
Abstract
The stromal interaction molecule (STIM)-calcium release-activated calcium channel protein (ORAI) and inositol 1,4,5-trisphosphate receptors (IP3Rs) play pivotal roles in the modulation of Ca2+-regulated pathways from gene transcription to cell apoptosis by driving calcium-dependent signaling processes. Increasing evidence has implicated the dysregulation of STIM–ORAI and IP3Rs in tumorigenesis and tumor progression. By controlling the activities, structure, and/or expression levels of these Ca2+-transporting proteins, malignant cancer cells can hijack them to drive essential biological functions for tumor development. However, the molecular mechanisms underlying the participation of STIM–ORAI and IP3Rs in the biological behavior of cancer remain elusive. In this review, we summarize recent advances regarding STIM–ORAI and IP3Rs and discuss how they promote cell proliferation, apoptosis evasion, and cell migration through temporal and spatial rearrangements in certain types of malignant cells. An understanding of the essential roles of STIM–ORAI and IP3Rs may provide new pharmacologic targets that achieve a better therapeutic effect by inhibiting their actions in key intracellular signaling pathways.
Similar content being viewed by others


Background
Calcium signals are widespread and rigorously regulate the majority of fundamentally important physiologic processes ranging from cell proliferation to cell apoptosis [1]. The precise and tightly controlled intracellular calcium ion concentration depends on finely tuned modulation by various calcium-transporting processes, including Ca2+ channels, pumps, and receptors [2]. These Ca2+-transporting molecules strictly regulate the transient or sustained waves, spikes, or oscillations of Ca2+ signaling in different cellular compartments and microdomains to maintain a delicate balance between feeding into the cytoplasm and releasing from internal Ca2+ stores [3]. Any perturbation and disorder of the delicate Ca2+ homeostasis may lead to long-ranging consequences. Therefore, it is not surprising that any derangement of Ca2+ channels and/or receptors will contribute to the establishment of many life-threatening diseases, such as cardiopathy [4], heart failure [5], neurodegenerative diseases [6], and cancer [7]. Remarkably, the altered expression or activity of these Ca2+ channels or receptors is characterized by the features of specific cancer subtypes, of which the most important is the protein complex consisting of the stromal interaction molecule (STIM), calcium release-activated calcium channel protein (ORAI), and inositol 1,4,5-trisphosphate (IP3) receptors (IP3Rs). STIM–ORAI is able to sense and respond to intracellular Ca2+ microenvironmental changes that occur during cancer development. This complex primarily mediates the Ca2+ influx, with STIM serving as the endoplasmic reticulum (ER) Ca2+ sensor and ORAI as the Ca2+-selective entry channel. Furthermore, the roles of IP3Rs are also discussed. There is emerging evidence that IP3Rs, which regulate the Ca2+ flux from the ER into the cytosol and mitochondria, play crucial roles in the apoptotic pathway; additionally, IP3Rs have been implicated in cellular senescence [3]. The last decade in clinical oncology has been noteworthy because of advances in our understanding of the derangement of Ca2+ channels/transporters that are thought to be responsible for the development of cancers [8]. Although enormous explorations have been performed, the molecular mechanisms by which these derangements affect tumorigenesis and tumor progression are far from being fully understood. In this review, we discuss recent progress in understanding the roles of STIM–ORAI and IP3Rs, with a focus on exploring the mechanism underlying the hijacking of the Ca2+-transporting molecules STIM-ORAI and IP3Rs by malignant cancer cells that leads to tumor onset, growth, and metastasis. The concomitant interaction between STIM–ORAI and IP3Rs is also discussed. Understanding the molecular basis and pathologic transformations of Ca2+-transporting molecules in cancer cells will offer an opportunity for pharmacologic modulation and therapeutic intervention.
The structure and function of the STIM and ORAI protein families
External stimulation leads to an increase in cytoplasmic Ca2+ from either the entry of extracellular Ca2+ across the plasma membrane or the release of Ca2+ from internal calcium stores in the ER and sarcoplasmic reticulum (SR) [9]. Both of these functions involve the permeable Ca2+ channels that are located on the plasma membrane. Upon the stimulation, extracellular Ca2+ can enter the cytoplasm through Ca2+ channel transport. Moreover, the plasma membrane is responsible for refilling the internal Ca2+ stores when they are depleted. The primary Ca2+ entry pathway is store-operated Ca2+ entry (SOCE), which includes two key components: a sensitive sensor of calcium store depletion (STIM) and an effective channel that can facilitate calcium entry into the cell (ORAI) [10–12].
STIM, which is predominantly located in the ER, was identified using an RNA interference screen in Drosophila S2 cells; then, two mammalian orthologs (STIM1 and STIM2) were found [13]. Both STIM1 and STIM2 act as sensors of Ca2+ store levels in the ER and control calcium refilling by forming connections with ORAI [14]. STIM1 contains an ER luminal N-terminus and a cytosolic C-terminus. The ER luminal portion consists of a canonical Ca2+-binding EF-hand (a conventional helix-loop-helix EF motif), a hidden EF hand, and a sterile α-motif (SAM) domain. The cytosolic strand includes three putative coiled-coil (CC1, CC2, and CC3) regions, calcium release-activated calcium (CRAC) modulatory domain (CAD) or a STIM-ORAI-activating region (SOAR), serine or proline-rich segments, and lysine-rich regions [15]. The low Ca2+-binding affinity of EF-SAM perfectly matches the detailed alteration of the Ca2+ concentration and enables the ER sensor protein to respond to changes in the Ca2+ concentration in ER. The Ca2+ depletion in the ER leads to the dissociation of Ca2+ from the EF hand, thereby destabilizing the entire EF-SAM entity. The CC regions and the serine/proline-rich region promote the oligomerization of STIM, thereby enabling its redistribution into multiple punctae and its localization at ER-plasma membrane junctions. The CC domain has been proven to control the exposure and oligomerization of the SOAR [16]. The structure of STIM2 is similar to that of STIM1; however, STIM1 is widely expressed at both the cell surface and the ER, whereas STIM2 is expressed only in the intracellular space. A growing number of studies have indicated that STIM2 is a potent inhibitor and a feedback regulator of STIM1 via preventing it from forming an aggregate to stabilize basal concentrations of Ca2+ in the cytosol and ER [17]. Thus, STIM2 is regarded as a critical regulator of basal Ca2+ levels in the human signaling proteome, whereas STIM1 seems to be more involved in the Ca2+ entry associated with more pronounced depletion [18, 19].
