
Abstract
Background
Congenital heart disease (CHD) is a well-known co-occurring condition in Down syndrome (DS). We aimed to review the literature to evaluate the current evidence to address key questions.
Methods
A series of key questions were formulated a priori to inform the search strategy and review process. These addressed the topics of prevalence, type of CHD, severity, and screening. Using the National Library of Medicine database, PubMed, detailed literature searches were performed. The quality of available evidence was then evaluated, the existing literature was summarized, and knowledge gaps were identified.
Results
Fifty-six relevant original articles were identified which addressed at least one key question. Study details, including: research design, internal validity, external validity, and relevant results are presented. The total prevalence of CHD reported in DS ranged from 20 to 57.9%. In later decades, the prevalence remained constant at 40—55%. The types and classification of CHD varied considerably between studies. Some studies indicate a trend towards a milder phenotype, but this was not consistent. Over time, some studies observed an improved prognosis for CHD in DS. Studies investigating screening for CHD by physical examination, chest X-ray, and electrocardiogram report sensitivities of 71–95%.
Conclusion
To further improve knowledge on CHD in DS, we suggest that future studies cover a wide range of nations and regions, with a longitudinal design, and account for potential confounding factors.
Similar content being viewed by others


Introduction
Down syndrome (DS) is present in one in 800 infants born in the United States, making it the most common chromosomal condition associated with a unique medical and developmental profile [1, 2]. Congenital heart disease (CHD) is one of the co-occurring medical diagnoses associated with DS [3, 4]. Among patients with DS, the presence of CHD is a known contributor to morbidity and mortality [5].
Although the association of CHD with DS has been known for decades, much has changed over time in terms of available diagnostics, medical care, and treatments. We conducted this project to present an updated literature review on CHD in DS. The overarching goals of this review were to: 1) Formulate key questions a priori and identify which original articles address these key questions. 2) Search PubMed to identify original research articles that address the cardiac phenotype of individuals with DS. 3) Assess these articles using United States Preventative Services Task Force (USPTF) methods [6]. 4) Summarize the published literature and identify gaps in evidence.
Methods
Key questions
In accordance with USPSTF practice we formulated a series of key questions as outlined in a prior review [7]. By consensus, the following key questions were formulated by the authors:
-
1.
What is the total prevalence of congenital heart disease in DS? And, has the prevalence of congenital heart disease in DS changed over time?
-
2.
What is the type of congenital heart disease in DS? And, has the type of congenital heart disease in DS changed over time?
-
3.
What is the severity of congenital heart disease in DS in terms of treatment and prognosis? And, has the severity of congenital heart disease in DS changed over time?
-
4.
What screening for congenital heart disease in DS is performed in studies? And, has the screening for congenital heart disease in DS changed in the published literature over time?
PubMed literature search
Literature searches were conducted in August – September 2020 using the National Library of Medicine (NLM) biomedical literature database PubMed (MEDLINE) (NCBI 1946–2020) to identify original research manuscripts addressing our prioritized topics. We used the Medical Subject Headings (MeSH) (the NLM controlled vocabulary thesaurus for indexing) to capture related entry terminology in our searches. For example, the MeSH term “Down syndrome” included the search entry terms: Downs syndrome, Down’s syndrome, Mongolism, Trisomy 21, Partial Trisomy 21. The MeSH term “Down syndrome” was combined using the Boolean operator ‘AND’ with the MeSH term “congenital heart defect” which included the search entry terms of specific cardiac malformations, to capture the unfiltered literature. Then, the limiters “Human”, “English” were applied to narrow the scope of the search to filtered literature. We did not use subject age as a limiter. We also did not exclude articles based on timing of study either prenatal or postnatal, and did not filter our search to only live births. Abstracts were reviewed and included according to their relevance to key questions. Whenever an abstract made mention of any key question (or there was doubt) the full article was procured. The methods and results sections were then reviewed to determine which articles met inclusion or exclusion criteria. A single reviewer conducted the literature searches, reviewed articles for inclusion, and extracted data. Article inclusion criteria included: data addresses at minimum one key question and supporting data is original (not previously published). Exclusion criteria included: data does not address at least one key question, study uses an uninterpretable methodology, case series < 5, does not provide supporting data, did not present data specific to DS, focused on a single type of CHD, is a unique subset of DS (e.g. surgical patients) which would not generalize to answer our key questions.
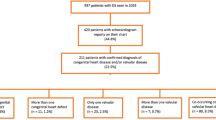
Using only the PubMed articles meeting inclusion, data pertaining to key questions were extracted from the abstract, methods, and results sections and entered into a preformatted Excel data template for analysis. For graphical representation of temporal changes, we grouped reported CHD’s according to [15]. to facilitate comparisons across studies [8]. See Fig. 1 for an overview of the identification, inclusion and exclusion process and Supplemental Table for details of extracted data.

PRISMA Diagram of Articles Identified, Screened, Eligible, and Included in Review of CHD Phenotype in Down Syndrome. Research design hierarchy in accordance with United States Preventative Services Task Force methods
Factors which may impact these key questions such as: publication year, population details (number of subjects studied, age of sample, location, source of subjects and demographics: gender, race, prenatal or postnatal diagnosis of CHD, prenatal or postnatal diagnosis of DS) if provided, and research methodology were recorded.
