
Abstract
Abnormal wound healing is likely to induce scar formation, leading to dysfunction, deformity, and psychological trauma in burn patients. Despite the advancement of medical care treatment, scar contracture in burn patients remains a challenge. Myofibroblasts play a key role in scar contracture. It has been demonstrated that myofibroblasts, as well as inflammatory cells, fibroblasts, endothelial cells, and epithelial cells, secrete transforming growth factor-β1 (TGF-β1) and other cytokines, which can promote persistent myofibroblast activation via a positive regulation loop. In addition to the cellular contribution, the microenvironments, including the mechanical tension and integrin family, are also involved in scar contracture. Most recently, eukaryotic initiation factor 6 (eIF6), an upstream regulator of TGF-β1, has been demonstrated to be involved in myofibroblast differentiation and contraction in both in vitro fibroblast-populated collagen lattice (FPCL) and in vivo external mechanical stretch models. Moreover, the data showed that P311 could induce the transdifferentiation of epidermal stem cells to myofibroblasts by upregulating TGF-β1 expression, which mediated myofibroblast contraction. In this review, we briefly described the most current progress on the biological function of myofibroblasts in scar contracture and subsequently summarized the molecular events that initiated contracture. This would help us better understand the molecular basis of scar contracture as well as to find a comprehensive strategy for preventing/managing scar contracture.
Similar content being viewed by others


Background
It is commonly accepted that scars are a pathologic wound healing response to burn injuries, traumatic injuries, and surgeries. Hypertrophic scars and keloids, which only occur in humans, present with exuberant scar formation [1]. Although these disorders do not pose a health risk, the scar contracture resulting in dysfunction and deformity remains a challenge in the clinic [2, 3]. The management of the scar contracture, such as surgical intervention, drugs, silicone materials, pressure therapy, splinting, lasers, and radiation, is used to control scar formation and contracture, but it is still far from achieving our expected outcomes [4]. Schneider found that 620 of the 1865 analyzed adult burn patients developed at least one joint contracture, which meant 33% of patients had dysfunction in their joints after burn injuries [5].
Wound healing proceeds through three overlapping stages. The inflammatory stage is triggered by injury, wherein platelets, neutrophils, and macrophages release inflammatory mediators and cytokines that participate in the recruitment of inflammatory cells, fibroblasts, endothelial cells, and epithelial cells. The proliferative stage involves fibroblast activation, myofibroblast differentiation, and extracellular matrix (ECM) deposition. In this phase, the myofibroblasts have acquired contractile properties that can contract the wound and promote re-epithelialization. The third healing stage is matrix remodeling, including scar tissue remodeling. During this stage, the persistent activation of myofibroblasts, imbalance of deposition and degradation of ECM, and poor arrangement of newly formed fibers can lead to scar formation.
Myofibroblasts, a type of cell differentiated from quiescent fibroblasts and other cells, have been demonstrated to play an essential role in the induction and maintenance of scar contracture. In normal acute wound healing, the myofibroblasts are temporally limited and cleared by apoptosis in the third healing phase when the tissues are repaired. However, in hypertrophic scars and keloids, these myofibroblasts persist at a high number for long periods and promote the synthesis of α-smooth muscle actin (α-SMA), transforming growth factor-β1 (TGF-β1), and other growth factors, and they have sustained contractile ability via the TGF-β1 positive loop [6].
Review
The origins and characteristics of myofibroblasts
In the inflammatory stage, fibrocytes and fibroblasts are believed to be activated in response to inflammatory factors; they then migrate to the location of injury based on a chemoattractant gradient and differentiate into myofibroblasts. In general, most myofibroblasts are derived from fibroblast differentiation around the local wound area [7]. In addition, other myofibroblasts may originate from pericytes [8], smooth muscle cells from the vasculature [9], fibrocytes from bone marrow-derived peripheral blood [10], epithelial cells through the epithelial-mesenchymal transition (EMT) [11], epidermal stem cells [12, 13], local mesenchymal stem cells, and bone marrow-derived mesenchymal stem cells [14].