The ORAI gene encodes a family consisting of three proteins (ORAI1, ORAI2, and ORAI3). Each of these proteins consists of 4 TM-spanning segments and 3 cytosolic strands, which include the N-terminus, the second loop con-necting TM2 and TM3, and the C-terminus [20]. The ORAI1 C-terminus forms the cytosolic extension, and the C-terminal putative CC domain is important for binding to the SOAR/CAD domain of STIM1. By binding to the intracellular C-terminus, STIM1 recruits ORAI to the puncta in the plasma membrane.
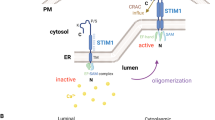
The primary intracellular hysiologic functions of STIM and ORAI are straightforward (Fig. 1a). STIM detects the decrease in Ca2+ stores in the ER and moves within the ER to ER-plasma membrane junctions. Then, STIM recruits ORAI to the ER-plasma membrane junctions, where the two proteins form a close contact. The formation of the active STIM–ORAI complex at the conformational gate of the SOCE channel allows Ca2+ entry [21].

Disrupted dynamic equilibrium of stromal interaction molecule 1 (STIM1)-calcium release-activated calcium channel protein 1 (ORAI1) and inositol 1,4,5-trisphosphate receptor (IP3R)-mediated Ca2+ signaling in tumor biology. a In normal cells, STIM1 exists as a single-transmembrane protein in the endoplasmic reticulum (ER). The STIM1 canonical Ca2+-binding EF-hand (a conventional helix-loop-helix EF motif) can sensitively detect the depletion of ER luminal Ca2+, leading to STIM1 oligomerization and interactions with the C-terminus of ORAI1. The STIM1–ORAI1 complex controls the opening of the Store-operated Ca2+ entry (SOCE) channel ORAI1, thereby allowing Ca2+ entry. The increased Ca2+ in the ER can enter into the mitochondria via IP3Rs, leading to mitochondrial Ca2+ overload and indirectly causing apoptosis. The mitochondrial outer membrane permeabilization (MOMP) is considered a critical step during the point-of-no-return apoptosis in the mitochondria. b In prostate cancer cells, an increase in the level of the endogenous ORAI3 protein causes the association of ORAI3 with ORAI1 to form a heteromultimeric channel that can alter the ORAI3-ORAI1 ratio. These functions represent an oncogenic switch that promotes prostate cancer cell proliferation and confers apoptosis resistance. c STIM1–ORAI1-mediated Ca2+ signaling accelerates tumor cell migration through controlling focal adhesion (FA) turnover and actomyosin contractility. The STIM1–ORAI1-mediated Ca2+ influx regulates actomyosin formation and increases its contractile force. STIM1–ORAI1 induces the Ca2+ influx and promotes the cleavage of FA proteins. The red represents all of the factors involved in resistance to apoptosis, and the blue represents all of the factors that promote apoptosis. d Bcl-2 is a representative anti-apoptotic protein that interacts with IP3R via its N-terminal BH4 domain. Then, Bcl-2 inhibits the Ca2+ flux into the mitochondria, leading to mitochondrial Ca2+ deficiency and preventing cancer cell apoptosis. The deficient Ca2+ can break MOMP and finally prevent cancer cell apoptosis
The structure and function of the IP3 receptor family
The IP3R is the most ubiquitous intracellular Ca2+ channel, and its isoforms (IP3R1, IP3R2, and IP3R3) have been identified in vertebrates [22]. The majority of cell types express more than one isoform but have a predominant one. The three IP3R isoforms have distinct but overlapping expression patterns. IP3R1 is expressed in neuronal cells, IP3R2 is expressed in liver and muscle cells, and IP3R3 is expressed in most cultured cell types [23]. The general domain structure of IP3Rs (which exist as tetramers) has been determined; IP3Rs contain a binding site for IP3 in the N-terminal region, the channel domain, and the determinants for tetramer formation in the C-terminus [24, 25].
IP3Rs predominantly reside in the ER. IP3 is produced by phospholipase C and binds to IP3Rs to induce calcium release from the ER upon cell activation by endogenous or exogenous hormones, growth factors, or neurotransmitters. Notably, IP3-induced Ca2+ release is typically regulated by the Ca2+ concentration in the cytosol and ER. Furthermore, the activities of IP3Rs are biphasically regulated by cytosolic Ca2+. The concentration–response relationship is a typical bell-shaped curve, indicating that the IP3-mediated Ca2+ release is potentiated at a low Ca2+ concentration and inhibited at a higher concentration [26]. The Ca2+ storage in the lumen of the ER also regulates IP3Rs, which can prevent excessive ER depletion at low levels of store filling [27]. IP3R-mediated Ca2+ elevation regulates fundamental cellular functions, such as fertilization, cell cycle entry, cell division, metabolism, and transcription [28]. An important function of IP3Rs is to decide the cell fate by controlling the mitochondrial Ca2+ elevation and mitochondrial metabolism. Cell survival or apoptosis is encoded in the frequency and amplitude of Ca2+ oscillations mediated by IP3Rs and decoded by different Ca2+-sensitive kinases or phosphatases that in turn regulate the target proteins. When IP3Rs transport appropriate amounts of Ca2+ from the ER to the mitochondria, they catalyze the conversation of pyruvate to acetyl-coenzyme A (CoA) to produce adenosine triphosphate (ATP) and nicotinamide adenine dinucleotide 2′-phosphate (NADPH). The insufficient transport of Ca2+ to the mitochondria induces cellular autophagy. Conversely, when activated IP3Rs excessively transport Ca2+ from the ER to the mitochondria, the mitochondrial Ca2+ is overloaded, which induces a dissipation of the mitochondrial potential, the opening of the permeability transition pore, and the release of pro-apoptotic factors such as cytochrome c [29]; this process ultimately triggers cell apoptosis. Therefore, IP3Rs play pivotal roles in the apoptotic process via controlling the cellular response to apoptotic signals and conferring oncogenic features to the cell [30].