Evidence ratings by condition
Included articles were critically appraised by a single reviewer to determine each study’s research design, subject ascertainment, total number of subjects, source of control subjects, and the extent of internal validity and external validity. The evaluation of internal validity considers study design factors such as ascertainment and selection bias, test procedures and consideration of confounding variables. For example, the internal validity of a cohort study is rated as good if it “Meets all criteria: comparable groups are assembled initially and maintained throughout the study (follow-up greater than or equal to 80%); reliable and valid measurement instruments are used and applied equally to all groups; interventions are spelled out clearly; all important outcomes are considered; and appropriate attention to confounders in analysis. In addition, intention-to-treat analysis is used for RCTs” (USPSTF Procedure Manual (2015), p. 70 [6]). External validity considers the generalizability of findings to a broader (more representative) population [6]. See appendix VII in the USPSTF report for criteria on research design hierarchy and the rating system used for scoring internal and external validity [6]. See Fig. 1 for summary of evidence rating, and the Supplemental Table for rating of each article.
Results
Original literature review
Through review of the literature, we identified 56 articles which fit our criteria of having original data, answering a key question about DS and types of CHD, in humans, and reported on more than 5 cases (Fig. 1, Table 1). These studies were published from 1950 to 2019, and used various study designs including: cohort studies [N = 22], cross-sectional studies [24], case-control studies [10], and case-series [2]. The study methods included: retrospective review [24] registries and databases [26], prospective screening programs [5], and parent survey [3]. Nearly all studies reported on a cohort, of which some were population-based samples [23] and some reported data longitudinally [5]. Some studies compared those with DS and CHD to those with DS without CHD. In addition to the bracketed number of studies, please see the Supplemental Table for specific details on each of the 56 studies.
Data extracted from original literature review which addressed one of our four key questions are presented below. We report studies based in the United States followed by international results.
-
1.
What is the total prevalence of congenital heart disease in DS? And, has the prevalence of congenital heart disease in DS changed over time?
Eighteen articles answered this key question from a variety of locations
In the United States, in national discharge data of 11,372 DS births, approximately 36% recorded selected cardiac malformations in 2007 [9]. Publications from the population-based National DS Project reported prevalence of CHD in infants with DS; in 2008, 649 of 1469 (44.2%), and in 2011, 483 of 1079 (44.8%) [10, 11]. Prior, data from the population-based registry, the California Birth Defects Monitoring Program, reported CHD in 385 of 687 (56%) infants with DS in one publication [12], and in another 1620 of 2894 (56%) infants with DS had a cardiovascular system birth defect [13].
Publications from the population-based Metropolitan Atlanta Congenital Defects Program (MACDP) found 227 DS cases, of which 44% had CHD in 1989 to 1995 [14], and in 1968 to 1989 saw that 173 of 552 DS cases (33%) had cardiovascular malformations [15]. Over time, the frequency of these defects increased dramatically from about 20% in the early 1970s to more than 50% in the late 1980s (p = 0.0001), which the authors attributed to improvement in the ascertainment of cardiovascular malformations among infants with DS in a surveillance population [15].
To describe prevalence in CHD over time, eight additional international studies provided longitudinal data. In Sweden, among 2588 singleton live-born infants with DS between 1992 and 2012, 1387 infants had a diagnosed congenital heart defect, giving an overall birth prevalence of 54% which was similar over time [8]. In a multi-site European study of 14,109 cases with DS, of whom 6738 were live births, 306 fetal deaths, and 7065 terminations of pregnancy for fetal anomaly in 2000–2010, the overall prevalence of cardiac anomaly was 43.6% (95% confidence interval (CI): 42.4–44.7%) and had remained nearly constant [16]. A study of birth defects registries in France, Italy and Sweden in 1978–1993, found that cardiac defects were registered in 26% of the 5581 infants with DS [17]. In a Norwegian study no apparent increasing or decreasing trend in the prevalence of CHD in live born infants with DS was observed during 1994–2009 [18]. No significant change in prevalence was seen in Thailand with 64 of 149 with DS born in 2009–2013 who had CHD (43%) compared with 112 in 295 (38.6%) DS patients born in 1992–2002 [19]. In the United Kingdom, 342 of 821 live born infants (42%) in 1985–2006 had CHD [20]. Present a figure showing an increasing prevalence from approx. 30% in the late 1980s to approx. 50% in the early 1990s following which the prevalence seemed to stabilize around 40–50% until 2006 [20]. In Egypt, a downward trend in the prevalence of CHD (from 56.2% in the birth cohort 1992–1996 to 25% in the birth cohort 2012–2016) was observed in one clinic (p < .001) [21], while in another, the proportion of infants with DS and CHD increased from 29.8% in 1995 to 48.2% in 2000 [22].
Prevalence of CHD was reported at single time points in two international studies, including: 207 of 482 (43%) children with DS in the Netherlands born 2003–2006 [23] and 224 of 394 (56.9%) infants with DS in Korea in 2005–2006 [24]. One article was initially included, but on critical review was identified to have exclusion criteria which would make results regarding prevalence not generalizable, and is excluded from Fig. 2 [25].

CHD total prevalence. Overview of studies (assessed as population-based) reporting total prevalence of CHD among all DS patients. Studies are ordered according to period of birth midpoint/study period midpoint. CHD: congenital heart defect. DS: Down syndrome
In summary, in the eighteen studies cited above, we found that the total prevalence of CHD in DS ranged from 20 to 57.9%, mean 44.8% in DS patients born from the early 1970s to 2015 (Fig. 2). Over time, studies show an increasing prevalence in the late 1980s-early1990s from around 30% to around 50% following which the prevalence seems to stabilize around 40–55% until 2015 (Fig. 2). The findings indicate an apparent increase in reported CHD prevalence in the first 10–15 years of this period from around 20–30% to around 40–55%. The total prevalence rates varied over time: increased in Atlanta from 1970s to 1980s due to increased CHD ascertainment [15], decreased in Egypt from 1992 to 1996 to 2012–2016 [21], and was unchanged in Sweden from 1992 to 2012, in Europe from 2000 to 2010, and in Thailand from 1992 to 2013 [8, 16, 19].
