
Abstract
Six strains of Enterococcus faecalis (S1, S12, S17, S18, S19 and S32) were isolated from copper fed pigs in Denmark. These Gram-positive bacteria within the genus Enterococcus are able to survive a variety of physical and chemical challenges by the acquisition of diverse genetic elements. The genome of strains S1, S12, S17, S18, S19 and S32 contained 2,615, 2,769, 2,625, 2,804, 2,853 and 2,935 protein-coding genes, with 41, 42, 27, 42, 32 and 44 genes encoding antibiotic and metal resistance, respectively. Differences between Cu resistant and sensitive E. faecalis strains, and possible co-transfer of Cu and antibiotic resistance determinants were detected through comparative genome analysis.
Similar content being viewed by others

Introduction
Copper is an essential trace element with an ubiquitous cellular distribution and performs several biological functions [1]. It serves as an important structural component or catalytic co-factor for a wide range of different enzymes in various important biochemical pathways in bacteria, plants and animals [2]. Because Cu, among many other micronutrients, is beneficial for growth promotion and feed efficiency of farm animals [3, 4], it is extensively used as an additive in swine feed. Normally, the concentration of Cu used in animal feed is in excess of the nutritional requirements of animals as it is used as an alternative to in-feed antibiotics for prevention of diarrheal disease [5]. Therefore, enteric bacteria, both commensal and pathogenic, in these animals have typically acquired several additional Cu resistance determinants to survive its toxicity [1, 6, 7].
Enterococci belong to the gastrointestinal flora of humans and animals, and have been known for more than a century for their pathogenicity to humans, causing urinary tract and surgical wound infections, bacteraemia and endocarditis [8]. Currently, more than 30 species within the genus Enterococcus have been described, and the two most studied enterococcal species are Enterococcus faecium and Enterococcus faecalis [9]. Over the last two decades, E. faecalis and E. faecium have become increasingly important nosocomial pathogens worldwide and are difficult treat due to their increasing multidrug resistance [10]. The intrinsic resistance of Enterococcus to many antibiotics and its acquisition of resistance determinants to other antimicrobial agents led to the emergence of Enterococcus as a nosocomial pathogen [11, 12]. Recently, the co-selection of MDR isolates by antibiotics, metals and biocides has been reported [13, 14], and the resistance of Enterococcus to both Cu and antibiotics has been established [15, 16]. However, few studies have addressed gene transfer and the underlying molecular mechanisms of the various Cu resistance determinants in E. faecalis [17]. Herein, we present the genome sequences along with the main features of six E. faecalis strains showing the differences between Cu resistant and sensitive strains of E. faecalis , and suggesting possible co-transfer of Cu and antibiotic resistance determinants in these bacteria.
Organism information
Classification and Features
Phylogenetic analysis was performed using the 16S rRNA gene sequences on the six strains S1, S12, S17, S18, S19 and S32 and related species. Sequences were aligned using Clustal W, and a phylogenetic tree was constructed using neighbor-joining (NJ) method implemented in MEGA version 6.0. The resultant tree topologies were evaluated by bootstrap analyses with 1,000 random samplings. Phylogenetic analysis based on 16S rRNA gene sequences showed that the six strains clustered together with E. faecalis ATCC 29212 and E. faecalis SFL with a high bootstrap value (100 %). All the E. faecalis are in a distinct branch with the other enterococci, such as E. casseliflavus , E. faecium , E. hirae and the another pig gut Firmicute, that is Streptococcus equinus NCDO 1037 (Fig. 1). The six strains could be classified as members of the genus Enterococcus based on their 16S rRNA gene phylogeny and phenotypic characteristics (Table 1).

Phylogenetic tree highlighting the position of the six E. faecalis strains relative to phylogenetically closely related type strains within the genus Enterococcus. The sequences were aligned using Clustal W, and the neighbor-joining tree was constructed based on kimura 2-parameter distance model using MEGA 6.0. Bootstrap values above 50 % are shown obtained from 1,000 bootstrap replications. Bar, 0.02 substitutions per nucleotide position. GenBank accession numbers are displayed in parentheses. Large triangles represent the six Enterococcus strains sequenced in this study
E. faecalis is a Gram-positive, oval-shaped, and often highly pathogenic bacterium classified as a member of the genus Enterococcus (Table 1 and Fig. 2) [18, 19]. It is a natural inhabitant of the mammalian gastrointestinal tract and is commonly found in soil, sewage, water and food [8]. E. faecalis is quite versatile and able to survive a variety of physical and chemical challenges by the acquisition of diverse genetic elements, which may contribute to their adaption to different hosts and environments [20, 21]. They are able to grow in temperatures ranging from 0 °C up to 50 °C, and can survive in the presence of 6.5 % NaCl and in broth at pH 9.6 [22]. They can also be resistant to heavy and transition metals [17], as well as many different antibiotics [23–25], especially vancomycin [20, 21].

