
Abstract
Sepsis-induced myocardial dysfunction is a common complication in septic patients and is associated with increased mortality. In the clinical setting, it was once believed that myocardial dysfunction was not a major pathological process in the septic patients, at least in part, due to the unavailability of suitable clinical markers to assess intrinsic myocardial function during sepsis. Although sepsis-induced myocardial dysfunction has been studied in clinical and basic research for more than 30 years, its pathophysiology is not completely understood, and no specific therapies for this disorder exist. The purpose of this review is to summarize our current knowledge of sepsis-induced myocardial dysfunction with a special focus on pathogenesis and clinical characteristics.
Similar content being viewed by others

Background
Sepsis is a systemic deleterious host response to infection or injury resulting in severe sepsis and septic shock. It is a leading cause of morbidity and mortality in intensive care units [1, 2]. Although the hospital mortality of septic patients decreased from 37 to 30.8 % during the 2 years after the introduction of the Surviving Sepsis Campaign guidelines for the management of sepsis, mortality remains high [3]. In 2012, a global study of the burden of sepsis estimated that the case-fatality rate for patients with severe sepsis approaches 50 % [4].
The cardiovascular system plays an important role in the pathogenesis of sepsis. Over the last 50 years, a large number of studies have demonstrated that myocardial dysfunction is a common finding in septic patients, and approximately 50 % of septic patients exhibit signs of myocardial dysfunction. Nevertheless, the exact clinical significance of sepsis-induced myocardial dysfunction (SIMD) is still elusive. Because the heart, as only one part of the circulatory system, constantly responds to changing peripheral hemodynamics, it is difficult to distinguish between cardiac responses to alterations in preload, afterload or/and neurohumoral activity during sepsis and the direct influence of sepsis on the heart in the clinical setting [5, 6]. Recently, many clinical studies have suggested that myocardial dysfunction was associated with increased mortality in septic patients [7–9]. An animal experimental study showed that myocardial depression was present at the early stage of sepsis, and early myocardial functional changes could predict outcomes in septic animals [10]. In particular, using transgenic mice with cardiomyocyte-specific expression of a constitutively active PI3K isoform that protects myocardial function, Li and coworkers demonstrated, for the first time, a causal relationship between the maintenance of myocardial function and survival in sepsis. They found that cardiac specific activation of PI3K/Akt-dependent signaling significantly attenuated myocardial dysfunction and, in turn, improved survival in cecal ligation and puncture (CLP)-induced sepsis [11]. Therefore, fully understanding the pathogenesis of SIMD and seeking specific therapy will provide beneficial effects on outcomes in septic patients.
The aim of the present review is to discuss the pathophysiology of SIMD, with a special focus on its clinical characteristics and pathogenesis.
The definition of SIMD
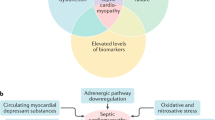
Although numerous studies have demonstrated evidence of cardiovascular impairments in patients with sepsis over the last 50 years, there is no universally accepted definition of SIMD [12]. The initial concept of SIMD came from the study by Parker and Parrillo et al. in 1984. They observed that 50 % of septic patients had a decreased initial left ventricle ejection fraction (EF) with increased mean end-systolic and end-diastolic volumes despite normal or elevated cardiac index found in all septic patients [13]. Since then, SIMD has been defined in numerous clinical investigations as a reversible decrease in EF of both ventricles, with ventricular dilation and less response to fluid resuscitation and catecholamines [14]. However, it is now well known that left ventricular EF is a load-dependent index that reflects the coupling between left ventricular afterload and contractility, rather than the intrinsic myocardial contractile function. During septic shock, although left ventricular intrinsic contractility is seriously impaired, left ventricular EF may be normal when the afterload is severely depressed [12, 15]. On the other hand, myocardial dysfunction has been demonstrated to be constant if determined by using the load-independent parameters of systolic and diastolic function in all animal experimental models of septic shock [16]. Thus, it has recently been suggested that SIMD can be defined as the intrinsic myocardial systolic and diastolic dysfunction of both the left and right sides of the heart induced by sepsis [12, 16].
The clinical characteristics of SIMD
Hemodynamic alterations during sepsis have been investigated for 60 years. Early animal studies performed by Weil et al. in 1956 showed that an injection of endotoxin could cause a sudden decrease in venous return, arterial blood pressure and cardiac output (CO) with increased systemic vascular resistance (SVR), leading to animal death [17]. Clinical observations by Clowes and McLean et al. demonstrated that cardiovascular disturbances during septic shock included two distinct clinical pictures. One was an early hyperdynamic phase (warm shock) characterized by increased CO and decreased SVR, as well as warm and perfused skin; another was late hypodynamic phase (cold shock), in which SVR increased and CO decreased, resulting in tissue hypoperfusion, cool skin, organ failure and ultimate death [18, 19]. These findings led to the belief that patients with septic shock initially went through an early hyperdynamic phase and eventually either recovered or deteriorated into the hypodynamic phase and even death. However, these studies used central venous pressure (CVP) to reflect left ventricular end-diastolic volume and adequacy of resuscitation. In fact, we now know that CVP is not a reliable index of cardiac preload in septic patients. With the introduction of pulmonary artery catheters, which allow simultaneous measurement of both CO and pulmonary artery wedge pressure at the bedside, many studies have shown that septic shock patients or animals with adequate fluid resuscitation have only a persistent hyperdynamic state, which usually persists until death even in nonsurvivors, and the hypodynamic state is very likely due to an inadequate fluid resuscitation [20]. It is now generally accepted that these hemodynamic alterations during sepsis are due to decreased preload, reduced afterload, myocardial dysfunction, blood flow redistribution between organs and microcirculatory impairments [12].