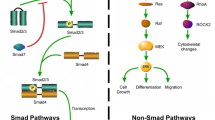
The myofibroblast cell is a phenotypically intermediate cell type between fibroblasts and smooth muscle cells. The myofibroblasts exhibit the characteristics of smooth muscle cells, containing high-contractile stress fibers. The stress fibers consist of α-SMA protein, which can be used to differentiate between myofibroblasts and fibroblasts in tissues. However, it remains difficult and complicated to discriminate between myofibroblasts and other contractile cells, such as smooth muscle cells, pericytes, and myoepithelial cells, even if the smooth muscle cells express smooth muscle myosin heavy chain, h-caldesmon (H-CAD), smoothelin, and the muscle intermediate filament protein, desmin, which are absent from myofibroblasts [15]. There are a lot of cytokines and mechanical microenvironment factors that contribute to scar contracture (Fig. 1).

The cytokines and mechanical environment contribute to myofibroblast contraction: The inflammatory factors and growth factors such as TGF-β1, CTGF, IGF, PDGF, VEGF, IL-6, IL-8, Fizz1, and YAP1 could upregulate the expression of TGF-β1, α-SMA, collagen I, collagen III, and fibronectin via a positive feedback loop. The exogenous mechanical force can also promote the expression of α-SMA via FAK, RAC, NADPH oxidase, MAPK/p38, and Rho signaling pathways, enhancing the contractile force. bFGF, EGF, IFN-γ, and IL-10 can inhibit the myofibroblasts differentiation, thereby decreasing the contraction. P311 could upregulate the TGF-β1 expression. In contrast, eIF6 inhibits the TGF-β1 expression as an upstream regulator
The cytokine contribution to scar contracture
Transforming growth factor-β family
It has been demonstrated that TGF-β1 is one of the most important factors controlling myofibroblast differentiation and function. TGF-β is found in all tissues, and it consists of three isoforms, β1, β2, and β3. Binding of active TGF-β1 to the TGF-β receptor type II leads to the phosphorylation and recruitment of TGF-β receptor type I. The heteromeric receptor complex induces the phosphorylation of Smad2/3, followed by association with Smad4. Subsequently, the Smad complex translocates into the nucleus to enhance gene transcription via cooperation with DNA transcription factors. Additionally, it induces overproduction of α-SMA, collagen I, collagen III, fibronectin (FN), and other cytokines [16]. TGF-β1 can also reduce matrix metalloproteinases (MMPs) activity via decreasing proteases, such as tissue inhibitor of metalloproteinases (TIMPs) I and II [17]. The TGF-β/Smads signaling pathway is a positive autocrine loop in both hypertrophic scar and keloid formation, which then increases the stress fiber stabilization and stiffness of the microenvironment.
Importantly, TGF-β1 is an inducer of myofibroblast differentiation, which is considered a potential therapeutic target for hypertrophic scars and keloids. It has been reported that peroxisome proliferator-activated receptor-γ (PPARγ) ligands, 15d-PGJ2 and GW7845, could inhibit the expression and phosphorylation of TGF-β1/Smads [18]. Either disruption or neutralization of TGF-β/Smads signaling by botulinum toxin type A, tetrandrine, baicalein, loureirin B, or the Uighur medicine ASMq can decrease the myofibroblast properties [19]. In addition, TGF-β1 could also promote myofibroblast differentiation independent of Smads signaling and instead act via the wnt, p38, and PI3K/PKA2 signaling pathways [20]. P311, identified by suppressive subtraction hybridization as potentially involved in smooth muscle (SM) myogenesis, was highly expressed in hypertrophic scars and could induce a TGF-β1-independent, nonfibrogenic myofibroblast phenotype [21, 22]. Furthermore, in the renal fibrosis model, it was found that overexpression of P311 was concurrent with the expression of α-SMA and TGF-β1 via the TGF-β1/Smad signaling pathway [23]. Eukaryotic initiation factor 6 (eIF6), acting as a key binding protein of P311 [24], has recently been demonstrated by our team as a novel upstream regulator to inhibit myofibroblast differentiation at the TGF-β1 transcription level via H2A.Z occupancy and Sp1 recruitment. Additionally, there is downregulation of α-SMA and collagen I expression [25]. In addition, our next study demonstrated that eIF6-mediated TGF-β1 can also be regulated by external mechanical stretch [26].