The emerging roles of STIM and ORAI in tumorigenesis and tumor progression
STIM and ORAI have been found to be abundantly expressed in human cancer tissues and multiple tumor cell lines. Abnormal spatial and temporal changes in these two proteins have been found to be involved in many aspects of tumorigenesis, including cancer cell proliferation, migration, and apoptosis resistance.
STIM and ORAI are overexpressed in tumors
Increasing evidence has shown that STIM and ORAI are overexpressed in many types of malignant tumors, including breast cancer [31], glioblastoma [32], prostate cancer [33], hepatocellular carcinoma [34], esophageal squamous cell carcinoma (ESCC) [35], and clear cell renal cell carcinoma (ccRCC) [36] (Table 1). An investigation of 24 patients with cervical cancer found that 71 % of the patients showed increased expression of STIM1 in primary cervical cancer tissues compared with non-cancerous tissues. Abnormal overexpression of STIM1 contributed to large tumor sizes and low 5-year survival rates. A similar association between STIM expression and tumor growth was also demonstrated in the study by Yang et al. [34]. The authors found that highly invasive CC-LM3 hepatocytes overexpressed STIM1 at a level approximately eightfold higher than normal LO2 hepatocytes in vitro. A study of 295 breast cancer patients obtained a similar result [37]. The survival of breast cancer patients with high STIM1 mRNA levels in tumors was significantly reduced compared with the control group. Additionally, STIM1 could also be used as a predictive marker for metastatic potential in patients with hepatocellular carcinoma [38]. The high expression of ORAI1 also indicated a poor prognosis and depressed recurrence-free survival. In line with these findings, Zhu et al. [35] demonstrated that malignant ESCC tissues displayed an ectopic overexpression of ORAI1 compared with neighboring non-tumorous esophageal tissues. A similar result for ORAI3 in breast cancer cell lines was reported by Faouzi et al. [39], who showed that the expression of the ORAI3 mRNA was increased in breast cancer tissues from the majority (76.9 %) of patients compared with healthy control tissues. Increased expression of ORAI3 in tumor tissues from 60 patients presenting non-small cell lung adenocarcinoma was also noted by Ay et al. [40]. Additionally, Schmidt et al. [41] showed that the expression levels of STIM and ORAI were significantly higher in cisplatin-resistant ovarian carcinoma cells than in cisplatin-sensitive cells (Table 1). These results provide evidence supporting an association between STIM–ORAI expression and poor outcomes in patients with malignant cells.
STIM and ORAI: pivotal roles in cancer development
The functions of STIM and ORAI in certain types of cancer have fascinated many investigators. The use of pharmacologic interference and small interfering RNA (siRNA)-mediated gene knockdown approaches to down-regulate STIM and ORAI at both the mRNA and protein levels inhibits tumor cell proliferation, promotes cell apoptosis, and reduces tumor size. These results revealed that STIM and ORAI promoted tumorigenesis and tumor progression through the following key events: elevated proliferation, enhanced migration, and increased resistance to apoptosis.
A study of STIM1 indicated that the gene locus encoding STIM1 on chromosome 11p15 was deleted in human rhabdomyosarcoma and rhabdoid tumor cell lines [42]. Ectopic overexpression of STIM1 in vitro could induce morphologic changes in rhabdomyosarcoma cells and ultimately lead to cell death. Therefore, STIM was a suspected tumor suppressor. However, Gueguinou et al. [43] demonstrated that knockdown of STIM1 did not inhibit the migration of breast cancer cells. Moreover, Zhu et al. [35] showed that there was no significant difference between tumor tissues and normal tissues from patients with ESCC. These results implied that STIM1 might play a nonessential role in cancer metastatic processes. These contradictory findings imply that the features and expression of STIM vary in different cancer tissues and stages.
Compared with STIM, the role of ORAI in tumorigenesis may be more explicit. The dysregulation of ORAI is affected by the activation of proto-oncogenes or the inactivation of tumor suppressors. Recently, compelling evidence has suggested that ORAI3 is closely related with c-Myc, which is a key proto-oncogene and is enhanced in most human cancers [43]. In this study, ORAI3 down-regulation specifically reduced the expression and activity of c-Myc via the mitogen-activated protein kinase (MAPK) pathway, leading to breast cancer cell arrest in the G1 phase. Ay et al. [40] found that high expression of ORAI3 promoted non-small cell lung adenocarcinoma cell proliferation via the phosphoinositide 3-OH kinase (PI3K)/Akt signaling pathway, which was constitutively activated in lung cancer cells and was central to cell proliferation and survival. Schmidt et al. [41] also demonstrated that ORAI overexpression induced the activity of the oncoprotein Akt, which contributed to therapy resistance in ovarian carcinoma cells.