Changes in systolic and diastolic function
In 1984, Parker and Parrillo et al. provided the first valuable clue for the SIMD [13]. Using serial radionucleotide ventriculograms and simultaneous evaluation of CO by thermodilution, they demonstrated that 20 septic shock patients had a high CO and low SVR. Importantly, they further found that 13 patients who survived had a depressed left ventricular EF and acute left ventricular dilatation, which were sustained for 4 days and then returned to normal within 7–10 days. However, nonsurvivors of septic shock maintained normal left ventricular EF and volume. Similarly, Parker et al. used the same methodology and observed right ventricular dysfunction in septic shock patients. They found that the survivors and nonsurvivors of septic shock maintained a reversible reduction in biventricular EF and increased end-diastolic and -systolic volumes in the study period [21]; these results are different from those found in their previous study.
During the same historical period, two-dimensional echocardiography was performed to evaluate myocardial function in septic patients [22, 23]. Because echocardiography is a first-line non-invasive technique for hemodynamic evaluation in patients with cardiovascular disorders and it can be performed at the bedside, various echocardiographic indices, such as EF and cardiac index, have been developed to assess cardiac function. Vieillard Baron and coworkers investigated 40 patients with septic shock by transesophageal echocardiography and observed that stroke index was strongly correlated with left ventricular EF, while left ventricular volume always remained in a normal range after adequate preload optimization [24]. In 2013, a meta-analysis that included more than 700 patients failed to find any evidence to support the above view that the survivors from severe sepsis or septic shock had a reduced EF. In addition, there were no significant differences between septic survivors and nonsurvivors in terms of biventricular EF and indexed biventricular dimensions [25]. In another meta-analysis, a cut-off of left ventricular EF of 50 % was used to identify patients with systolic dysfunction, and no significant difference in mortality rates was found in septic patients with reduced EF compared to patients with normal EF [8]. Evidently, these studies revealed a complicated and contradictory picture about myocardial dysfunction in septic patients. It is now known that these inconsistent results from the above studies are very likely due to the limitations of currently used indices of ventricular function because cardiac index and EF are load-dependent indices that do not reflect the intrinsic myocardial contractile function during sepsis [16].
To detect subtle myocardial dysfunction during sepsis, some investigators examined myocardial function during sepsis using advanced echocardiographic techniques, such as speckle tracking and Doppler tissue echocardiography. Speckle tracking echocardiography is more sensitive than the conventional echocardiographic technique and is able to detect ventricular strain that reflects segmental myocardial deformation. It was demonstrated that peak left ventricular global longitudinal systolic strain, determined by using speckle tracking echocardiography at the time of admission in septic patients, correlated with mortality rate, whereas left ventricular EF had no prognostic relevance [9]. Another clinical investigation also suggested that strain imaging by speckle tracking echocardiography might be useful in the early detection of myocardial dysfunction in sepsis. It showed that 50 % of septic patients with preserved left ventricular EF had a depressed left ventricular global longitudinal function compared to non-septic patients. In patients with sepsis and preserved left ventricular EF, both left ventricular global and right ventricular free wall strain were lower than in non-septic patients with preserved left ventricular EF. These findings indicate that left ventricular and right ventricular systolic dysfunction in patients with early septic shock and preserved left ventricular EF can be detected by speckle tracking echocardiography [26]. It is noteworthy that the clinical features of segmental ventricular dysfunction during SIMD are sometimes consistent with Takotsubo cardiomyopathy, in which the contractile function of the middle-to-apical segments of the left ventricle is depressed and there is hyperkinesis of the basal walls, inducing the balloon-like appearance of the distal ventricle [27].
In contrast to systolic dysfunction, diastolic dysfunction is often ignored, and its role in determining early mortality from sepsis has not been adequately investigated. Recently, it has been demonstrated that the mitral annular early diastolic peak velocity (e’ wave) obtained by tissue Doppler imaging is one of the most load-independent measures of diastolic dysfunction. The ratio of the early mitral inflow velocity (E), recorded with pulsed-wave Doppler, to the e’ wave (E/e’) correlates with left ventricular end-diastolic pressure, and a high E/e’ ratio represents low left ventricular compliance in numerous cardiac conditions [28, 29]. Using the reduced e’ wave or the increased E/e’ ratio to identify left ventricular diastolic dysfunction, some investigators found that diastolic dysfunction was common in septic patients. A reduced mitral annular e’-wave was the strongest predictor of mortality, and E/e’ was an independent predictor of hospital survival, which offered better discrimination between hospital survivors and non-survivors than cardiac biomarkers such as cardiac-specific troponins (cTn) and N-terminal proB-type natriuretic peptide (NT-proBNP) [7, 30, 31].
