
Abstract
Bacillus thuringiensis is the most widely used biological pesticide in the world. It belongs to the Bacillus cereus sensu lato group, which contains six species. Among these six species, B. thuringiensis, B. anthracis, and B. cereus have a low genetic diversity. B. thuringiensis strain HD521 shows maroon colony which is different from most of the B. thuringiensis strains. Strain HD521 also displays an ability to inhibit plant sheath blight disease pathogen (Rhizoctonia solani AG1 IB) growth and can form bipyramidal parasporal crystals consisting of three cry7 genes. These crystals have an insecticidal activity against Henosepilachna vigintioctomaculata larva (Coleoptera). Here we report the complete genome sequence of strain HD521, which has one chromosome and six circular plasmids.
Similar content being viewed by others


Introduction
The B. cereus sensu lato group has low genetic diversity when they are measured by multilocus sequence typing and 16S sequencing and some gene contents [1–3]. B. thuringiensis , B. anthracis , and B. cereus are considered one lineage of the B. cereus group [4]. The classification of these organisms is based on the differences in their phenotypes and in their pathological effects. The virulence genes are generally located on the plasmids, which obtained them through horizontal gene transfer. These genes give them different phenotypes and pathologies [1]. B. thuringiensis is a rod-shaped, Gram-positive, spore-forming bacterium. It produces parasporal protein crystals that show different insecticidal activities against multifarious insect larvae, and some of them exhibit cytocidal activity against cancer cells [5, 6]. B. thuringiensis can also produce antibiotics such as Zwittermycin A, which is used to enhance its insecticidal activity and inhibit pathogens fungi, oomycetes, and similar organisms [7–9]. The complete antibiotic biosynthesis gene cluster was first identified in the strain B. cereus UW85 [10]. The specific pathology against insects makes B. thuringiensis a mainstay of microbial insect control. Although 42 B. thuringiensis strains have been sequenced, gapless chromosomes and plasmids have only been obtained from 15 strains’ [11]. Here the complete genome sequence of B. thuringiensis strain HD521 is reported and an annotation and description of its genome features is provided. This may provide insight into the genomic diversity among B. thuringiensis , B. anthracis , and B. cereus and the mechanism by which the Zwittermycin A gene cluster was transferred between B. cereus and B. thuringiensis .
Organism information
Classification and features
B. thuringiensis strain HD521 was first isolated from soil sample of the United States [12]. It was obtained from Bacillus Genetic Stock Center (BGSC). Strain HD521 likes the majority of the B. thuringiensis strains, cells are Gram-positive and rod-shaped [5]. It is an aerobic, facultative anaerobic, motile and spore-forming bacterium, with growth temperatures from 10 to 48 °C and optimal growth at 28–35 °C and pH 4.9–8.0 with an optimal pH 7.0 [12–15]. Baumann [16] showed that B. thuringiensis strain HD521 utilizes D-glucose, D-ribose, trehalose, pyruvate, glycerol and L-serine and produces extracellular of amylase and gelatinase. Hydrolysis study shows that it has ability to hydrolyze starch, gelatin, glycogen and N-acetyl-glucosamine [17]. It exhibits maroon colonies and produces bipyramidal parasporal crystals during the stationary phase of its growth cycle, which consisted of three cry7 genes (Fig. 1a). Strain HD521 showed an ability to inhibit R. solani AG1 IB growth (Fig. 1b). SDS-PAGE analysis of spores and crystals mixtures showed the strain HD521 expression a major protein band of 130 kDa, which is consistent with the following analysis of its parasporal crystal gene (Fig. 1c). The key features of HD521 are showed in Table 1.

General characteristics of Bacillus thuringiensis strain HD521. a Scanning electron microscope (SEM) analysis of HD521 spores and parasporal crystals. b Antagonism assay of HD521 against Rhizoctonia solani subgroup AG1 IB on PDA medium. c SDS-PAGE analysis of spore-crystal suspension of HD521: lane M, molecular mass standard; lane 1, HD521
Fourteen strains and HD521 were chosen for phylogenetic analysis. They showed a sequence similarity of more than 97 % based on blast analysis [18]. A 16 s rRNA sequence from B. subtilis 168 was selected as outgroup. The maximum likelihood method was used to construct the phylogenetic tree and the phylogenetic relationship of these 15 strains is shown in Fig. 2. Phylogenetic tree shows that strain HD521 has a close genetic relationship to strain HD771. The bootstrap value of this Phylogenetic tree is very low because of the 16S rRNA nucleotide sequence divergence of the chosen strains is low which is accordance to the previous studies. Ash showed that 16S rRNA nucleotide sequences among B. cereus , B. thuringiensis and B. anthracis were high similar and exhibit more than 99 % similarity [19], and they are considered as a single species [4, 20, 21].