-
2.
What is the type of congenital heart disease in DS? And, has the type of congenital heart disease in DS changed over time?
Fifteen studies in the United States specified the types of CHD
With the study population in a single city [2] of Atlanta [14, 15], specific states [7] including Alabama [26], California [12, 13], Massachusetts [27], Minnesota [28], Pennsylvania [29] and Texas [30], regions [2] of Maryland / Washington DC / Virginia [31], and Maryland / Pennsylvania [32], or multiple states [4] [5, 10, 11, 25]. These studies used databases, registries, and clinical records, and were all retrospective reviews. None of these fifteen studies described the type of CHD longitudinally.
Thirty-nine articles outside the United States provided data on the type of CHD in DS
Five studies in North America including: Canada [33], Mexico [34], Jamaica [35, 36], and Guatemala [37]. Two studies in South America, including: Brazil [38, 39]. Eleven in Europe including: Italy [40], Norway [18], Germany [41], United Kingdom [20], Netherlands [23], Europe (multiple countries) [42], Ireland [43], Portugal [44], France [45], England [46, 47]. Eleven studies in Asia including: Malaysia [48], Thailand [19], Saudi Arabia [49], Pakistan [50], Korea [24], Singapore [51], India [52], Oman [53], Turkey [54], Turkey [55], Japan [56]. Seven studies in Africa including: Ethiopia [57], Algeria [58], Egypt [21], Morocco [59], Libya [60], Sudan [61], Egypt [22]. One study from Australia [62]. The type of CHD for all studies is reported in the Supplemental Table and summarized in Fig. 3 (including studies where CHD types are reported as proportion of DS patients) and Fig. 4 (including studies where CHD types are reported as proportion of all CHD types found). The figures show variable proportions of types of defects across periods of birth/study periods. In Fig. 3, the majority of studies covering the period approx. Late 1960s to mid 1990s observe a greater proportion of complex defects (atrioventricular septal defect (AVSD), aortic arch abnormalities, tetralogy of Fallot, transposition of the great arteries, and single ventricle hearts) compared with shunt defects (isolated ventricular septal defect (VSD), isolated atrial septal defect (ASD), and isolated patent ductus arteriosus). In contrast, studies covering later years more often find the proportion of shunt defects exceeding the proportion of complex defects. In studies reporting on the number of CHD types out of all CHDs covering the period 1968–2018 (Fig. 4) shunt defects were consistently reported at higher proportions than complex defects.

Distribution of CHD types among DS patients. Proportion of DS patients with a given CHD grouped according to [15, 8]: complex defects (AVSD, aortic arch abnormalities, tetralogy of Fallot, transposition of the great arteries, and single ventricle hearts), valve defects (aortic, pulmonary, and mitral-tricuspid valve defects), shunt defects (isolated VSD, isolated ASD, and isolated patent ductus arteriosus), and other defects [15]. do not report prevalence rates for the latter three groups according to year of birth, so for this study only proportions of complex defects are displayed in the figure. CHD: congenital heart defect. DS: Down syndrome. AVSD: atrioventricular septal defect. VSD: ventricular septal defect. ASD: atrial septal defect

CHD types as proportion of all CHDs. Frequencies of CHD types out of the total number of CHDs grouped according to [15, 8]: complex defects (AVSD, aortic arch abnormalities, tetralogy of Fallot, transposition of the great arteries, and single ventricle hearts), valve defects (aortic, pulmonary, and mitral-tricuspid valve defects), shunt defects (isolated VSD, isolated ASD, and isolated patent ductus arteriosus), and other defects. CHD: congenital heart defect. DS: Down syndrome. AVSD: atrioventricular septal defect. VSD: ventricular septal defect. ASD: atrial septal defect
Six articles described the type of CHD longitudinally
In the United States, one study found an increasing prevalence during the 1980s of ascertained patent ductus arteriosus, endocardial cushion defects and ASD [15]. In Sweden, the risk of complex CHD (as defined in Figs. 3 and 4) decreased over time: compared with 1992 to 1994, the risk in 2010 to 2012 was reduced by almost 40% (adjusted risk ratio 0.62, 95% confidence interval 0.48–0.79) [8]. In contrast, chances for isolated VSD or ASD showed significant increases during later years, and although AVSD was far more common than VSD in 1992 to 1994, they were equally common in 2010 to 2012 [8]. Results from the United Kingdom, may support this finding [20]. Here, the proportions of ASD increased from approx. 9% in 1985–1989 to approx. 19% in 2000–2006 (our estimates based on the Fig. 3 in the article by Irving et al. [20]).
However, a 28-country, population-based study using congenital anomaly registries in Europe in 2000–2010 found no evidence of a trend in the proportions of births with DS with a severe cardiac anomaly (either of: single ventricle, hypoplastic left heart, hypoplastic right heart, Ebstein anomaly, tricuspid atresia, pulmonary valve atresia, common arterial truncus, AVSD, aortic valve atresia/stenosis, transposition of great vessels, tetralogy of Fallot, total anomalous pulmonary venous return, and coarctation of aorta) since 2000 [16]. There was no observed change in prevalence of ASD and VSD among births with DS over the 10 years of study [16]. The authors suggested that population screening for DS and subsequent terminations has not influenced the prevalence of specific congenital anomalies in these European infants [16]. Similarly, there was no apparent trend towards lower prevalence of AVSD among live born DS patients in Norway in 1994–2009 [18]. An Egyptian study found a decreasing prevalence of isolated CHD (as opposed to multiple CHD) from 56.2% in the birth cohort 1992–1996 to 19.8% in the birth cohort 2012–2016 [21].