Positive growth factors and cytokines
In addition, many other growth factors show positive roles in myofibroblast differentiation, such as connective tissue growth factor (CTGF), platelet-derived growth factor (PDGF), insulin growth factor (IGF), and vascular endothelial growth factor (VEGF) [27, 28]. CTGF could synergize the action of TGF-β, promoting ECM production and scar contracture. Inhibition of the expression of CTGF can reduce the formation of hypertrophic scars. Likewise, PDGF is released into the wound and induces myofibroblast activation directly or in synergy with TGF-β1. PDGF stimulates fibroblast proliferation and regulates collagen synthesis via extracellular signaling-regulated kinase (ERK) and the PI3K/JNK signaling pathway. Blocking of PDGF receptors α and β was shown to inhibit myofibroblast formation. IGF acts as a mitogenic factor to enhance the expression of collagens I and III while also reducing the release of collagenase. VEGF could induce the expression of collagen I and promote scar formation [27].
Moreover, interleukin-6 (IL-6) and interleukin-8 (IL-8) increase α-SMA transcription in fibroblast cultures [29, 30]. Similarly, nerve growth factor enhanced α-SMA expression in fibroblasts [31]. Fizz1 induced the expression of α-SMA [32]. YAP1 contributes to the maintenance of a synthetic and contractile phenotype in fibrosis [33]. Agonists of myofibroblast contraction, such as angiotensin-II, endothelin-1, and thrombin, can upregulate the expression of α-SMA [34,35,36]. As a result, these inflammatory cytokines play roles in myofibroblast differentiation and scar contracture [37].
Negative growth factors and cytokines
In contrast, some negative factors act against myofibroblast differentiation. Basic fibroblast growth factor (bFGF, FGF2), epidermal growth factor (EGF), interferon-γ (IFN-γ), interleukin-10 (IL-10), prostaglandin E2 (PGE2), eIF6, and TGF-β3 have been shown to suppress the expression of α-SMA and ECM synthesis [25, 38, 39]. bFGF has been shown to suppress myofibroblast function and α-SMA expression by antagonizing TGF-β1. EGF could negatively regulate the role of TGF-β1 in inducing myofibroblast contraction through attenuating autologous release of TGF-β1. IFN-γ and IL-1β induced apoptosis in myofibroblasts and antagonized TGF-β1 regulation and production [40, 41]. Additionally, TGF-β3 exerted suppressive effects on myofibroblasts in a 3-D repair model [42].
The mechanical microenvironment contribution to scar contracture
Mechanical tension
Hypertrophic scars frequently occur at particular sites, including the anterior chest wall, auricle, scapula, and suprapubic regions, which are frequently subjected to the high stretch tension from the natural daily movements of the body [43]. The activity of myofibroblasts depends on the mechanical microenvironment. The stress fibers, fibronectin, and smooth muscle actin appear earlier in the inflammatory stage, which can increase the mechanical tension by association with the ECM. With mechano-sensitive ion channels in the plasma membrane, integrin-mediated stress perception and geometrical changes of myofibroblasts can sense stress [44]. Fibrosis tissue exerts greater forces of 20–100 kPa with a collagen-dense tendon [45]. In contractile wound granulation tissue and myofibroblasts cultured on elastic substrates, the threshold stiffness for the expression of α-SMA in stress fibers is approximately 20 kPa [46]. In liver fibrosis, hepatic stellate cells can be activated with 15 kPa of pressure and then differentiate into α-SMA-positive myofibroblasts [47].
α-SMA has been demonstrated as a mechano-sensitive protein that induces a rapid mechanism to control the myofibroblast contractile function. There is positive regulation between stress and α-SMA expression level. The exogenous mechanical force, when applied through integrins, activates the Rho or mitogen-activated protein kinase (MAPK)/p38 signaling pathway, which then enhances the activation of serum response factor (SRF) and increases α-SMA transcription and incorporation into actin filaments. The persistent upregulation of α-SMA increases the intracellular tension and induces a higher force compared to the cytoplasmic actin stress fiber, which stimulates ECM contraction [48]. A decrease in the intracellular stress will make the myofibroblasts insensitive to external mechanical factors via interfering with α-SMA, inhibiting the Rho/Rho-associated kinase pathway effect on myosin activity, interfering with mega karyoblastic leukemia factor 1 (MKL1) that is linked with mechanical stress, and interfering with YAP/TAZ transcription factors that mediate mechano-responses [49,50,51,52].