Neighbor-joining phylogenetic tree highlighting the position of B. thuringiensis HD521 relative to B. thuringiensis, B. anthracis, and B. cereus. The strains and their 16 s rRNA corresponding to the GenBank accession numbers given below: A) BTALH (B. thuringiensis str. AL Hakam) (CP000485.1): 9309–10762; B) BTYBT020 (B. thuringiensis serovar finitimus YBT-020) (CP002508.1): 9307–10866; C) BASterne (B. anthracis str. Sterne ) (AE017225.1): 9336–10845; D) BAAmesAncestor (B. anthracis str. ‘Ames Ancestor’) (AE017334.2): 29129–30635; E) BACDC684 (B. anthracis str. CDC684) (CP001215.1): 9207–10715; F) BCE33L (B. cereus E33L) (CP_000001.1): 9337–10846; G) BT9727 (B. thuringiensis serovar konkukian str. 97–27) (AE017355.1): 9337–10846; H) BCATCC14579 (B. cereus ATCC14579) (AE016877.1): 9186–10741; I) BTBMB171 (B. thuringiensis BMB171) (CP001903.1): 9197–10736; J) BTHD771 (B. thuringiensis HD-771) (CP003752.1): 4786891–4788450; K) BTHD521 (B. thuringiensis subsp. indiana HD521) (CP010106): 9198–10737; L) BCQ1 (B. cereus Q1) (CP000227.1): 9338–10843; M) BTCT43 (B. thuringiensis serovar chinensis CT-43) (CP001907.1):9201–10740; N) BTMC28 (B. thuringiensis MC28) (CP003687.1):1369440–1370979; O) BS168 (B. sublitis subsp. str. 168) (CM000487.1): 9808–11362
Genome sequencing information
Genome project history
Studies of cytological and biological activity have provided three reasons to select it for sequencing of its whole genome: 1) Strain HD521 produces maroon colonies, unlike most of the B. thuringiensis strains. It can also form bipyramidal parasporal crystals and shows insecticidal activity against the larva of Henosepilachna vigintioctomaculata (Coleoptera). 2) Strain HD521 shows an ability to inhibit the growth of the pathogenic fungus R. solani AG1 IB and to provide information regarding the mechanism of antibiotic gene cluster transfer between B. thuringiensis and B. cereus . 3) Until now, the genomes of only 15 strains of B. thuringiensis have been completed. No B. thuringiensis serovar Indiana strain has been fully sequenced. The complete sequence of HD521 may contribute to the evolution and comparative genomics of the B. thuringiensis and Bacillus cereus sensu lato group. The complete gapless chromosome sequence and sequences of 6 plasmids sequence have been deposited in GenBank under the accession numbers of CP010106, CP010107, CP010108, CP010109, CP010110, CP010111 and CP010112. A summary of the genome sequencing project information has been deposited in the Joint Genome Institute with MIGS version 2.0 under the ID of Gp0111431 [22]. The summary of the detail information is shown in Table 2.
Growth conditions and DNA preparation
One colony was picked from LB plate medium and growth in 50 ml LB fluid medium overnight at 180 rpm, 30 °C. Cells were collected by centrifugation and washed with 20 ml cold TES buffer twice (30 mM Tris base, 5 mM EDTA, 50 mM NaCl; pH = 8.0) and then resuspended in 7.2 ml TES buffer with 20 % sucrose, lysozyme (20 mg/ml) and RNase A (1 μl/ml) and then incubated at 37 °C for 3–4 h. 7.2 ml TES with 8 % sodium dodecyl sulfate (SDS) was added in the spheroplast suspension and incubated at 68 °C for 10 min. Then 3.6 ml of 3 M sodium acetate (PH = 4.8) was added and the total suspension was incubate at −20 °C for 30 min. The suspension was centrifuged at 18,000 × g for 20 min at 4 °C. Supernatant was transferred into a new centrifuge tube and then centrifuged at 18,000 × g for 20 min at 4 °C again. Two volumes of cold absolute ethanol were added to the supernatant and incubated at −20 °C for about 12 h. DNA was pelleted at 18,000 × g for 20 min at 4 °C and pellet was dissolved in 300 μl sterile double distilled water and stored at −20 °C for further use. All of the operations were according to the previous report [23].