In summary, in the included articles, the type of CHD reported varies greatly (Fig. 3). The studies which provided longitudinal data differed in the location and year; there was no clear consensus if, or how, the prevalence of specific types (complex, or severe) of CHD were changing over time.
-
3.
What is the severity of congenital heart disease in DS in terms of treatment and prognosis? And, has the severity of congenital heart disease in DS changed over time?
Seven articles addressed this key question regarding CHD severity
In the United States during 2000–2014, neonates with DS who died were significantly more likely to have the diagnosis of complete transposition of the great vessels (37.5 vs 7.5%, respectively), double outlet right ventricle (17.7 vs 7.4%, respectively), Ebstein’s anomaly (29.4 vs 7.4%, respectively), left-sided obstructive lesion (14.9 vs 6.9%), or pulmonary venous abnormality (26.1 vs 7.5%, respectively) compared to those who survived [5]. Rates of surgical management were reported in two studies: in one study, all with AVSD underwent surgery [49], and in a second study, surgery was the most common treatment modality (54.3%) [59].
In Norway, the five-year hazard ratio for death was highest for children with conotruncal defects, then AVSD and then other CHDs [18]. Mortality was especially high for children with DS who had extracardiac malformations with 93% dying in first year [18]. In Malaysia, one study reported on children with DS and CHD of which 30% of lesions closed spontaneously, 35% underwent surgery / intervention, 9% died before surgery / intervention, and 10% were treated with comfort care [48]. The authors assess proportions of cases in each DS patient category for each birth year in 2006–2015, however, the small numbers limit interpretation of potential changes in time. The overall 1-, 5-, and 10-year survival rates for cases with DS and CHD were 85.5, 74.6, and 72.9%, respectively, with 31% of deaths being cardiac related [48]. One-year survival was reported comparable in 2006 (87%) and 2015 (84%) [48]. A German study found a temporal change in treatment: the likelihood of surgical treatment increased from 0% for DS patients born in the 1950s/1960s to 2.1% in the 1970s and 85.6% among patients born after 2000 [41]. Further, the authors report the proportion of patients developing Eisenmenger syndrome decreasing from 53.3% in the earliest birth cohort to 0.5% in the latest birth cohort. Results from the United Kingdom supports the improved prognosis: compared with 1985–1995, DS infants in 1996–2006 more often underwent surgery (62% vs. 72%), had lower mortality following surgery (30% vs. 5%) including lower early postoperative mortality [20]. Overall, the 1-year survival among DS infants with CHD improved from 82 to 94% from the early to the late cohort [20].
-
4.
What screening for congenital heart disease in DS is performed in studies? And, has the screening for congenital heart disease in DS changed in the published literature over time?
Some studies specified that all had an echocardiogram [49, 59], while others relied on retrospective review and were limited by documentation and the possibility that echocardiogram may not have been performed for all. Echocardiogram appeared to be generally accepted as the diagnostic standard. One study evaluated if screening with physical examination, ECG, and chest X-ray is an effective method of identifying which infants with DS should have an echocardiogram, and found that this method would have resulted in 69 (17%) fewer echocardiograms without missing infants with major CHD, but missing cases of patent ductus arteriosus and ASD [25]. In a similar study investigating the ability of clinical examination, chest X-ray, and ECG soon after birth separately and in combination to detect CHD, the three modalities combined showed a sensitivity of 71% and a specificity of 91% [43]. Another study assessed the accuracy of physical examination alone for identifying CHD in neonates with DS and report a sensitivity of 80% and specificity of 56%, concluding that physical examination is not a sufficient screen for CHD [29].
Discussion
Through literature review using MeSH terms in PubMed, we identified 56 articles which provided original data about DS to answer one of our key questions on prevalence, types of CHD, severity, and screening. We found that:
-
1)
The total prevalence of CHD in DS ranged from 20 to 57.9% in 18 studies; the earliest studies indicated an increase in prevalence, while in later decades, the reported prevalence appeared constant around 40–50% (Fig. 2) The total prevalence rates over time were reported in 9 studies and were: increased in Atlanta from 1970s to 1980s due to increased CHD ascertainment [15], increased in the United Kingdom from the late 1980s to early 1990s [20], decreased and increased at two sites in Egypt from 1992 to 1996 to 2012–2016 and 1995 to 2000, respectively [21, 22], and unchanged in Sweden from 1992 to 2012, in Europe from 2000 to 2010, and in Thailand from 1992 to 2002 to 2009–2013 [8, 16, 19].
-
2)
The types of CHD identified varied considerably between studies (Figs. 3 and 4). The six studies which provided longitudinal data differed in location and year; there was no clear consensus if the prevalence of specific types of CHD in DS changed over time, although some studies indicated a trend towards increasing relative proportion of milder lesions.
-
3)
Seven articles addressed the key question of CHD severity. These showed links to mortality for specific types of CHD in DS, and some reported on rates of surgical (and non-surgical) treatment. Generally, the last decades have shown improvements in treatment outcomes and mortality.
-
4)
Echocardiogram remains the accepted diagnostic approach, though some have evaluated additional approaches and timing and frequency of echocardiogram.