Integrin family
Integrin is an essential mechano transducer that is connected with stress fibers in cells and the ECM surrounding the cells [53]. These signal mediators are cell surface receptors that consist of two isoforms, α and β subunits. Integrin is involved in the activation of latent TGF-β1 and production of collagen, α-SMA and connective tissue growth factor (CTGF) via reactive FAK, RAC, and NADPH oxidase as well as an oxygen species (ROS)-dependent mechanism [54]. Fibroblasts with integrin β1 knockout are less able to adhere to and contract the ECM [55]. Integrins α1β1, α2β1, α3β1, αvβ5, α5β1, αvβ3, αvβ6, αvβ8, and α11β1 were demonstrated to partially participate in fibroblast proliferation, collagen contraction, and myofibroblast differentiation [56]. Deletion of integrin α3β1 decreased the accumulation of myofibroblasts and collagen I, which decreased the fibrosis [57]. Moreover, some other integrins, such as integrins αvβ5, αvβ3, and α8β1, were shown to bind to LAP-TGF-β1 and are involved in TGF-β1 activation [58, 59].
Conclusions
After injury, the quiescent fibroblasts and other original cells are activated in response to inflammatory signals, such as TGF-β1. Following the TGF-β/Smad signaling cascade, TGF-β1 enhances gene transcription, as demonstrated by the upregulation of α-SMA, collagen I/III, and other fibrotic genes. Meanwhile, the expression of TGF-β1 is also increased by a positive feedback loop. Furthermore, eIF6 and P311 were involved in myofibroblast differentiation and contraction via regulating the TGF-β1 expression. This indicated that eIF6 and P311 may be new potential target genes for treating scar contracture. In addition, there are other cytokines, such as growth factors and inflammatory relative factors, which can up/downregulate myofibroblast contracture. Continuing ECM alignment creates larger surfaces for adhesion formation, which connects TGF-β-LAP-LTBP and integrins. The larger adhesions permit the development of stronger stress fibers and generation of a higher contractile force.
In the wound repair phase, contraction can close the original wound and reduce the surface area. However, the continuation of contraction after wound healing can have high clinical morbidity of the joint contractures, functional loss, delayed return to work, and poor cosmetic results. The molecular basis/pathogenesis of scar contracture is complicated and includes cellular factors and environmental contributions. No single treatment method has been demonstrated to be effective [60]. To find the appropriate treatment for scar contractures, we prefer to consider a comprehensive strategy, including cytokines and environmental aspects, and then translate the basic discoveries into potential therapies.
Abbreviations
- bFGF:
-
Basic fibroblast growth factor
- CTGF:
-
Connective tissue growth factor
- EGF:
-
Epidermal growth factor
- eIF6:
-
Eukaryotic initiation factor 6
- EMT:
-
Epithelial-mesenchymal transition
- ERK:
-
Extracellular signaling-regulated kinase
- FN:
-
fibronectin
- FPCL:
-
Fibroblast-populated collagen lattice
- H-CAD:
-
h-Caldesmon
- IGF:
-
Insulin growth factor
- MAPK:
-
Mitogen-activated protein kinase
- MKL:
-
Megakaryoblastic leukemia factor
- MMP:
-
Matrix metalloproteinase
- PDGF:
-
Platelet-derived growth factor
- PG:
-
Prostaglandin
- PPARγ:
-
Peroxisome proliferator-activated receptor-γ
- SRF:
-
Serum response factor
- TGF-β1:
-
Transforming growth factor-β1
- TIMPs:
-
Tissue inhibitor of metalloproteinases
- VEGF:
-
Vascular endothelial growth factor
- α-SMA:
-
α-Smooth muscle actin
References
Oosterwijk AM, Mouton LJ, Schouten H, Disseldorp LM, van der Schans CP, Nieuwenhuis MK. Prevalence of scar contractures after burn: A systematic review. Burns. 2017;43(1):41–49.
Martin P. Wound healing—aiming for perfect skin regeneration. Science. 1997;276(5309):75–81.
Heng MC. Wound healing in adult skin: aiming for perfect regeneration. Int J Dermatol. 2011;50(9):1058–66.
Berman B, Viera MH, Amini S, Huo R, Jones IS. Prevention and management of hypertrophic scars and keloids after burns in children. J Craniofac Surg. 2008;19(4):989–1006.
Goverman J, Mathews K, Goldstein R, Holavanahalli R, Kowalske K, Esselman P, et al. Adult Contractures in Burn Injury: A Burn Model System National Database Study. J Burn Care Res. 2017;38(1):e328–e336.
Bochaton-Piallat ML, Gabbiani G, Hinz B. The myofibroblast in wound healing and fibrosis: answered and unanswered questions. F1000Res. 2016;5:1–8.
Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol. 2003;200(4):500–3.