Genome sequencing and assembly
The genome of HD521 was sequenced at Beijing Genomics Institute (BGI; Shenzhen, China) using Illumina HiSeq 2000 platform with two paired-End Modules. Sequencing of 500 bp paired-end modules gathered 3.88 M reads, about 58 fold coverage (0.36 Gb); sequencing of 180 bp paired-end modules gather 14.76 M reads, about 319 fold coverage (2 Gb). These data were de novo assembled with Velvet, version 1.2.10 [24]. The assembly finally resulted in 77 scaffolds. Possible circular scaffolds were verified by PCR. The precedence relationships among the remainder scaffolds were predicted by using Nucleotide BLAST with the beginning and the end sequences of each scaffold. A Fosmid library was constructed and used to confirm some long-distance connected relations between corresponding scaffolds. Gap closing was using primer walking. Finally, 186 correct sub-clones were used to close gaps among the possible connected scaffolds. Anteroposterior sequences of the gaps were used for primer design directly for inner gap closing, 13 sub-clones were used for inner gap closing. Finally one gapless chromosome and six plasmids were obtained.
Genome annotation
Open reading frames were called used GeneMarkS with the model parameter trained on the complete sequence [25]. The predicted ORFs were translated and searched in the National Center for Biotechnology Information non-redundant database and then annotated to PFAM, GO, KEGG, Swiss-Prot, COG, and TrEMBL databases. The NR, KEGG, Swiss-Prot, and TrEMBL databases were annotated using Blast and e-values of 1e-50, and each protein was selected using the best hit. PFAM was annotated using InterProScan, and GO was annotated using Blast2GO with the NR database annotation. The rRNA, tRNA, and sRNA were predicted using rRNAmmer, tRNAscan, and Rfam, respectively [26–28]. Genes with signal peptides and transmembrane helices were predicted using SignalP, version 3.0 and TMHMM, version 2.0 [29, 30]. CRISPER repeats were predicted by using CRISPRfinder [31, 32].
Genome properties
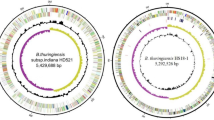
The genome of HD521 consisted of 7 replicons: a circular chromosome with a length of 5,429,688 bp (Fig. 3, Table 3). The G + C content of the circular chromosomes was 35.28 %. It included a predicted 5,538 genes 138 are RNA genes. Of these 5,400 genes, with a collective length of 4,544,493 bp, were protein-encoding genes. Table 4 displays the six circular plasmids pBTHD521-1, pBTHD521-2, pBTHD521-3, pBTHD521-4, pBTHD521-5 (Fig. 4a), and pBTHD521-6 (Fig. 4b). The G + C contents of the six plasmids ranged from 29.45 to 35.91 % and contained a total of 772 predicted genes. The plasmid pBTHD521-5 contained three cry7 genes, which can form bipyramidal parasporal crystals (data not shown). Among all the predicted genes, 3,323 were placed in 25 general COG function gene catalogs. The distribution of the predicted genes, which are annotated with COG functional categories, is presented in Table 5.