Any structural remodeling and functional changes of ORAI3 may trigger a switch to a more aggressive cell phenotype. Dubois et al. [44] showed that enhanced ORAI3 expression favored heteromerization with ORAI1 to form a novel channel in in vitro models; the remodeled ORAI1–ORAI3 complex might serve as the oncogenic switch in prostate cancer (Fig. 1b). Additionally, the authors found that the relative expression level of the ORAI3 protein in cancer tissues was obviously higher than the level in noncancerous tissues. Overexpressed ORAI3 was shown to encode SOCE in a subset of breast cancer cells that partially substituted for functional ORAI1 channels [45]. Importantly, elevated expression level of the ORAI3 favored the association with ORAI1 to form heteromultimeric, store-independent, arachidonic, acid-regulated channels at the expense of “classical” homomultimeric ORAI1-based SOCE. The “nonclassical” association of ORAI3 and ORAI1 crippled the functions of SOCE, leading to the resistance of malignant cells to apoptosis due to the declining infusion of Ca2+. Furthermore, the remodeled ORAI channels promoted cancer cell proliferation via activation of the transcription factor nuclear factor of activated T cells (NFAT), followed by the stimulation of cyclin D1 expression, which is a key rate-limiting controller of the G1/S phase transition. Faouzi et al. [39] demonstrated that ORAI3 contributed to the regulation of the cell cycle by the estrogen receptor expressed on breast cancer cells but not normal breast epithelial cells. These authors reported that knockdown of ORAI3 caused a surprising increase in the levels of the well-established tumor suppressors P53 and P21, leading to cell cycle arrest.
STIM and ORAI have also been found to affect the migration of cancer cells. Increasing evidence has shown that tumor migration can be viewed as a Ca2+-dependent signaling process, and STIM–ORAI is hijacked by malignant cancer cells to drive the biological functions required for tumor development [46]. In other words, although tumor migration is a complicated and multistep process, STIM–ORAI participates in almost every aspect of tumor cell migration, including the formation of lamellipodia/membrane protrusions at the front edge, cycles of adhesion and detachment, cell body contraction, and tail retraction [47]. Blocking STIM-ORAI with its inhibitor, SKF-96365/2-aminoethoxydiphenyl borate (2-APB), or siRNA-mediated gene knockdown can obviously restrain the migration of hepatocarcinoma [34], breast cancer [37], glioblastoma multiforme [48], pancreatic adenocarcinoma [49], and melanoma cells [50]. The STIM-ORAI-mediated Ca2+ influx accelerates focal adhesion (FA) turnover through the constitutively active forms of the small GTPase RAC1 and the Ca2+-dependent proline-rich tyrosine kinase 2 (Pyk2) [51]. The efficiency of the assembly and disassembly of FAs decides the speed of cancer cell migration (Fig. 1c). Assembled FAs serve as anchorage points for actomyosin to provide the traction force that moves the cell body forward [52, 53]. At the same time, the STIM-mediated Ca2+ signaling enhances contractile forces by regulating the actomyosin reorganization. Actomyosin is a complex of actin filaments and non-muscle myosin II. The actomyosin generates cortical tension with the extracellular matrix or neighboring cells and transmits the contraction to FAs that move the cell body [54]. These findings have corroborated that the STIM1-ORAI-mediated Ca2+ signaling exerted comprehensive and crucial functions to promote tumor cell migration by interacting with FA and actomyosin [55]. Moreover, knockdown of ORAI3 reduced the expression levels of cycle D and E1 and finally inhibited the transcriptional activity of NFAT [56]. NFAT is a constitutively active form of the Ca2+-dependent transcription factor that plays a critical role in the tissue invasion of tumor cells by promoting the expression of autotaxin and cyclooxygenase 2 (COX2); these factors participate in the epithelial-to-mesenchymal transition [57].
The burgeoning field of IP3R in cancer biology
Growing attention has been paid to the special role of IP3Rs in tumorigenesis and tumor metastasis. Over the last 20 years, IP3Rs have been regarded as key regulators that control cell death and survival in a variety of cellular systems. Interfering with the amount of IP3R-mediated Ca2+ transport from the ER to the mitochondria determines the susceptibility of cells to apoptotic stimulation. Because IP3Rs can promote senescence and/or apoptosis, the available evidence indicates that down-regulating IP3Rs or dampening their activities can decrease cellular sensitivity to apoptotic signaling, finally resulting in the survival of cells with oncogenic features. An in vitro study showed that knockdown of IP3R1 prevented apoptosis in bladder cancer cells and rendered them resistant to chemotherapeutics [58]. Conversely, overexpression of IP3Rs might increase the sensitivity of cancer cells to cisplatin [59]. As the molecular bridge between the ER and mitochondria, IP3Rs are also hijacked by different proto-oncogenes to give rise to cells with oncogenic features [60], such as Akt/protein kinase B (PKB) [61], Bcl-2 family members [62, 63], Bax inhibitor-1 (BI-1) [64, 65], and K-ras-induced actin-interacting protein (KRAP) [66]. Recently, it has become clear that Bcl-2 directly targets the central modulation domain of IP3Rs through its tetrahydrobiopterin (BH4) domain to inhibit their functions [61]. The spatiotemporal interaction of BH4 and IP3Rs hindered mitochondrial Ca2+ accumulation by abrogating Ca2+ transport from the ER to the outer mitochondrial membrane (Fig. 1d), therefore, the IP3R-BH4 complex counteracted the pressure of pro-apoptotic proteins to protect tumor cells [67]. The Bcl-2 family was also shown to enhance basal Ca2+ leakage through sensitization of IP3Rs to basal IP3 levels lower than the Ca2+ concentration in the ER [68, 69]. The low levels of Ca2+ in the ER destroyed the mitochondrial Ca2+ overload and decreased the susceptibility of the cells to apoptosis. Importantly, a peptide tool that was designed to disrupt the IP3R-BH4 complex could effectively induce an intracellular Ca2+ overload and provoke cell death in diffuse large B cell lymphoma (DLBCL) cells [70]. However, Kang et al. [71] reported that the invasion and migration of tumor cells were suppressed by caffeine, which is a well-known inhibitor of IP3Rs. The expression levels of IP3R3 in colon cells were directly related to tumor aggressiveness [72]. These results suggest that the regulatory mechanisms of IP3Rs may vary in different types of cancer, and many mechanisms are not fully understood.