Micrograph of E. faecalis strains obtained by scanning electron microscopy. Scale bar, 4 m
Genome sequencing information
Genome project history
The E. faecalis strains (S1, S12, S17, S18, S19 and S32) were isolated from Cu-fed pigs as part of the Danish Integrated Antimicrobial Resistance Monitoring (DANMAP) surveillance program [23]. The isolates were collected from healthy animals at or just prior to slaughter. Those whole-genome shotgun projects have been deposited in DDBJ/EMBL/GenBank under the accession number JTKS00000000, JTKT00000000, JTKU00000000, JTKV00000000, JTKW00000000 and JTKX00000000. Table 2 presents the project information and its association with MIGS version 2.0 compliance [26]. Cu resistant strains are E. faecalis strains S1, S18, S32, while the other three strains are Cu sensitive.
Growth conditions and genomic DNA preparation
E. faecalis were streaked on Slanetz agar (BD Difco) plates and grown for 48 h at 42 °C. Each strain was inoculated separately into 25 ml of brain heart infusion broth at 37 °C for 24 h. Genomic DNA was purified from the isolates using the Easy-DNA extraction kit (Invitrogen), and DNA concentrations were determined by the Qubit dsDNA BR assay kit (Invitrogen).
Genome sequencing and assembly
Whole genome sequencing of E. faecalis strains S1, S12, S17, S18, S19 and S32 was carried out on an Illumina Miseq platform (Illumina, Inc., San Diego, CA). Genomic libraries were prepared by the Nextera XT DNA sample preparation kit (Illumina, cat. No. FC-131-1024), and then sequenced using v3, 2 × 300 bp chemistry on the Illumina MiSeq platform. Genomic assemblies were constructed using Velvet version 1.1.04, generating 24, 57, 20, 103, 34 and 89 contigs, respectively.
Genome annotation
The resulting contigs were uploaded onto the Rapid Annotation using Subsystem Technology server databases and the gene-caller GLIMMER 3.02 [27, 28] to predict open reading frames. The predicted ORFs were translated and annotated by searching against clusters of orthologous groups using the SEED databases [29], as well as NCBI databases. RNAmmer 1.2 [30] and tRNAscan SE 1.23 [31] were used to identify rRNA genes and tRNA genes, respectively. CRISPR repeats were examined using CRISPR recognition tool (CRT) [32].
Genome properties
Whole genome sequencing of E. faecalis strains S1, S12, S17, S18, S19 and S32 resulted in 156, 162, 240, 84, 172 and 200 fold coverage of the genomes, respectively. The draft genome sizes were 2,762,808, 2,896,725, 2,786,673, 2,888,656, 2,969,229 and 3,037,709 bp in length, with an average GC content of 37.6, 37.4, 37.5, 37.4, 37.2 and 37.2 %, respectively, and comprises 2,615; 2,769; 2,625; 2,804; 2,853 and 2,935 protein coding sequences, respectively. Of the protein coding genes, 2,002; 2,006; 1,949; 2,001; 2,058 and 2,073 were genes with function predictions, with 41, 42, 27, 42, 32 and 44 genes responsible for antibiotics and toxic compounds resistant, respectively. There are 52 (4 rRNA genes and 48 tRNA genes), 54 (3 rRNA genes and 51 tRNA genes), 48 (3 rRNA genes and 45 tRNA genes), 52 (4 rRNA genes and 48 tRNA genes), 53 (3 rRNA genes and 50 tRNA genes) and 55 (5 rRNA genes and 50 tRNA genes) RNA genes for strains S1, S12, S17, S18, S19 and S32, respectively. The properties and statistics for the genome are summarized in Table 3. The distribution of genes into COG functional categories is presented in Table 4 and Fig. 3.