Circular representation of chromosome of HD521 performing relevant genome features. From outside to center; Genes on forward strand (colored genes annotated into COG categories), Genes on reverse strand (colored genes annotated into COG categories), G + C content (black), and G + C skew

Circular representation of plasmids pBTHD521-5 and pBTHD521-6. a and b Circular representation of plasmid pBTHD521-5 and pBTHD521-6 displaying relevant genome features. From outside to center: Genes on forward strand (dark red by COG categories), genes on reverse strand (green by COG categories), G + C content (black) and G + C skew. Red regions of pBTHD521-5 represent three cry7 genes: cry7Fb3 (KF672184), cry7Ga2, and cry7Da1; red region of pBTHD521-6 represents the Zwittermycin A gene cluster. c comparison of Zwittermycin A gene cluster between B. cereus UW85 and B. thuringiensis HD521 [10]. The figure of B. cereus UW85 Zwittermycin A gene cluster is cited from reference paper 10
Conclusions
B. thuringiensis , B. cereus , and B. anthracis have a close relationship as indicated by genomes and they are considered the same species [4]. Through the 15 complete B. thuringiensis genomes, 15 complete B. cereus genomes and 15 B. anthracis genomes, the B. thuringiensis , B. cereus , and B. anthracis genomes ranged in size from 5.3 Mbp to 6.7 Mbp (G + C content ranged from 34.91 to 35.41 %), 5.2 Mbp to 5.8 Mbp (G + C content 35.05 to 35.54 %), and 5.2 Mbp to 5.5 Mbp (G + C content 35.26 to 35.4 %), respectively [11, 33, 34]. B. thuringiensis has a larger genome than B. cereus and B. anthracis . The genome of B. thuringiensis strain HD521 consisted of a circular chromosome 5.4 Mbp in length and six plasmids ranging from 7,042 bp to 314,882 bp in size for a total genome 6.2 Mbp in length. There were 5,400 protein-encoding genes, 3,323 of which could be assigned to COG functional categories. Among these categories, 9.62 % of the genes were annotated to amino acid transport and metabolism, 9.27 % to transcription, 7.21 % to signal transduction mechanisms, 6.43 % to inorganic ion transport and metabolism, 5.78 % to inorganic ion transport and metabolism carbohydrate transport and metabolism, and 5.13 % to cell wall and membrane biogenesis. B. thuringiensis is an insect pathogen, B. cereus is an opportunistic human pathogen, and B. anthracis is a mammalian pathogen. The differences in their pathogenicity were caused by virulent components located in the plasmids. These were acquired by horizontal gene transfer [1, 4]. The pathogenicity of B. anthracis is caused by two plasmids, pXO1 and pXO2 [35]. B. thuringiensis and B. cereus are more similar to each other, the determinate difference is the insecticidal toxin genes, which are usually located on plasmids [4]. B. thuringiensis HD521 contains six plasmids, named pBTHD521-1 through pBTHD521-6. These plasmids each contain 11, 70, 89, 103, 243, and 256 protein-coding genes. The G + C content of these six plasmids ranged from 29.45 to 35.91 %. G + C contents of plasmid pBTHD521-1 and pBTHD521-4 were 29.45 and 29.79 %, which were markedly lower than the general G + C content (34.91 to 35.41 %) of B. thuringiensis . It is postulated that pBTHD521-1 and pBTHD521-4 were obtained by B. thuringiensis HD521 through horizontal gene transfer.
B. thuringiensis strain HD521 forms bipyramidal parasporal crystals and has an insecticidal activity against Henosepilachna vigintioctomaculata larva (Coleoptera). SDS-PAGE analysis of spore-crystal suspension showed HD521 express one major protein band of 130 kDa which is encoded by three cry7 genes located on plasmid pBTHD521-5. One cry7-like gene showed 99 % identity to cry7Fb genes. It was named cry7Fb3 by Delta-Endotoxin Nomenclature Committee. The other two cry7-like genes have a 100 % homology to cry7Ga2 and cry7Da1. B. thuringiensis strain HD521 also has an ability to inhibit the growth of plant sheath blight disease pathogen (R. solani AG1 IB). A Zwittermycin A gene cluster was found on plasmid pBTHD521-6. There were more than 56 kbp sequences matched to Zwittermycin B. cereus UW85 was found to produce a specific gene cluster sequence (FJ430564.1, 65 kbp) and this sequence included its main gene components. This indicated that strain HD521 has utility as a biocontrol agent not only against insect larva but also against plant disease. The complete genome sequence of HD521 may provide another model to study pathogenicity against pests, plant disease, and phylogenesis among Bacillus cereus sensu lato group.
References
Zwick ME, Joseph SJ, Didelot X, Chen PE, Bishop-lilly KA, Stewart AC, et al. Genomic characterization of the Bacillus cereus sensu lato species: Backdrop to the evolution of Bacillus anthracis. Genome Res. 2012;22:1512–24.
Priest FG, Barker M, Baillie LW, Holmes EC, Maiden MCJ. Population structure and evolution of the Bacillus cereus group. J Bacteriol. 2004;186:7959–70.