The interaction between STIM/ORAI and IP3Rs in cancer biology
The binding of IP3 to IP3Rs releases intracellular Ca2+, leading to a reduction in the Ca2+ concentration in the lumen of ER, which in turn activates the STIM sensor to allow extracellular Ca2+ to refill the empty ER Ca2+ stores across the ORAI in the plasma membrane. In rapidly growing cancers, IP3Rs are blocked by a variety of anti-apoptosis proteins, resulting in Ca2+ overload in the ER. The harsh microenvironment perturbs the STIM-ORAI functions and induces the accumulation of misfolded proteins in the ER. This triggers an adaptation program referred to as “ER stress.” Chronic ER stress kills normal cells but can contribute to tumor cell dormancy, thereby permitting survival in the stressed environment until more favorable conditions are encountered. Overexpression of STIM could reverse ER stress, implying that Ca2+ overload restrains the STIM functions in cancer cells [73]. Oncogenic KRAS mutations could reduce the Ca2+ store content in the ER via promoting IP3R1 overexpression to suppress agonist-induced Ca2+ release and mitochondrial Ca2+ accumulation in cancer cells [74]. Nevertheless, the relationships between STIM–ORAI and IP3Rs are not completely understood, and further investigations are needed to elucidate the mechanisms by which cancer cells control the functions of STIM-ORAI and IP3Rs.
STIM–ORAI and IP3Rs in cancer therapy
Multiple roles of STIM–ORAI and IP3Rs in several types of human cancer have made them attractive drug targets for tumor therapy. Inhibiting ORAI1 by pharmacologic antagonists in cultured epithelial cells derived from ESCC patients impeded ESCC cell proliferation, invasion, and migration [75]. Importantly, the growth of ESCC in vivo was significantly suppressed when ORAI1-mediated SOCE was knocked down by siRNA or blocked by pharmacologic inhibitors in xenografted nude mice. SKF-96365 and 2-APB, which are inhibitors of store-operated Ca2+ entry, inhibited the growth and metastasis of tumor cells after 1 week of treatment [76, 77]. No increase in metastasis was observed in mouse cancer models [78], even 2 weeks after withdrawal of SKF-96365. Similar phenomena have been found in cervical and esophageal cancer mouse models.
To date, “proof of principle” studies of the Ca2+ signal channels have shown that STIM-ORAI and IP3Rs either do not differ or are overexpressed in tumor tissues compared to those in normal tissues. However, the roles of STIM-ORAI and IP3Rs may be over- or underestimated depending on the use of the pharmacologic inhibitors or siRNA-mediated gene knockdown approaches in cancer cells. Moreover, only a relatively limited amount of information concerning STIM-ORAI and IP3Rs is available to date due to their complicated and comprehensive functions in tumor cells. Despite a wealth of data describing their functions, the elucidation of their roles in cancer is still at the beginning stages. How STIM-ORAI and IP3Rs affect carcinogenesis in vivo, the relationship between these proteins and Ca2+ oscillations in cancer cells, and whether the participation of these proteins in the cancer procedure is a general mechanism need to be investigated.
References
Carafoli E. The calcium-signalling saga: tap water and protein crystals. Nat Rev Mol Cell Biol. 2003;4(4):326–32. doi:10.1038/nrm1073.
Kippert F. Endocytobiotic coordination, intracellular calcium signaling, and the origin of endogenous rhythms. Ann NY Acad Sci. 1987;503:476–95.
Tymianski M, Tator CH. Normal and abnormal calcium homeostasis in neurons: a basis for the pathophysiology of traumatic and ischemic central nervous system injury. Neurosurgery. 1996;38(6):1176–95.
Mijares A, Altamirano F, Kolster J, Adams JA, Lopez JR. Age-dependent changes in diastolic Ca(2+) and Na(+) concentrations in dystrophic cardiomyopathy: role of Ca(2 +) entry and IP3. Biochem Biophys Res Commun. 2014;452(4):1054–9. doi:10.1016/j.bbrc.2014.09.045.
Ljubojevic S, Radulovic S, Leitinger G, Sedej S, Sacherer M, Holzer M, et al. Early remodeling of perinuclear Ca2+ stores and nucleoplasmic Ca2+ signaling during the development of hypertrophy and heart failure. Circulation. 2014;130(3):244–55. doi:10.1161/CIRCULATIONAHA.114.008927.
Vigont VA, Zimina OA, Glushankova LN, Kolobkova JA, Ryazantseva MA, Mozhayeva GN, et al. STIM1 protein activates store-operated calcium channels in cellular Model of huntington’s disease. Acta Naturae. 2014;6(4):40–7.
Elsholz F, Harteneck C, Muller W, Friedland K. Calcium—a central regulator of keratinocyte differentiation in health and disease. Eur J Dermatol. 2014. doi:10.1684/ejd.2014.2452.
Pan Z, Ma J. Open Sesame: treasure in store-operated calcium entry pathway for cancer therapy. Sci China Life Sci. 2015;58(1):48–53. doi:10.1007/s11427-014-4774-3.
Berridge MJ. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium. 2002;32(5–6):235–49.
Putney JW. Alternative forms of the store-operated calcium entry mediators, STIM1 and Orai1. Curr Top Membr. 2013;71:109–23. doi:10.1016/B978-0-12-407870-3.00005-6.
Collins HE, Zhu-Mauldin X, Marchase RB, Chatham JC. STIM1/Orai1-mediated SOCE: current perspectives and potential roles in cardiac function and pathology. Am J Physiol Heart Circ Physiol. 2013;305(4):H446–58. doi:10.1152/ajpheart.00104.2013.
Dingsdale H, Voronina S, Haynes L, Tepikin A, Lur G. Cellular geography of IP3 receptors, STIM and Orai: a lesson from secretory epithelial cells. Biochem Soc Trans. 2012;40(1):108–11. doi:10.1042/BST20110639.