Gokcinar-Yagci B, Uckan-Cetinkaya D, Celebi-Saltik B. Pericytes: properties, functions and applications in tissue engineering. Stem Cell Rev. 2015;11(4):549–59.
Hinz B. Myofibroblasts. Exp Eye Res. 2016;142:56–70.
Sun YB, Qu X, Caruana G, Li J. The origin of renal fibroblasts/myofibroblasts and the signals that trigger fibrosis. Differentiation. 2016;92(3):102–7.
Yan C, Grimm WA, Garner WL, Qin L, Travis T, Tan N, et al. Epithelial to mesenchymal transition in human skin wound healing is induced by tumor necrosis factor-alpha through bone morphogenic protein-2. Am J Pathol. 2010;176(5):2247–58.
Li H, Yao Z, He W, Gao H, Bai Y, Yang S, et al. P311 induces the transdifferentiation of epidermal stem cells to myofibroblast-like cells by stimulating transforming growth factor beta1 expression. Stem Cell Res Ther. 2016;7(1):175.
Yao Z, Li H, He W, Yang S, Zhang X, Zhan R, et al. P311 accelerates skin wound reepithelialization by promoting epidermal stem cell migration through Rho A and Rac1 activation. Stem Cells Dev. 2017;26(6):451–460.
Markeson D, Pleat JM, Sharpe JR, Harris AL, Seifalian AM, Watt SM. Scarring, stem cells, scaffolds and skin repair. J Tissue Eng Regen Med. 2015;9(6):649–68.
Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. 2007;127(3):526–37.
Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12(6):325–38.
Robert S, Gicquel T, Bodin A, Lagente V, Boichot E. Characterization of the MMP/TIMP imbalance and collagen production induced by IL-1beta or TNF-alpha release from human hepatic stellate cells. PLoS One. 2016;11(4):e0153118.
Zhang GY, Cheng T, Zheng MH, Yi CG, Pan H, Li ZJ, et al. Activation of peroxisome proliferator-activated receptor-gamma inhibits transforming growth factor-beta1 induction of connective tissue growth factor and extracellular matrix in hypertrophic scar fibroblasts in vitro. Arch Dermatol Res. 2009;301(7):515–22.
Chen M, Yan T, Ma K, Lai L, Liu C, Liang L, et al. Botulinum toxin type A inhibits alpha-smooth muscle actin and myosin II expression in fibroblasts derived from scar contracture. Ann Plast Surg. 2016;77(3):e46–9.
Bowley E, O’Gorman DB, Gan BS. Beta-catenin signaling in fibroproliferative disease. J Surg Res. 2007;138(1):141–50.
Pan D, Zhe X, Jakkaraju S, Taylor GA, Schuger L. P311 induces a TGF-beta1-independent, nonfibrogenic myofibroblast phenotype. J Clin Invest. 2002;110(9):1349–58.
Tan J, Peng X, Luo G, Ma B, Cao C, He W, et al. Investigating the role of P311 in the hypertrophic scar. PLoS One. 2010;5(4):e9995.
Yao Z, Yang S, He W, Li L, Xu R, Zhang X, et al. P311 promotes renal fibrosis via TGFbeta1/Smad signaling. Sci Rep. 2015;5:17032.
Peng X, Yuan S, Tan J, Ma B, Bian X, Xu C, et al. Identification of ITGB4BP as a new interaction protein of P311. Life Sci. 2012;90(15–16):585–90.
Yang SS, Tan JL, Liu DS, Loreni F, Peng X, Yang QQ, et al. Eukaryotic initiation factor 6 modulates myofibroblast differentiation at transforming growth factor-beta1 transcription level via H2A.Z occupancy and Sp1 recruitment. J Cell Sci. 2015;128(21):3977–89.
Shu Q, Tan J, Ulrike VD, Zhang X, Yang J, Yang S, et al. Involvement of eIF6 in external mechanical stretch-mediated murine dermal fibroblast function via TGF-beta1 pathway. Sci Rep. 2016;6:36075.
Lian N, Li T. Growth factor pathways in hypertrophic scars: molecular pathogenesis and therapeutic implications. Biomed Pharmacother. 2016;84:42–50.
Mattyasovszky SG, Wollstadter J, Martin A, Ritz U, Baranowski A, Ossendorf C, et al. Inhibition of contractile function in human joint capsule myofibroblasts by targeting the TGF-beta1 and PDGF pathways. PLoS One. 2016;11(1):e0145948.