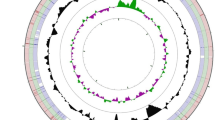
Graphical circular map of the genome comparison of E. faecalis S32 with the other five strains. Labeling from the outside to the inside circle: ring 1 and 4 show the protein coding genes on the forward/reverse strand (colored by COG categories); ring 2 and 3 show the denote genes on the forward/reverse strand; ring 5, 6, 7, 8 and 9 show the CDS vs CDS BLAST results of E. faecalis S32 with S1, S18, S12, S19 and S17, respectively; ring 10 shows the G + C content (peaks out/inside the circle indicate values higher or lower than the average G + C content, respectively); ring 11 shows GC skew (calculated as (G - C)/(G + C), peaks out/inside the circle indicates values higher or lower than 1, respectively). Ring 5–9 were arranged based on the CDS BLAST results, with the similarity rank from high to low, that is S1 and S18 were more similar to the reference strain S32 than the other three strains
Insights from the genome sequence
All of the six strains contain a four gene operon, copYZAB, encoding a Cu resistance determinant (Table 5), which was initially observed in the Gram-positive bacterium E. hirae [33]. CopA and CopB are P-type ATPases responsible for ATP-dependent Cu+ transport across the cytoplasmic membranes. The Cu chaperone CopZ binds two Cu+ atoms in a solvent accessible manner, presumably to facilitate their transfer to the transcriptional regulator CopY. Upon binding Cu+, CopY undergoes a conformational change and is released from the copA operator allowing expression of the copYZAB operon [1]. A gene encoding the cytoplasmic Cu homeostasis protein CutC was identified in all six strains (Table 5), and CutC has been demonstrated to be involved in Cu homeostasis in E. faecalis [34]. In addition, another possible gene encoding a putative Cu+-translocating P-type ATPase, was identified in all six strains named ctpA in this study (Table 5). The genome comparisons of the six E. faecalis strains using E. faecalis S32 as the reference strain by CGview comparison tool [35] indicated that S1 and S18 were more similar to the reference strain S32 than the other three strains (Fig. 3).
The tcrYAZB operon was initially identified on the pA17sv1 plasmid in E. faecium , which also carried genes encoding resistance to erythromycin (ermB) and vancomycin (vanA) [17, 36]. High toxic Cu levels could be tolerated due to the presence of tcrB in E. faecium or E. faecalis which encodes a Cu+-translocating P-type ATPase homologous to CopB encoded on copYZAB operon [37]. Comparing these six E. faecalis strains against others previously identified with increased Cu resistance, the tcrYAZB operon and adjacent cueO encoding a multicopper oxidase were only identified in E. faecalis S1, S18 and S32 (Table 5). Blasting of the tcrYAZB operon against the contigs of the other three strains verified that they were indeed lacking Cu resistance genes. The cueO gene identified in putative copper resistant strains encodes a multicopper oxidase that is transported across the cytoplasmic membrane and oxidizes Cu(I) to Cu(II) and so aids protection from high Cu concentrations in Enterococcus [9] or other Gram-positive strains [16]. The approximate 20-gene copper pathogenicity/fitness island present in E. faecalis S1, S18 and S32, show cueO is located in close vicinity of tcrYAZB and probably regulated by an adjacent two-component regulator system (Cu(I)-sensing regulator (cusR) and Cu(I)-sensing sensor (cusS)) (Fig. 4). Transposase and mobile element protein genes were also identified on this pathogenicity/fitness island next to tcrYAZB, indicating mobility. Moreover, genes encoding prolipoprotein diacylglyceryl transferase, which is responsible for oxidative stress tolerance potentially also caused by Cu+, could be identified on these potential pathogenicity and/or fitness islands as well. For the other three Cu sensitive E. faecalis S12, S17 and S19, tcrYAZB, cueO, cusR, cusS or genes encoding a prolipoprotein diacylglyceryl transferase could not be detected.

Cu pathogenicity island in E. faecalis S1, S18 and S32. a: prolipoprotein diacylglyceryl trPropertyansferase, b: intergral membrane protein, c: chaperone, d: hypothetical protein, e:transposase, f: disrupted P-type ATPase, g: integrase, h: adenylate kinase, i: resolvase, copY: CopY family transcriptional regulator, cueO: multicopper oxidase, cusR: Cu(I)-sensing regulator, cusS: Cu(I)-sensing sensor, tcrY: tcrYAZB operon regulator, tcrA: putative copper-efflux CPx-type ATPase, tcrB: Cu+-translocating CPx-type ATPase, tcrZ: putative chaperone
The antibiotic resistance gene tetM (resistance to tetracycline) could be identified in the three Cu resistant E. faecalis S1, S18, S32, and Cu sensitive E. faecalis S12; vanA (encoding vancomycin resistance) was identified only in Cu resistant E. faecalis S32; streptothricin acetyltransferase gene was identified in the Cu resistant E. faecalis S1, S18, S32; and aminoglycoside adenylyltransferase gene was identified in two Cu resistant E. faecalis S1 and S18 (Table 5).
