etidronate on the progression of vascular calcification in hemodialysis patients. Ther Apher Dial. 2004;8:241–7.
Toussaint ND, Lau KK, Strauss BJ, Polkinghorne KR, Kerr PG. Effect of alendronate on vascular calcification n CKD stages 3 and 4: a pilot randomized controlled trial. Am J Kidney Dis. 2010;56:57–68.
Elmarish S, Delaney JA, Bluemke DA, Budoff MJ, O’Brien KD, Fuster V, et al. Associations of LV hypertrophy with prevalent and incident valve calcification: multi-ethnic study of atherosclerosis. JACC Cardiovasc Imaging. 2012;5:781–8.
Wheeler DC, Becker GJ. Summary of KDIGO guideline. What do we really know about management of blood pressure in patients with chronic kidney disease? Kidney Int. 2013;83:377–83.
Fukagawa M, Yokoyama K, Koiwa F, Taniguchi M, Shoji T, Kazama JJ, et al. Clinical practice guideline for the management of chronic kidney disease-mineral and bone disorder. Ther Apher Dial. 2013;17:247–88.
Mathews SJ, de Las FL, Podaralla P, Cabellon A, Zheng S, Bierhals A, et al. Effects of sodium thiosulfate on vascular calcification in end-stage renal disease: a pilot study of feasibility, safety and efficacy. Am J Nephrol. 2011;33:131–8.
Nigwekar SU, Brunelli SM, Meade D, Wang W, Hymes J, Lacson E. Sodium thiosulfate therapy for calcific uremic arteriolopathy. Clin J Am Soc Nephrol. 2013;8:1162–70.
Pasch A, Schffner T, Huynh-Do U, Frey BM, Frey FJ, Farese S. Sodium thiosulfate prevents vascular calcification in uremic rats. Kidney Int. 2008;74:1444–53.
Schlieper G, Brandenburg V, Ketteler M, Floege J. Sodium thiosulfate in the treatment of calcific uremic arteriolopathy. Nat Rev Nephrol. 2009;5:539–43.
Adirekkiat S, Sumethkul V, Ingsathit A, Domrongkitchaiporn S, Phakdeekitcharoen B, Kantachuvesiri S, et al. Sodium thiosulfate delays the progression of coronary artery calcification in haemodialysis patients. Nephrol Dial Transplant. 2010;25:1923–9.
Acknowledgements
Not applicable.
Funding
Not applicable.
Availability of data and materials
Not applicable.
Authors’ contributions
TO wrote the whole manuscript, and SK read and advised about the content of the manuscript to the final version. Both authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
All authors gave consent for publication.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ohtake, T., Kobayashi, S. Impact of vascular calcification on cardiovascular mortality in hemodialysis patients: clinical significance, mechanisms and possible strategies for treatment. Ren Replace Ther 3, 13 (2017). https://doi.org/10.1186/s41100-017-0094-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41100-017-0094-y