In conducting this literature review, a number of confounding factors of the studies arose. First, the source of information differed between studies with some reviewing medical records and others employing registry data. Varying data quality may have been an issue – for example the extent to which a CHD diagnosis was captured in databases/registries. We initially coded CHD as published by authors, without additional interpretation or modification. For subsequent figures CHD types were grouped according to [15]. and we considered endocardial cushion defect and complete atrioventricular canal as AVSD [8](Figs. 3 and 4). Differences in classification of CHD among the studies could lead to validity issues as we try to summarize the available literature. As such, Figs. 3 and 4 should be interpreted with caution. Additionally, the method in which CHD was counted in studies differed, with some studies presenting results in number of defects (with multiple defects possible for a single patient) while some report results in number of patients. Also, in many studies the specific CHD was listed, but some studies grouped CHD in a variety of ways, including: right- or left-sided CHD, severity of CHD, primary or secondary CHD, isolated or complex CHD, or size of the VSD. In defining CHD some studies include isolated PDA, while others only include if remains open at given age [29]. There may be a selection bias in studies: for example, if severe cases of CHD in DS are more likely to have a clinic visit, the prevalence of severe CHD in a single-center study from this clinic could be falsely high.
Over the years, several factors may have impacted on prevalence, diagnosis, and management of CHD in DS. These factors are important to take into account when answering our key questions based on the results from 56 studies. In one study, the prevalence of CHD increased from 1970s to 1980s due to improvement in the ascertainment of cardiovascular malformations among infants with DS [15]. Both improved echocardiography techniques and availability of cardiac testing could impact the reported prevalence rates over time. Additionally, improved cardiac care, and surgical outcomes for CHD as a whole over time could impact neonatal mortality and prevalence of CHD in children with DS. The impact of elective terminations could impact how many infants are born with DS [2]; and it is possible that those with DS and prenatal diagnosis of CHD could undergo elective terminations at a rate different from those with DS and no CHD. Over the last decade, prenatal diagnosis of DS has become more widely available through use of cell-free fetal DNA (cff-DNA). A prenatal diagnosis of DS through cff-DNA could lead to additional fetal testing and identification of CHD prenatally. Increased elective terminations due to the advent of cff-DNA and fetal cardiac testing, could impact both the overall prevalence of CHD in DS, as well as the type of CHD. Most of the 56 studies we identified focused on CHD postnatally, but a multi-site European study of 14,109 cases with DS reported the proportion with any cardiac anomaly in live births and fetal deaths (43.6% of 3068 (95% CI: 42.4–44.7%)), and in all terminations of pregnancy for fetal anomaly (8.1% of 570 (95% CI: 7.4–8.7%)), and in terminations of pregnancy for fetal anomaly who had a postmortem examination (18.1% of 220 (95% CI: 16.0–20.4%)), but had under-reporting of medium and low mortality cardiac anomalies in TOPFAs [16].
Location of study may impact findings, and how we summarize results. For example, local resources may differ by location and impact some of the confounding factors described above, such as: the timing of diagnosis of DS or CHD, the availability of echocardiography and cardiac testing, the availability and uptake of prenatal diagnosis of DS (including cff-DNA testing in later years), the quality and availability of pediatric cardiac care, and the availability and outcomes of surgical management. In addition, there may be differences in racial and ethnic composition of the population by location. In one study of infants with DS, CHD was the most frequently reported cause of death from death certificates and the case fatality rate among infants with DS was significantly higher among blacks than whites, with the greatest racial disparity observed among infants without CHD who died in the post-neonatal period [63]. The genetic makeup of the population may lead to differences in the prevalence of CHD in DS, for example, in instances of consanguinity, as found in one study in which parental consanguinity was an independent predictors of CHD in children with DS, with adjusted odds ratio (OR) of 1.9 [21]. Altogether, local trends in CHDs in DS are potentially overshadowed in studies including data from different populations and may impact on our ability to assess general trends.
There may be other covariates which differ among these 56 studies, and influence the answers to our key questions. Lack of maternal folic acid supplementation was more frequent among infants with DS and atrioventricular septal defects (OR 1.69; 95% CI: 1.08–2.63; p 5 0.011) or atrial septal defects (OR 1.69; 95% CI, 1.11–2.58; p 5 0.007) than among infants with Down syndrome and no heart defect [10]. Parental origin of chromosome 21 may be relevant; one study found that CHD were more frequent in cases with a maternally derived extra chromosome 21 [64].
This literature review is limited by the information presented and published in the existing medical literature. For some of the confounding factors such as naming, diagnosing, counting, grouping and selecting cases, it would be useful to have a standard nomenclature or protocol when describing CHD in DS to allow studies to be compared. Also, when including results in Figs. 2, 3 and 4, we sorted studies based on midpoint of birth period/study period though some span a great number of years with overlapping time periods, which limits the interpretation of our figures. In addition, the 56 articles were identified through searching with MeSH terms and the PubMed database; relevant articles may have been missed and we have been made aware of three such articles [65,66,67]. Additionally, changes over time and by location, complicate our ability to combine results of studies and draw broad conclusions. To address this, we present original data from studies and our full review data in the Supplemental Table.
Future studies to update current findings of CHD in DS could address some of the gaps in the literature which we have highlighted, including: considering use of a standard nomenclature and protocols to increase consistency across sites. Ideally, an international population-based database could be created to focus on CHD in DS, and could begin to collect prospective information from time of initial diagnosis, including prenatal diagnoses, then tracking diagnostic outcomes and treatments longitudinally. Regional changes could also be due to local issues, and more longitudinal studies assessing changes in time and taking local factors into consideration may also provide important insights.