Michaelis M, Nieswandt B, Stegner D, Eilers J, Kraft R. STIM1, STIM2, and Orai1 regulate store-operated calcium entry and purinergic activation of microglia. Glia. 2015;63(4):652–63. doi:10.1002/glia.22775.
Kurosaki T, Baba Y. Ca2+ signaling and STIM1. Prog Biophys Mol Biol. 2010;103(1):51–8. doi:10.1016/j.pbiomolbio.2010.02.004.
Choi S, Maleth J, Jha A, Lee KP, Kim MS, So I, et al. The TRPCs-STIM1-Orai interaction. Handb Exp Pharmacol. 2014;223:1035–54. doi:10.1007/978-3-319-05161-1_13.
Fahrner M, Derler I, Jardin I, Romanin C. The STIM1/Orai signaling machinery. Channels (Austin). 2013;7(5):330–43. doi:10.4161/chan.26742.
Derler I, Fritsch R, Schindl R, Romanin C. CRAC inhibitors: identification and potential. Expert Opin Drug Discov. 2008;3(7):787–800. doi:10.1517/17460441.3.7.787.
Brandman O, Liou J, Park WS, Meyer T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell. 2007;131(7):1327–39. doi:10.1016/j.cell.2007.11.039.
Baba Y, Matsumoto M, Kurosaki T. Calcium signaling in B cells: regulation of cytosolic Ca2+ increase and its sensor molecules, STIM1 and STIM2. Mol Immunol. 2014;62(2):339–43. doi:10.1016/j.molimm.2013.10.006.
Shim AH, Tirado-Lee L, Prakriya M. Structural and functional mechanisms of CRAC channel regulation. J Mol Biol. 2015;427(1):77–93. doi:10.1016/j.jmb.2014.09.021.
Stathopulos PB, Ikura M. Structural aspects of calcium-release activated calcium channel function. Channels (Austin). 2013;7(5):344–53. doi:10.4161/chan.26734.
Sharp AH, Nucifora FJ, Blondel O, Sheppard CA, Zhang C, Snyder SH, et al. Differential cellular expression of isoforms of inositol 1,4,5-triphosphate receptors in neurons and glia in brain. J Comp Neurol. 1999;406(2):207–20.
Vermassen E, Parys JB, Mauger JP. Subcellular distribution of the inositol 1,4,5-trisphosphate receptors: functional relevance and molecular determinants. Biol Cell. 2004;96(1):3–17. doi:10.1016/j.biolcel.2003.11.004.
Michikawa T, Hamanaka H, Otsu H, Yamamoto A, Miyawaki A, Furuichi T, et al. Transmembrane topology and sites of N-glycosylation of inositol 1,4,5-trisphosphate receptor. J Biol Chem. 1994;269(12):9184–9.
Boehning D, Mak DO, Foskett JK, Joseph SK. Molecular determinants of ion permeation and selectivity in inositol 1,4,5-trisphosphate receptor Ca2+ channels. J Biol Chem. 2001;276(17):13509–12. doi:10.1074/jbc.C100094200.
Van Breemen C, Saida K. Cellular mechanisms regulating [Ca2+]i smooth muscle. Annu Rev Physiol. 1989;51:315–29. doi:10.1146/annurev.ph.51.030189.001531.
Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene. 2008;27(50):6407–18. doi:10.1038/onc.2008.308.
Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4(7):517–29. doi:10.1038/nrm1155.
Newmeyer DD, Ferguson-Miller S. Mitochondria: releasing power for life and unleashing the machineries of death. Cell. 2003;112(4):481–90.
Chambers KT, Unverferth JA, Weber SM, Wek RC, Urano F, Corbett JA. The role of nitric oxide and the unfolded protein response in cytokine-induced beta-cell death. Diabetes. 2008;57(1):124–32. doi:10.2337/db07-0944.
Motiani RK, Abdullaev IF, Trebak M. A novel native store-operated calcium channel encoded by Orai3: selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J Biol Chem. 2010;285(25):19173–83. doi:10.1074/jbc.M110.102582.
Motiani RK, Hyzinski-Garcia MC, Zhang X, Henkel MM, Abdullaev IF, Kuo YH, et al. STIM1 and Orai1 mediate CRAC channel activity and are essential for human glioblastoma invasion. Pflugers Arch. 2013;465(9):1249–60. doi:10.1007/s00424-013-1254-8.
Boutin B, Tajeddine N, Monaco G, Molgo J, Vertommen D, Rider M, et al. Endoplasmic reticulum Ca content decrease by PKA-dependent hyperphosphorylation of type 1 IP3 receptor contributes to prostate cancer cell resistance to androgen deprivation. Cell Calcium. 2015. doi:10.1016/j.ceca.2015.02.004.
Yang N, Tang Y, Wang F, Zhang H, Xu D, Shen Y, et al. Blockade of store-operated Ca(2 +) entry inhibits hepatocarcinoma cell migration and invasion by regulating focal adhesion turnover. Cancer Lett. 2013;330(2):163–9. doi:10.1016/j.canlet.2012.11.040.
Zhu H, Zhang H, Jin F, Fang M, Huang M, Yang CS, et al. Elevated Orai1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma. Oncotarget. 2014;5(11):3455–71.
Kim JH, Lkhagvadorj S, Lee MR, Hwang KH, Chung HC, Jung JH, et al. Orai1 and STIM1 are critical for cell migration and proliferation of clear cell renal cell carcinoma. Biochem Biophys Res Commun. 2014;448(1):76–82. doi:10.1016/j.bbrc.2014.04.064.
McAndrew D, Grice DM, Peters AA, Davis FM, Stewart T, Rice M, et al. ORAI1-mediated calcium influx in lactation and in breast cancer. Mol Cancer Ther. 2011;10(3):448–60. doi:10.1158/1535-7163.MCT-10-0923.