Daffonchio D, Cherif A, Brusetti L, Rizzi A, Mora D, Boudabous A, et al. Nature of polymorphisms in 16S-23S rRNA gene intergenic transcribed spacer fingerprinting of Bacillus and related genea. Appl Environ Microbiol. 2003;169:5128–37.
Helgason E, Økstad OA, Caugant DA, Johansen HA, Fouet A, Mock M, et al. Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis—One specis on the basis of genetic evidence. Appl Environ Microbiol. 2000;66:2627–30.
Schnepf E, Crickmore N, Rie JV, Lereclus D, Baum J, Feitelson J, et al. Bacillus thuringiensis and its pesticidal crystal proteins. Microbiol Mol Biol Rev. 1998;62:775–806.
Yamashita S, Akao T, Mizuki E, Saitoh H, Higuchi K, Park YS, et al. Characterization of the anti-cancer-cell parasporal protein of a Bacillus thuringiensis isolate. Can J Microbiol. 2000;46:913–9.
Shao TM, Bai L, Zhang J, Wang GG, Liu DQ, Li ZF, et al. A Nonribosomal Peptide synthetase gene tzw 1 is involved in Zwittermicin A biosynthesis in Bacillus thuringiensis G03. Curr Microbiol. 2008;57:61–5.
Broderick NA, Goodman RM, Raffa KF, Handelsman J. Synergy between zwittermicin A and Bacillus thuringiensis subsp. kurstakiagainst gypsy moth (Lepidoptera: Lymantriidae). Environ Entomol. 2000;29:101–7.
Silo-Suh LA, Stabb EV, Raffel SJ, Handelsman J. Target range of zwittermicin A, an aminopolyol antibiotic from Bacillus cereus. Curr Microbiol. 1998;37:6–11.
Kevany BM, Rasko DA, Thomas MG. Characterization of the complete Zwittermicin A biosynthesis gene cluster from Bacillus cereus. Appl Environ Microbiol. 2009;75:1144–55.
National Center for Biotechnology Information of Bacillus thuringiensis Genome Database [http://www.ncbi.nlm.nih.gov/genome/genomes/486]
Delucca II AJ, Simonson JG, Larson AD. Bacillus thuringiensis distribution in soil of the United States. Can J Microbiol. 1981;27:865–70.
Vilas-Bôas DT, Peruca APS, Arantes OMN. Biology and taxonomy of Bacillus cereus, Bacillus anthracis, and Bacillus thuringiensis. Can J Microbiol. 2007;53:673–87.
Barjac HD, Frachon E. Classification of Bacillus thuringiensis strains. Entomophaga. 1990;35:233–40.
West AW, Burges HD, Dixon TJ, Wyborn CH. Survival of Bacillus thuringiensis and Bacillus cereus spore inocula in soil: effects of pH, moisture, nutrient availability and indigenous microoganisms. Soil Biol Biochem. 1985;17:657–65.
Baumann L, Okamoto K, Unterman BM, Lynch M, Baumann P. Phenotypic characterization of Bacillus thuringiensis and Bacillus cereus. J Invertebr Pathol. 1984;44:329–41.
Lecadet MM, Frachon E, Dumanoir VC, Ripouteau H, Hamon S, Laurent P, et al. Updating the H-antigen classification of Bacillus thuringiensis. J Appl Microbiol. 1999;86:660–72.
Altschul SF, Gish W, Miller W, Myers E, Lipman DJ. Basic Local aligment search tool. J Mol Biol. 1990;215:403–10.
Ash C, Farrow JAE, Dorsch M, Stackebrandt E, Collins M. Comparative analysis of Bacillus anthracis, Bacillus cereus, and related species on the basis of reverse transcriptase sequencing of 16S RNA. Int J Syst Bacteriol. 1991;41:343–6.
Bavykin SG, Lysov YP, Zakhariev V, Kelly JJ, Jackman J, Stahl DA, et al. Use of 16S rRNA, 23S rRNA, and gyrB gene sequence analysis to determine phtlogenetic relationships of Bacillus cereus group microorganisms. J Clin Microbiol. 2004;42:3711–30.