Shintani Y, Fujiwara A, Kimura T, Kawamura T, Funaki S, Minami M, et al. IL-6 secreted from cancer-associated fibroblasts mediates chemoresistance in NSCLC by increasing epithelial-mesenchymal transition signaling. J Thorac Oncol. 2016;11(9):1482–92.
Clement S, Pascarella S, Conzelmann S, Gonelle-Gispert C, Guilloux K, Negro F. The hepatitis C virus core protein indirectly induces alpha-smooth muscle actin expression in hepatic stellate cells via interleukin-8. J Hepatol. 2010;52(5):635–43.
Yorifuji M, Sawaji Y, Endo K, Kosaka T, Yamamoto K. Limited efficacy of COX-2 inhibitors on nerve growth factor and metalloproteinases expressions in human synovial fibroblasts. J Orthop Sci. 2016;21(3):381–8.
Martins V, De Los Santos FG, Wu Z, Capelozzi V, Phan SH, Liu T. FIZZ1-induced myofibroblast transdifferentiation from adipocytes and its potential role in dermal fibrosis and lipoatrophy. Am J Pathol. 2015;185(10):2768–76.
Piersma B, de Rond S, Werker PM, Boo S, Hinz B, van Beuge MM, et al. YAP1 is a driver of myofibroblast differentiation in normal and diseased fibroblasts. Am J Pathol. 2015;185(12):3326–37.
Yang X, Wang Y, Yan S, Sun L, Yang G, Li Y, et al. Effect of testosterone on the proliferation and collagen synthesis of cardiac fibroblasts induced by angiotensin II in neonatal rat. Bioengineered. 201;8(1):14–20.
Kiya K, Kubo T, Kawai K, Matsuzaki S, Muramatsu R, Maeda D, et al. Endothelial cell-derived endothelin-1 is involved in abnormal scar formation by dermal fibroblasts through RhoA/Rho-kinase pathway. Exp Dermatol. 2016. doi:10.1111/exd.13264.
Yang WH, Deng YT, Hsieh YP, Wu KJ, Kuo MY. Thrombin activates latent TGFbeta1 via integrin alphavbeta1 in gingival fibroblasts. J Dent Res. 2016;95(8):939–45.
Bianchi E, Taurone S, Bardella L, Signore A, Pompili E, Sessa V, et al. Involvement of pro-inflammatory cytokines and growth factors in the pathogenesis of Dupuytren’s contracture: a novel target for a possible future therapeutic strategy? Clin Sci (Lond). 2015;129(8):711–20.
Park JS, Kim JY, Cho JY, Kang JS, Yu YH. Epidermal growth factor (EGF) antagonizes transforming growth factor (TGF)-beta1-induced collagen lattice contraction by human skin fibroblasts. Biol Pharm Bull. 2000;23(12):1517–20.
Shi J, Li J, Guan H, Cai W, Bai X, Fang X, et al. Anti-fibrotic actions of interleukin-10 against hypertrophic scarring by activation of PI3K/AKT and STAT3 signaling pathways in scar-forming fibroblasts. PLoS One. 2014;9(5):e98228.
Poosti F, Bansal R, Yazdani S, Prakash J, Beljaars L, van den Born J, et al. Interferon gamma peptidomimetic targeted to interstitial myofibroblasts attenuates renal fibrosis after unilateral ureteral obstruction in mice. Oncotarget. 2016;7(34):54240–52.
Bronnum H, Eskildsen T, Andersen DC, Schneider M, Sheikh SP. IL-1beta suppresses TGF-beta-mediated myofibroblast differentiation in cardiac fibroblasts. Growth Factors. 2013;31(3):81–9.
Chang Z, Kishimoto Y, Hasan A, Welham NV. TGF-beta3 modulates the inflammatory environment and reduces scar formation following vocal fold mucosal injury in rats. Dis Model Mech. 2014;7(1):83–91.
Ogawa R, Mitsuhashi K, Hyakusoku H, Miyashita T. Postoperative electron-beam irradiation therapy for keloids and hypertrophic scars: retrospective study of 147 cases followed for more than 18 months. Plast Reconstr Surg. 2003;111(2):547–53. discussion 554–5.
Van De Water L, Varney S, Tomasek JJ. Mechanoregulation of the myofibroblast in wound contraction, scarring, and fibrosis: opportunities for new therapeutic intervention. Adv Wound Care (New Rochelle). 2013;2(4):122–41.