Stone RL, Baggerly KA, Armaiz-Pena GN, Kang Y, Sanguino AM, Thanapprapasr D, et al. Focal adhesion kinase: an alternative focus for anti-angiogenesis therapy in ovarian cancer. Cancer Biol Ther. 2014;15(7):919–29. doi:10.4161/cbt.28882.
Faouzi M, Kischel P, Hague F, Ahidouch A, Benzerdjeb N, Sevestre H, et al. ORAI3 silencing alters cell proliferation and cell cycle progression via c-myc pathway in breast cancer cells. Biochim Biophys Acta. 2013;1833(3):752–60. doi:10.1016/j.bbamcr.2012.12.009.
Ay AS, Benzerdjeb N, Sevestre H, Ahidouch A, Ouadid-Ahidouch H. Orai3 constitutes a native store-operated calcium entry that regulates non small cell lung adenocarcinoma cell proliferation. PLoS One. 2013;8(9):e72889. doi:10.1371/journal.pone.0072889.
Schmidt S, Liu G, Liu G, Yang W, Honisch S, Pantelakos S, et al. Enhanced Orai1 and STIM1 expression as well as store operated Ca2+ entry in therapy resistant ovary carcinoma cells. Oncotarget. 2014;5(13):4799–810.
Sabbioni S, Barbanti-Brodano G, Croce CM, Negrini M. GOK: a gene at 11p15 involved in rhabdomyosarcoma and rhabdoid tumor development. Cancer Res. 1997;57(20):4493–7.
Gueguinou M, Chantome A, Fromont G, Bougnoux P, Vandier C, Potier-Cartereau M. KCa and Ca(2+) channels: the complex thought. Biochim Biophys Acta. 2014;1843(10):2322–33. doi:10.1016/j.bbamcr.2014.02.019.
Dubois C, Vanden AF, Lehen’Kyi V, Gkika D, Guarmit B, Lepage G, et al. Remodeling of channel-forming ORAI proteins determines an oncogenic switch in prostate cancer. Cancer Cell. 2014;26(1):19–32. doi:10.1016/j.ccr.2014.04.025.
Motiani RK, Stolwijk JA, Newton RL, Zhang X, Trebak M. Emerging roles of Orai3 in pathophysiology. Channels (Austin). 2013;7(5):392–401. doi:10.4161/chan.24960.
Krishnan K, Khanna C, Helman LJ. The molecular biology of pulmonary metastasis. Thorac Surg Clin. 2006;16(2):115–24. doi:10.1016/j.thorsurg.2005.12.003.
Motiani RK, Zhang X, Harmon KE, Keller RS, Matrougui K, Bennett JA, et al. Orai3 is an estrogen receptor alpha-regulated Ca(2)(+) channel that promotes tumorigenesis. Faseb J. 2013;27(1):63–75. doi:10.1096/fj.12-213801.
Sobradillo D, Hernandez-Morales M, Ubierna D, Moyer MP, Nunez L, Villalobos C. A reciprocal shift in transient receptor potential channel 1 (TRPC1) and stromal interaction molecule 2 (STIM2) contributes to Ca2+ remodeling and cancer hallmarks in colorectal carcinoma cells. J Biol Chem. 2014;289(42):28765–82. doi:10.1074/jbc.M114.581678.
Kondratska K, Kondratskyi A, Yassine M, Lemonnier L, Lepage G, Morabito A, et al. Orai1 and STIM1 mediate SOCE and contribute to apoptotic resistance of pancreatic adenocarcinoma. Biochim Biophys Acta. 2014;1843(10):2263–9. doi:10.1016/j.bbamcr.2014.02.012.
Umemura M, Baljinnyam E, Feske S, De Lorenzo MS, Xie LH, Feng X, et al. Store-operated Ca2+ entry (SOCE) regulates melanoma proliferation and cell migration. PLoS One. 2014;9(2):e89292. doi:10.1371/journal.pone.0089292.
Chen YF, Chou CY, Wilkins RJ, Ellory JC, Mount DB, Shen MR. Motor protein-dependent membrane trafficking of KCl cotransporter-4 is important for cancer cell invasion. Cancer Res. 2009;69(22):8585–93. doi:10.1158/0008-5472.CAN-09-2284.
Chen YF, Chiu WT, Chen YT, Lin PY, Huang HJ, Chou CY, et al. Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proc Natl Acad Sci USA. 2011;108(37):15225–30. doi:10.1073/pnas.1103315108.
Yang S, Zhang JJ, Huang XY. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell. 2009;15(2):124–34. doi:10.1016/j.ccr.2008.12.019.
Pathak A, Kumar S. Biophysical regulation of tumor cell invasion: moving beyond matrix stiffness. Integr Biol (Camb). 2011;3(4):267–78. doi:10.1039/c0ib00095g.
Stock C, Ludwig FT, Hanley PJ, Schwab A. Roles of ion transport in control of cell motility. Compr Physiol. 2013;3(1):59–119. doi:10.1002/cphy.c110056.
Faouzi M, Hague F, Potier M, Ahidouch A, Sevestre H, Ouadid-Ahidouch H. Down-regulation of Orai3 arrests cell-cycle progression and induces apoptosis in breast cancer cells but not in normal breast epithelial cells. J Cell Physiol. 2011;226(2):542–51. doi:10.1002/jcp.22363.
Mancini M, Toker A. NFAT proteins: emerging roles in cancer progression. Nat Rev Cancer. 2009;9(11):810–20. doi:10.1038/nrc2735.
Tsunoda T, Koga H, Yokomizo A, Tatsugami K, Eto M, Inokuchi J, et al. Inositol 1,4,5-trisphosphate (IP3) receptor type1 (IP3R1) modulates the acquisition of cisplatin resistance in bladder cancer cell lines. Oncogene. 2005;24(8):1396–402. doi:10.1038/sj.onc.1208313.