Conclusions
Since the co-transfer of genes encoding antibiotic resistance along with Cu tolerance genes in one transconjugant has been demonstrated [14], the results in this study might provide valuable information corroborating the co-transfer of genes encoding additional Cu resistance and genes encoding numerous antibiotic resistances. Also, the identified antibiotic resistance gene tetM in all the Cu resistant strains is consistent with the MDR Enterococcus strains observed in the environment [13–16].
Abbreviations
- MDR:
-
Multidrug-resistant
References
Samanovic MI, Ding C, Thiele DJ, Darwin KH. Copper in microbial pathogenesis: meddling with the metal. Cell Host Microbe. 2012;11(2):106–15.
Yazdankhah S, Rudi K, Bernhoft A. Zinc and copper in animal feed–development of resistance and co-resistance to antimicrobial agents in bacteria of animal origin. Microb Ecol Health Dis. 2014;25.
Cunha T. Swine feeding and nutrition. New York: Elsevier; 2012.
Jacob ME, Fox JT, Nagaraja T, Drouillard JS, Amachawadi RG, Narayanan SK. Effects of feeding elevated concentrations of copper and zinc on the antimicrobial susceptibilities of fecal bacteria in feedlot cattle. Foodborne Pathogens Dis. 2010;7(6):643–8.
Monteiro SC, Lofts S, Boxall A. Pre-assessment of environmental impact of zinc and copper used in animal nutrition. 2010.
Hodgkinson V, Petris MJ. Copper homeostasis at the host-pathogen interface. J Biol Chem. 2012;287(17):13549–55.
Amachawadi R, Shelton N, Shi X, Vinasco J, Dritz S, Tokach M, et al. Selection of fecal enterococci exhibiting tcrB-mediated copper resistance in pigs fed diets supplemented with copper. Appl Env Microbiol. 2011;77(16):5597–603.
Murray BE. The life and times of the Enterococcus. Clin Microbiol Rev. 1990;3(1):46–65.
van Schaik W, Top J, Riley DR, Boekhorst J, Vrijenhoek JE, Schapendonk CM, et al. Pyrosequencing-based comparative genome analysis of the nosocomial pathogen Enterococcus faecium and identification of a large transferable pathogenicity island. BMC Genomics. 2010;11(1):239.
Willems RJ, Top J, van Schaik W, Leavis H, Bonten M, Sirén J, et al. Restricted gene flow among hospital subpopulations of Enterococcus faecium. Mbio. 2012;3(4):e00151–00112.
Paulsen I, Banerjei L, Myers G, Nelson K, Seshadri R, Read T, et al. Role of mobile DNA in the evolution of vancomycin-resistant Enterococcus faecalis. Science. 2003;299(5615):2071–4.
de Regt MJ, van Schaik W, van Luit-Asbroek M, Dekker HA, van Duijkeren E, Koning CJ, et al. Hospital and community ampicillin-resistant Enterococcus faecium are evolutionarily closely linked but have diversified through niche adaptation. PLoS One. 2012;7(2):1–9.
Novais C, Freitas AR, Silveira E, Antunes P, Silva R, Coque TM, et al. Spread of multidrug-resistant Enterococcus to animals and humans: an underestimated role for the pig farm environment. J Antimicrob Chemother. 2013;1–9.
Silveira E, Freitas AR, Antunes P, Barros M, Campos J, Coque TM, et al. Co-transfer of resistance to high concentrations of copper and first-line antibiotics among Enterococcus from different origins (humans, animals, the environment and foods) and clonal lineages. J Antimicrob Chemother. 2014;69(4):899–906.
Hasman H, Kempf I, Chidaine B, Cariolet R, Ersbøll AK, Houe H, et al. Copper resistance in Enterococcus faecium, mediated by the tcrB gene, is selected by supplementation of pig feed with copper sulfate. Appl Environ Microbiol. 2006;72(9):5784–9.
Solioz M, Abicht HK, Mermod M, Mancini S. Response of Gram-positive bacteria to copper stress. JBIC J Biological Inorganic Chem. 2010;15(1):3–14.
Hasman H. The tcrB gene is part of the tcrYAZB operon conferring copper resistance in Enterococcus faecium and Enterococcus faecalis. Microbiol. 2005;151(9):3019–25.
Schleifer K, Kraus J, Dvorak C, Kilpper-Bälz R, Collins M, Fischer W. Transfer of Streptococcus lactis and related streptococci to the genus Lactococcus gen. nov. Syst Appl Microbiol. 1985;6(2):183–95.
Devriese L, Baele M, Butaye P. The genus Enterococcus. In: The Prokaryotes: Volume 4: Bacteria: Firmicutes, Cyanobacteria. 2006. p. 163–74.
Arias CA, Murray BE. The rise of the Enterococcus: beyond vancomycin resistance. Nat Rev Microbiol. 2012;10(4):266–78.