Helgason E, Caugant DA, Olsen I, Kolstø AB. Genetic structure of population of Bacillus cereus and B. thuringiensis isolates associated with periodontitis and other human infections. J Clin Microbiol. 2000;38:1615–22.
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, et al. Minimum Information about a Genome Sequence (MIGS) specification. Nat Biotechnol. 2008;26:541–7.
Reyes-Ramirez A, Ibarra JE. Plasmid patterns of Bacillus thuringiensis type strains. Appl Environ Microbiol. 2008;74:125–9.
Zerbino DR, Birney E. Velvet: Algorithms for de novo short read assembly using de Bruiin graphs. Genome Res. 2008;18:821–9.
Besemer J, Lomsadze A, Borodovsky M. GeneMarks: a self-traning method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001;29:2607–18.
Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–8.
Lowe TM, Eddy SR. tRNAscanSE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–64.
Griffiths-Jones S, Moxon S, Marshall M, Khanna A, Eddy SR, Bateman A. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005;33:D121–4.
Bendtsen JD, Nielsen H, Heijine GV, Brunak S. Improved prediction of signal peptides: signal 3.0. J Mol Biol. 2004;340:783–95.
Krogh A, Larsson B, Heijine GV, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden markov model: application to complete genomes. J Mol Biol. 2001;305:567–80.
Grissa I, Vergnaud G, Pourcel C. CRISPRFinder:a web tool to identify clutered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35:W52–7.
CRISPRfinder [http://crispr.u-psud.fr/Server/]
National Center for Biotechnology Information of Bacillus cereus Genome Database [http://http://www.ncbi.nlm.nih.gov/genome/genomes/157]
National Center for Biotechnology Information of Bacillus anthracis Genome Database [http://www.ncbi.nlm.nih.gov/genome/genomes/181]
Okinaka RT, Cloud K, Hampton O, Hoffmaster AR, Hill KK, Peim P, et al. Sequence and organization of pXO1, the large Bacillus anthracis plasmid harboring the anthrax toxin genes. J Bacteriol. 1999;181:6509–15.
Woese CR, Kandler O, Wheelis ML. Towards a nature system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A. 1990;87:4576–9.
Gibbons NE, Murray RGE. Proposals Concerning the Higher Taxa of Bacteria. Int J Syst Bacteriol. 1978;28:1–6.
List Editor. Validation of the publication of new names and new combinations previously effectively published outside the IJSB. List No. 25. Int J Syst Bacteriol. 1988;38:220–2.
Murray RGE. The Higher Taxa, or, a Place for Everything. In: Holt JG, editor. Bergey's Manual of Systematic Bacteriology, vol. 1. 1st ed. Baltimore: The Williams and Wilkins Co.; 1984. p. 31–4.
Skerman VBD, McGowan V, Sneath PHA. Approved Lists of Bacterial Names. Int J Syst Bacteriol. 1980;30:225–420.
Prévot AR. In: Hauderoy P, Ehringer G, Guillot G, Magrou J, Prévot AR, Rosset D, Urbain A, editors. Dictionnaire des Bactéries Pathogènes. Paris: Masson et Cie; 1953. p. 1–692.
Fischer A. Untersuchungen über Bakterien. Jahrbücher für Wissenschaftliche Botanik. 1895;27:1–163.
Cohn F. Untersuchungen über Bakterien. Beitr Biol Pflanz. 1872;1:127–224.
Berliner E. Über die Schlaffsucht der Mehlmottenraupe (Ephestia kuhniella Zell) und ihren Erreg er Bacillus thuringiensis n. sp. Zeitschrift für angewandte Entomologie Berlin. 1915;2:29–56.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25–9.
Acknowledgements
This study was supported by the Chinese Major Project to Create New Crop Varieties Using Gene Transfer Technology (2011ZX08009-003-001-009) for transgenic research and the Creation of New Security insect-resistant transgenic rice materials (2014ZX08001001-002-003).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
The experiments and the writing of the manuscript were performed by QL. The genome assembling, annotation and analysis and the editing of the manuscript was performed by LZX, TZ, AP and HGH. The design and support of all the manuscript was performed by PL and APZ. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Li, Q., Xu, L.Z., Zou, T. et al. Complete genome sequence of Bacillus thuringiensis strain HD521. Stand in Genomic Sci 10, 62 (2015). https://doi.org/10.1186/s40793-015-0058-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40793-015-0058-1