Conclusion
CHD has remained a consistently common co-occurring condition in DS for decades. Recent studies show there may be trends in specific types of CHD with increases in isolated, less severe types and decreased types which are complex, more severe, however not all studies support this. Future studies would ideally be international, population-based, longitudinal, use consistent nomenclature, and account for factors which impact prevalence and severity of CHD.
Availability of data and materials
The data that support the findings of this study were derived from the following resources available in the public domain: PubMED at https://www.ncbi.nlm.nih.gov/pubmed/ and the full data from our review is listed in the Supplemental Table.
References
de Graaf G, Buckley F, Skotko BG. Estimation of the number of people with Down syndrome in the United States. Genet Med. 2017;19(4):439–47.
de Graaf G, Buckley F, Skotko BG. Estimates of the live births, natural losses, and elective terminations with Down syndrome in the United States. Am J Med Genet A. 2015 Apr;167(4):756–67.
Lin AE, Santoro S, High FA, Goldenberg P, Gutmark-Little I. Congenital heart defects associated with aneuploidy syndromes: new insights into familiar associations. Am J Med Genet C Semin Med Genet. 2020;184(1):53–63.
Versacci P, Di Carlo D, Digilio MC, Marino B. Cardiovascular disease in Down syndrome. Curr Opin Pediatr. 2018;30(5):616–22.
Cua CL, Haque U, Santoro S, Nicholson L, Backes CH. Differences in mortality characteristics in neonates with Down’s syndrome. J Perinatol. 2017;37(4):427–31.
United States Preventative Services Task Force. Procedure Manual. 2015 [cited 2019 Dec 30]. Available from: https://www.uspreventiveservicestaskforce.org/uspstf/procedure-manual.
Capone GT, Chicoine B, Bulova P, Stephens M, Hart S, Crissman B, et al. Co-occurring medical conditions in adults with Down syndrome: A systematic review toward the development of health care guidelines. Am J Med Genet A. 2018 Jan;176(1):116–33.
Cleves MA, Hobbs CA, Cleves PA, Tilford JM, Bird TM, Robbins JM. Congenital defects among liveborn infants with Down syndrome. Birth Defects Res Part A Clin Mol Teratol. 2007;79(9):657–63.
Bean LJH, Allen EG, Tinker SW, Hollis ND, Locke AE, Druschel C, et al. Lack of maternal folic acid supplementation is associated with heart defects in Down syndrome: a report from the National Down syndrome Project. Birth Defects Res Part A Clin Mol Teratol. 2011;91(10):885–93.
Freeman SB, Bean LH, Allen EG, Tinker SW, Locke AE, Druschel C, et al. Ethnicity, sex, and the incidence of congenital heart defects: a report from the National Down syndrome Project. Genet Med. 2008 Mar;10(3):173–80.
Torfs CP, Christianson RE. Maternal risk factors and major associated defects in infants with Down syndrome. Epidemiology. 1999 May;10(3):264–70.
Torfs CP, Christianson RE. Anomalies in Down syndrome individuals in a large population-based registry. Am J Med Genet. 1998 Jun 5;77(5):431–8.
Freeman SB, Taft LF, Dooley KJ, Allran K, Sherman SL, Hassold TJ, et al. Population-based study of congenital heart defects in Down syndrome. Am J Med Genet. 1998 Nov 16;80(3):213–7.
Khoury MJ, Erickson JD. Improved ascertainment of cardiovascular malformations in infants with Down’s syndrome, Atlanta, 1968 through 1989. Implications for the interpretation of increasing rates of cardiovascular malformations in surveillance systems. Am J Epidemiol. 1992;136(12):1457–64.
Bergstrom S, Carr H, Petersson G, Stephansson O, A-KE B, Dahlstro MA, et al. Trends in congenital heart defects in infants with Down syndrome. Pediatrics. 2016;138(1):e20160123.
Morris JK, Garne E, Wellesley D, Addor M-C, Arriola L, Barisic I, et al. Major congenital anomalies in babies born with Down syndrome: a EUROCAT population-based registry study. Am J Med Genet A. 2014 Dec;164A(12):2979–86.
Källén B, Mastroiacovo P, Robert E. Major congenital malformations in Down syndrome. Am J Med Genet. 1996 Oct 16;65(2):160–6.
Brodwall K, Greve G, Leirgul E, Klungsøyr K, Holmstrøm H, Vollset SE, et al. The five-year survival of children with Down syndrome in Norway 1994-2009 differed by associated congenital heart defects and extracardiac malformations. Acta Paediatr. 2018 May;107(5):845–53.
Jaruratanasirikul S, Limpitikul W, Dissaneevate P, Booncharoen P, Tantichantakarun P. Comorbidities in Down syndrome livebirths and health care intervention: an initial experience from the birth defects registry in southern Thailand. World J Pediatr. 2017 Apr;13(2):152–7.
Irving CA, Chaudhari MP. Cardiovascular abnormalities in Down’s syndrome: spectrum, management and survival over 22 years. Arch Dis Child. 2012;97(4):326–30.
El-Gilany A-H, Yahia S, Wahba Y. Prevalence of congenital heart diseases in children with Down syndrome in Mansoura, Egypt: a retrospective descriptive study. Ann Saudi Med. 2017 Sep;37(5):386–92.
Mokhtar MM, Abdel-Fattah M. Major birth defects among infants with Down syndrome in Alexandria, Egypt (1995-2000): trends and risk factors. East Mediterr Health J. 2001 May;7(3):441–51.