Cattoir V, Leclercq R. Twenty-five years of shared life with vancomycin-resistant enterococci: is it time to divorce? J Antimicrob Chemother. 2013;68(4):731–42.
Gardini F, Martuscelli M, Caruso MC, Galgano F, Crudele MA, Favati F, et al. Effects of pH, temperature and NaCl concentration on the growth kinetics, proteolytic activity and biogenic amine production of Enterococcus faecalis. Int J Food Microbiol. 2001;64(1):105–17.
DMAMAP. Use of antimicrobial agents and the occurrence of antimicrobial resistance in bacteria from food animals, foods and humans in Denmark. 2005.
Mazaheri Nezhad Fard R, Heuzenroeder MW, Barton MD. Antimicrobial and heavy metal resistance in commensal enterococci isolated from pigs. Vet Microbiol. 2011;148(2):276–82.
Kim J, Lee S, Choi S. Copper resistance and its relationship to erythromycin resistance in Enterococcus isolates from bovine milk samples in Korea. J Microbiol. 2012;50(3):540–3.
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26(5):541–7.
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9(1):75.
Meyer F, Paarmann D, D'Souza M, Olson R, Glass EM, Kubal M, et al. The metagenomics RAST server–a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics. 2008;9(1):386.
Hemmerich C, Buechlein A, Podicheti R, Revanna KV, Dong Q. An Ergatis-based prokaryotic genome annotation web server. BMC Bioinformatics. 2010;26(8):1122–4.
Lagesen K, Hallin P, Rødland EA, Stærfeldt H-H, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35(9):3100–8.
Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):0955–64.
Bland C, Ramsey TL, Sabree F, Lowe M, Brown K, Kyrpides NC, et al. CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics. 2007;8(1):209.
Odermatt A, Suter H, Krapf R, Solioz M. An ATPase operon involved in copper resistance by Enterococcus hirae. Ann NY Acad Sci. 1992;671:484.
Latorre M, Olivares F, Reyes-Jara A, López G, González M. CutC is induced late during copper exposure and can modify intracellular copper content in Enterococcus faecalis. Biochem Bioph Res Co. 2011;406(4):633–7.
Grant JR, Arantes AS, Stothard P. Comparing thousands of circular genomes using the CGView Comparison Tool. BMC Genomics. 2012;13(1):202.
Hasman H, Aarestrup FM. tcrB, a gene conferring transferable copper resistance in Enterococcus faecium: occurrence, transferability, and linkage to macrolide and glycopeptide resistance. Antimicrob Agents Chemother. 2002;46(5):1410–6.
Amachawadi RG, Shelton NW, Jacob ME, Shi X, Narayanan SK, Zurek L, et al. Occurrence of tcrB, a transferable copper resistance gene, in fecal enterococci of swine. Foodborne Pathog Dis. 2010;7(9):1089–97.
Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci. 1990;87(12):4576–9.
Schleifer K-H. Phylum XIII. Firmicutes Gibbons and Murray. In: Bergey’s Manual of Systematic Bacteriology. New York: Springer; 2009: 19–1317.
Ludwig W, Schleifer K, Whitman W. Class I. Bacilli class nov. Bergey's Manual of Systematic Bacteriology. 2009;3:19–20.
Ludwig W, Schleifer K, Whitman W. Order II. Lactobacillales ord nov Bergeys Manual of Syst Bacteriol. 2009;3:463–513.
Amyes SG. Enterococci and streptococci. Int J Antimicrob Agents. 2007;29:S43–52.
Rôças IN, Siqueira Jr JF, Santos K. Association of Enterococcus faecalis With Different Forms of Periradicular Diseases. J Endod. 2004;30(5):315–20.
Stuart CH, Schwartz SA, Beeson TJ, Owatz CB. Enterococcus faecalis: Its role in root canal treatment failure and current concepts in retreatment. J Endod. 2006;32(2):93–8.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–9.
Acknowledgements
This work was supported by the Center for Environmental and Agricultural Microbiology (CREAM) funded by the Villum Kann Rasmussen Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SZ drafted the manuscript, performed laboratory experiments, and analyzed the data. DW and YW performed the comparative genome analysis. HH, FA and HA sequenced, assembled, and annotated the genome. YZ revised the manuscript. CR organized the study and revised the manuscript. All authors read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Zhang, S., Wang, D., Wang, Y. et al. Genome sequences of copper resistant and sensitive Enterococcus faecalis strains isolated from copper-fed pigs in Denmark. Stand in Genomic Sci 10, 35 (2015). https://doi.org/10.1186/s40793-015-0021-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40793-015-0021-1