Jan CR, Yu CC, Huang JK. Clomiphene, an ovulation-inducing agent, mobilizes intracellular Ca2+ and causes extracellular Ca2+ influx in bladder female transitional carcinoma cells. Horm Res. 2000;54(3):143–8.
Olive PL, Durand RE. Apoptosis: an indicator of radiosensitivity in vitro? Int J Radiat Biol. 1997;71(6):695–707.
Chan TO, Tsichlis PN. PDK2: a complex tail in one Akt. Sci STKE. 2001;2001(66):e1. doi:10.1126/stke.2001.66.pe1.
Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T, et al. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc Natl Acad Sci USA. 2005;102(1):105–10. doi:10.1073/pnas.0408352102.
Letai AG. Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. Nat Rev Cancer. 2008;8(2):121–32. doi:10.1038/nrc2297.
Li C, Wang X, Vais H, Thompson CB, Foskett JK, White C. Apoptosis regulation by Bcl-x(L) modulation of mammalian inositol 1,4,5-trisphosphate receptor channel isoform gating. Proc Natl Acad Sci USA. 2007;104(30):12565–70. doi:10.1073/pnas.0702489104.
Eckenrode EF, Yang J, Velmurugan GV, Foskett JK, White C. Apoptosis protection by Mcl-1 and Bcl-2 modulation of inositol 1,4,5-trisphosphate receptor-dependent Ca2+ signaling. J Biol Chem. 2010;285(18):13678–84. doi:10.1074/jbc.M109.096040.
Fujimoto T, Machida T, Tsunoda T, Doi K, Ota T, Kuroki M, et al. KRAS-induced actin-interacting protein regulates inositol 1,4,5-trisphosphate-receptor-mediated calcium release. Biochem Biophys Res Commun. 2011;408(2):214–7. doi:10.1016/j.bbrc.2011.03.112.
Monaco G, Beckers M, Ivanova H, Missiaen L, Parys JB, De Smedt H, et al. Profiling of the Bcl-2/Bcl-X(L)-binding sites on type 1 IP(3) receptor. Biochem Biophys Res Commun. 2012;428(1):31–5. doi:10.1016/j.bbrc.2012.10.002.
Zhong F, Davis MC, McColl KS, Distelhorst CW. Bcl-2 differentially regulates Ca2+ signals according to the strength of T cell receptor activation. J Cell Biol. 2006;172(1):127–37. doi:10.1083/jcb.200506189.
Distelhorst CW, Lam M, McCormick TS. Bcl-2 inhibits hydrogen peroxide-induced ER Ca2+ pool depletion. Oncogene. 1996;12(10):2051–5.
Vervloessem T, Yule DI, Bultynck G, Parys JB. The type 2 inositol 1,4,5-trisphosphate receptor, emerging functions for an intriguing Ca(2+)-release channel. Biochim Biophys Acta. 2015;1853(9):1992–2005. doi:10.1016/j.bbamcr.2014.12.006.
Kang SS, Han KS, Ku BM, Lee YK, Hong J, Shin HY, et al. Caffeine-mediated inhibition of calcium release channel inositol 1,4,5-trisphosphate receptor subtype 3 blocks glioblastoma invasion and extends survival. Cancer Res. 2010;70(3):1173–83. doi:10.1158/0008-5472.CAN-09-2886.
Shibao K, Fiedler MJ, Nagata J, Minagawa N, Hirata K, Nakayama Y, et al. The type III inositol 1,4,5-trisphosphate receptor is associated with aggressiveness of colorectal carcinoma. Cell Calcium. 2010;48(6):315–23. doi:10.1016/j.ceca.2010.09.005.
Selvaraj S, Sun Y, Sukumaran P, Singh BB. Resveratrol activates autophagic cell death in prostate cancer cells via downregulation of STIM1 and the mTOR pathway. Mol Carcinog. 2015. doi:10.1002/mc.22324.
Fujimoto T, Shirasawa S. Identification of KRAP-expressing cells and the functional relevance of KRAP to the subcellular localization of IP3R in the stomach and kidney. Int J Mol Med. 2012;30(6):1287–93. doi:10.3892/ijmm.2012.1126.
Xie R, Xu J, Wen G, Jin H, Liu X, Yang Y, et al. The P2Y2 nucleotide receptor mediates the proliferation and migration of human hepatocellular carcinoma cells induced by ATP. J Biol Chem. 2014;289(27):19137–49. doi:10.1074/jbc.M113.540047.
Xie R, Xu J, Wen G, Jin H, Liu X, Yang Y, et al. The P2Y2 nucleotide receptor mediates the proliferation and migration of human hepatocellular carcinoma cells induced by ATP. J Biol Chem. 2014;289(27):19137–49. doi:10.1074/jbc.M113.540047.
Yang S, Zhang JJ, Huang XY. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell. 2009;15(2):124–34. doi:10.1016/j.ccr.2008.12.019.
Pierro C, Cook SJ, Foets TC, Bootman MD, Roderick HL. Oncogenic K-Ras suppresses IP(3)-dependent Ca(2)(+) release through remodelling of the isoform composition of IP(3)Rs and ER luminal Ca(2)(+) levels in colorectal cancer cell lines. J Cell Sci. 2014;127(Pt 7):1607–19. doi:10.1242/jcs.141408.
Authors’ contributions
KQX organized the writing and revised the manuscript. JW drafted and revised the manuscript. YCH designed the figure. HHX and ZMS participated in revising the manuscript and the figure. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wen, J., Huang, YC., Xiu, HH. et al. Altered expression of stromal interaction molecule (STIM)-calcium release-activated calcium channel protein (ORAI) and inositol 1,4,5-trisphosphate receptors (IP3Rs) in cancer: will they become a new battlefield for oncotherapy?. Chin J Cancer 35, 32 (2016). https://doi.org/10.1186/s40880-016-0094-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40880-016-0094-2