Hinz B. Tissue stiffness, latent TGF-beta1 activation, and mechanical signal transduction: implications for the pathogenesis and treatment of fibrosis. Curr Rheumatol Rep. 2009;11(2):120–6.
Goffin JM, Pittet P, Csucs G, Lussi JW, Meister JJ, Hinz B. Focal adhesion size controls tension-dependent recruitment of alpha-smooth muscle actin to stress fibers. J Cell Biol. 2006;172(2):259–68.
Wells RG. The role of matrix stiffness in hepatic stellate cell activation and liver fibrosis. J Clin Gastroenterol. 2005;39(4 Suppl 2):S158–61.
Wang J, Zohar R, McCulloch CA. Multiple roles of alpha-smooth muscle actin in mechanotransduction. Exp Cell Res. 2006;312(3):205–14.
Gates DH, Lee JS, Hultman CS, Cairns BA. Inhibition of rho-kinase impairs fibroblast stress fiber formation, confluence, and contractility in vitro. J Burn Care Res. 2007;28(3):507–13.
Sandbo N, Lau A, Kach J, Ngam C, Yau D, Dulin NO. Delayed stress fiber formation mediates pulmonary myofibroblast differentiation in response to TGF-beta. Am J Physiol Lung Cell Mol Physiol. 2011;301(5):L656–66.
Abe M, Sogabe Y, Syuto T, Yokoyama Y, Ishikawa O. Evidence that PI3K, Rac, Rho, and Rho kinase are involved in basic fibroblast growth factor-stimulated fibroblast-collagen matrix contraction. J Cell Biochem. 2007;102(5):1290–9.
Dai X, She P, Chi F, Feng Y, Liu H, Jin D, et al. Phosphorylation of angiomotin by Lats1/2 kinases inhibits F-actin binding, cell migration, and angiogenesis. J Biol Chem. 2013;288(47):34041–51.
DiPersio CM, Zheng R, Kenney J, Van De Water L. Integrin-mediated regulation of epidermal wound functions. Cell Tissue Res. 2016;365(3):467–82.
Chen R, Zhang Z, Xue Z, Wang L, Fu M, Lu Y, et al. Focal adhesion kinase (FAK) siRNA inhibits human hypertrophic scar by suppressing integrin alpha, TGF-beta and alpha-SMA. Cell Biol Int. 2014;38(7):803–8.
Leask A. Integrin beta1: a mechanosignaling sensor essential for connective tissue deposition by fibroblasts. Adv Wound Care (New Rochelle). 2013;2(4):160–6.
Hu X, Li N, Tao K, Fang X, Liu J, Wang Y, et al. Effects of integrin alphanubeta3 on differentiation and collagen synthesis induced by connective tissue growth factor in human hypertrophic scar fibroblasts. Int J Mol Med. 2014;34(5):1323–34.
Kim KK, Wei Y, Szekeres C, Kugler MC, Wolters PJ, Hill ML, et al. Epithelial cell alpha3beta1 integrin links beta-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J Clin Invest. 2009;119(1):213–24.
Sarrazy V, Koehler A, Chow ML, Zimina E, Li CX, Kato H, et al. Integrins alphavbeta5 and alphavbeta3 promote latent TGF-beta1 activation by human cardiac fibroblast contraction. Cardiovasc Res. 2014;102(3):407–17.
Bouzeghrane F, Mercure C, Reudelhuber TL, Thibault G. Alpha8beta1 integrin is upregulated in myofibroblasts of fibrotic and scarring myocardium. J Mol Cell Cardiol. 2004;36(3):343–53.
Junker JP, Philip J, Kiwanuka E, Hackl F, Caterson EJ, Eriksson E. Assessing quality of healing in skin: review of available methods and devices. Wound Repair Regen. 2014;22 Suppl 1:2–10.
Acknowledgements
None.
Funding
This work was supported by NSFC (81401603).
Availability of data and materials
Not applicable.
Authors’ contributions
JT was a major contributor in writing the manuscript and revising it critically for important intellectual content. JW supervised the writing of the manuscript and revised it critically for important intellectual content. Both authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Tan, J., Wu, J. Current progress in understanding the molecular pathogenesis of burn scar contracture. Burn Trauma 5, 14 (2017). https://doi.org/10.1186/s41038-017-0080-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41038-017-0080-1