Weijerman ME, van Furth AM, van der Mooren MD, van Weissenbruch MM, Rammeloo L, Broers CJM, et al. Prevalence of congenital heart defects and persistent pulmonary hypertension of the neonate with Down syndrome. Eur J Pediatr. 2010 Oct;169(10):1195–9.
Kim M-A, Lee YS, Yee NH, Choi JS, Choi JY, Seo K. Prevalence of congenital heart defects associated with Down syndrome in Korea. J Korean Med Sci. 2014 Nov;29(11):1544–9.
Bogarapu S, Pinto NM, Etheridge SP, Sheng X, Liesemer KN, Young PC, et al. Screening for congenital heart disease in infants with Down syndrome: is universal echocardiography necessary? Pediatr Cardiol. 2016 Oct;37(7):1222–7.
Wells GL, Barker SE, Finley SC, Colvin EV, Finley WH. Congenital heart disease in infants with Down’s syndrome. South Med J. 1994;87(7):724–7.
Greenwood RD, Nadas AS. The clinical course of cardiac disease in Down’s syndrome. Pediatrics. 1976 Dec;58(6):893–7.
Tandon R, Edwards JE. Cardiac malformations associated with Down’s syndrome. Circulation. 1973 Jun;47(6):1349–55.
McElhinney DB, Straka M, Goldmuntz E, Zackai EH. Correlation between abnormal cardiac physical examination and echocardiographic findings in neonates with Down syndrome. Am J Med Genet. 2002;113(3):238–41.
Spahis JK, Wilson GN. Down syndrome: perinatal complications and counseling experiences in 216 patients. Am J Med Genet. 1999 Jun 25;89(2):96–9.
Ferencz C, Neill CA, Boughman JA, Rubin JD, Brenner JI, Perry LW. Congenital cardiovascular malformations associated with chromosome abnormalities: an epidemiologic study. J Pediatr. 1989 Jan;114(1):79–86.
Park SC, Mathews RA, Zuberbuhler JR, Rowe RD, Neches WH, Lenox CC. Down syndrome with congenital heart malformation. Am J Dis Child. 1977 Jan;131(1):29–33.
Rowe RD, Uchida IA. Cardiac malformation in mongolism: a prospective study of 184 mongoloid children. Am J Med. 1961;31:726–35.
Corona-Rivera JR, Nieto-García R, Gutiérrez-Chávez AS, Bobadilla-Morales L, Rios-Flores IM, Corona-Rivera A, et al. Maternal risk factors for congenital heart defects in infants with Down syndrome from Western Mexico. Am J Med Genet A. 2019;179(9):1857–65.
Livingstone-Sinclair K, Scott C, Trotman H. Adherence to health management guidelines for children with Down’s syndrome at the Bustamante Hospital for Children, Jamaica. Trop Doct. 2018;48(4):301–5.
Tomlinson TW, Scott CH, Trotman HLM. Congenital cardiovascular lesions in children with trisomy 21 at the Bustamante Hospital for Children. Cardiol Young. 2010;20(3):327–31.
Vida VL, Barnoya J, Larrazabal LA, Gaitan G, de Maria GF, Castañeda AR. Congenital cardiac disease in children with Down’s syndrome in Guatemala. Cardiol Young. 2005;15(3):286–90.
Sica CD, Cesa CC, Pellanda LC. Growth curves in Down syndrome with congenital heart disease. Rev Assoc Med Bras. 2016;62(5):414–20.
Bermudez BEBV, Medeiros SL, Bermudez MB, Novadzki IM, Magdalena NIR. Down syndrome: prevalence and distribution of congenital heart disease in Brazil. Sao Paulo Med J. 2015 Dec 8;133(6):521–4.
Santoro M, Coi A, Spadoni I, Bianchi F, Pierini A. Sex differences for major congenital heart defects in Down syndrome: A population based study. Eur J Med Genet. 2018 Sep;61(9):546–50.
Körten M-A, Helm PC, Abdul-Khaliq H, Baumgartner H, Kececioglu D, Schlensak C, et al. Eisenmenger syndrome and long-term survival in patients with Down syndrome and congenital heart disease. Heart. 2016 Oct 1;102(19):1552–7.
Stoll C, Garne E, Clementi M, EUROSCAN Study Group. Evaluation of prenatal diagnosis of associated congenital heart diseases by fetal ultrasonographic examination in Europe. Prenat Diagn. 2001 Apr;21(4):243–52.
Tubman TR, Shields MD, Craig BG, Mulholland HC, Nevin NC. Congenital heart disease in Down’s syndrome: two year prospective early screening study. BMJ. 1991 Jun 15;302(6790):1425–7.
Pinto FF, Nunes L, Ferraz F, Sampayo F. Down’s syndrome: different distribution of congenital heart diseases between the sexes. Int J Cardiol. 1990 May;27(2):175–8.
Stoll C, Alembik Y, Dott B, Roth MP. Study of Down syndrome in 238,942 consecutive births. Ann Genet. 1998;41(1):44–51.
Liu MC, Corlett K. A study of congenital heart defects in mongolism. Arch Dis Child. 1959 Oct;34:410–9.
Evans PR. Cardiac anomalies in mongolism. Br Heart J. 1950 Jul;12(3):258–62.
Zahari N, Mat Bah MN, A Razak H, Thong M-K, et al. Eur J Pediatr. 2019;178(8):1267–74.
Morsy MM, Algrigri OO, Salem SS, Abosedera MM, Abutaleb AR, Al-Harbi KM, et al. The spectrum of congenital heart diseases in Down syndrome. A retrospective study from Northwest Saudi Arabia. Saudi Med J. 2016 Jul;37(7):767–72.
Yaqoob M, Manzoor J, Hyder SN, Sadiq M. Congenital heart disease and thyroid dysfunction in Down syndrome reported at Children’s hospital, Lahore, Pakistan. Turk J Pediatr. 2019;61(6):915–24.
Tan M, Xu C, Sim SKR, Seow ALH, Tan TH, Quek SC. Types and distribution of congenital heart defects associated with trisomy 21 in Singapore. J Paediatr Child Health. 2013 Mar;49(3):223–7.
Narayanan DL, Yesodharan D, Kappanayil M, Kuthiroly S, Thampi MV, Hamza Z, et al. Cardiac spectrum, cytogenetic analysis and thyroid profile of 418 children with Down syndrome from South India: a cross-sectional study. Indian J Pediatr. 2014 Jun;81(6):547–51.
Venugopalan P, Agarwal AK. Spectrum of congenital heart defects associated with Down syndrome in high consanguineous Omani population. Indian Pediatr. 2003;40(5):398–403.
Nisli K, Oner N, Candan S, Kayserili H, Tansel T, Tireli E, et al. Congenital heart disease in children with Down’s syndrome: Turkish experience of 13 years. Acta Cardiol. 2008;63(5):585–9.
Aynaci FM, Orhan F, Celep F, Karagüzel A. Frequency of cardiovascular and gastrointestinal malformations, leukemia and hypothyroidism in children with Down syndrome in Trabzon, Turkey. Turk J Pediatr. 1998;40(1):103–9.
Matsuo N, Oshima M, Masuyoshi N, Shimizu K, Okada R. Major and minor anomalies in Japanese children with Down’s syndrome. Jpn Heart J. 1972;13(4):307–16.
Muntha A, Moges T. Congenital cardiovascular anomalies among cases of Down syndrome: A hospital based review of cases in TikurAnbessa specialized hospital, Ethiopia. Ethiop J Health Sci. 2019;29(2):165–74.
Boussouf K, Zaidi Z, Amrane M, Hammoudi N, Mebarki M, Amalou SA. Study of congenital heart diseases in patients with Down syndrome in Algeria. East Mediterr Health J. 2017;23(9):632–6.
Benhaourech S, Drighil A, Hammiri AE. Congenital heart disease and Down syndrome: various aspects of a confirmed association. Cardiovasc J Afr. 2016 Oct;27(5):287–90.
Elmagrpy Z, Rayani A, Shah A, Habas E, Aburawi EH. Down syndrome and congenital heart disease: why the regional difference as observed in the Libyan experience? Cardiovasc J Afr. 2011 Dec;22(6):306–9.
Ali SK. Cardiac abnormalities of Sudanese patients with Down’s syndrome and their short-term outcome. Cardiovasc J Afr. 2009 Apr;20(2):112–5.
Selikowitz M. Health problems and health checks in school-aged children with Down syndrome. J Paediatr Child Health. 1992 Oct;28(5):383–6.
Shin M, Kucik JE, Correa A. Causes of death and case fatality rates among infants with Down syndrome in metropolitan Atlanta. Birth Defects Res Part A Clin Mol Teratol. 2007;79(11):775–80.
Muranjan M, Chaudhari T, Vundinti BR. Phenotypic heterogeneity and parental origin of extra chromosome 21 in Down syndrome. Indian Pediatr. 2010 May;47(5):429–32.
Espinola-Zavaleta N, Soto ME, Romero-Gonzalez A, Gómez-Puente LDC, Muñoz-Castellanos L, Gopal AS, et al. Prevalence of congenital heart disease and pulmonary hypertension in Down’s syndrome: an echocardiographic study. J Cardiovasc Ultrasound. 2015;23(2):72–7.
Mourato FA, Villachan LRR, Mattos S da S. Prevalence and profile of congenital heart disease and pulmonary hypertension in Down syndrome in a pediatric cardiology service. Rev Paul Pediatr. 2014;32(2):159–63.
Matthiesen NB, Agergaard P, Henriksen TB, Bach CC, Gaynor JW, Hjortdal V, et al. Congenital Heart Defects and Measures of Fetal Growth in Newborns with Down Syndrome or 22q11.2 Deletion Syndrome. J Pediatrics. 2016;175:116–122.e4.
Acknowledgements
Not applicable.
Funding
The authors do not have funding related to the work to write this manuscript.
Author information
Authors and Affiliations
Contributions
SLS conceptualized the project, conducted the literature reviews, critically-reviewed articles, and analyzed, interpreted the data, and drafted first version of the manuscript. EHS reviewed literature review data, interpreted articles, created figures and tables, revised manuscript. All authors read and approved the final manuscript.
Authors’ information
Dr. Santoro receives research funding from the National Institutes of Health. Dr. Santoro receives research funding from the LuMind IDSC Down Syndrome Foundation to conduct clinical trials for people with DS and serves on the Professional Advisory Board for the Massachusetts Down Syndrome Congress. The other authors have no conflicts of interest relevant to this article to disclose.
Ellen Steffensen has received funding from Aarhus University, the Health Research Foundation of Central Denmark Region, and the Health Foundation (Helsefonden).
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Santoro, S.L., Steffensen, E.H. Congenital heart disease in Down syndrome – A review of temporal changes. J Congenit Heart Dis 5, 1 (2021). https://doi.org/10.1186/s40949-020-00055-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40949-020-00055-7