Alterations in electrocardiogram
In 1982, Terradellas et al. reported the acute elevation of the ST segment in bacterial shock patients with no history of heart disease [32]. Other investigators then found that endotoxin induced initial tachycardia followed by significant bradycardia and elevation of the ST segment in rats [33]. However, it was also reported that ST-segment elevations were rare in septic shock patients in the setting of normal coronary angiography. In addition, the electrocardiographic changes during septic shock also include a loss of QRS amplitude, increase in QT interval, development of narrowed QRS intervals with deformed bundle branch blocks [34] and new-onset atrial fibrillation, especially in older septic shock patients [35].
Biomarkers of SIMD
B-type natriuretic peptide (BNP) is a hormone synthesized in the myocardium. It is produced in the prohormone form, and before secretion is split into the inactive NT-proBNP and the active BNP [36]. Numerous studies demonstrated markedly elevated plasma levels of BNP and NT-proBNP in septic patients [37, 38], and the increased plasma BNP and NT-proBNP concentrations were associated with myocardial depression and increased mortality in patients with sepsis [39, 40]. These findings suggest that plasma BNP and NT-proBNP levels represent reliable markers for the identification of SIMD. However, other studies indicate that the relationship between BNP and both left ventricular EF and left-sided filling pressures is weak and data on the prognostic impact of high BNP levels in septic patients are conflicting [41]. In fact, many factors, including right ventricular overload, catecholamine therapy and increased cytokine production, may contribute to BNP release during sepsis. Thus, it is suggested that cTn may be integrated into the monitoring of SIMD [41]. Plasma cTnI and cTnT levels have also been proven to be highly sensitive and specific markers of SIMD [14]. Although the mechanisms underlying cTnI release during sepsis are still unclear, increased plasma concentrations of cTnI and cTnT were found in septic patients, and both cTnI and cTnT were exclusively associated with left ventricular dysfunction [42]. A meta-analysis showed that plasma troponin elevation in septic patients was also a predictor of mortality [43]. In addition, serum heart-type fatty acid-binding protein concentration was also found to be a useful diagnostic marker for organ dysfunction and 28-day mortality in septic patients [44, 45]. Zhang et al. found that serum heart-type fatty acid-binding protein was frequently elevated in septic patients and appeared to be associated with SIMD [46]; large prospective clinical trials on its role in identifying SIMD are now warranted.
Cardiac structural changes
Human autopsies and animal experimental studies have revealed that sepsis-induced myocardial changes are classified within inflammatory cardiomyopathy. The major cardiac pathological changes during sepsis include myocardial infiltration by immune cells (especially macrophages and neutrophils), subendocardial hemorrhage, interstitial and intracellular edema, endothelial cell edema, microcirculatory fibrin deposition, as well as focal myofibrillar dissolution, cardiomyocyte necrosis and interstitial fibrosis. Intracytoplasmic lipid accumulation in cardiomyocytes is also observed in septic hearts. Immunohistochemical examination shows that high diffuse expression of tumor necrosis factor-α (TNF–α) is localized to cardiomyocytes, macrophages, smooth muscle cells and endothelial cells [12, 47–49]. As mentioned above, SIMD is thought to be completely reversible. However, we do not know whether the histological myocardial alterations in sepsis are reversible. Therefore, the possibility that myocardial dysfunction is not completely reversible in septic patients should be further examined.
Pathogenesis of SIMD
Based on an animal study, the first hypothesis on the mechanism of SIMD was global myocardial ischemia resulting from inadequate coronary blood flow. However, coronary blood flow was later found to be either preserved or increased in septic shock patients with myocardial dysfunction, disproving the above hypothesis. Although some studies demonstrated that impairment in cardiac microcirculation was present during sepsis due to significant maldistribution of coronary blood flow, endothelial damage, intravascular fibrin depositions and neutrophil infiltration, which might result in focal myocardial ischemia and decreased cardiac function, no myocardial hypoxia was confirmed in septic animals. It is now suggested that the elevation of plasma cardiac troponins may be attributed to an increase in cardiomyocyte membrane permeability rather than myocardial ischemic necrosis. These findings indicate that coronary circulation alterations are less important in the mechanisms of SIMD [6, 50]. According to the current evidence, it is generally accepted that SIMD may be a result of the interaction of many factors, including inflammation, metabolism and neuroimmunomodulation.
Myocardium-depressing factors
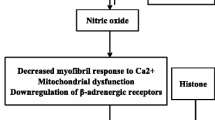
In the 1960s, many investigators reported the presence of myocardium-depressing factors that could cause SIMD [12]. During sepsis, various pathogen-associated molecular patterns (PAMPs), such as lipopolysaccharide (LPS), and endogenous damage-associated molecular patterns (DAMPs), including high mobility group box 1 (HMGB1) and extracellular histones, interact with Toll-like receptors (TLRs) on immune cells and other cells. All TLRs, except TLR3, signal through the myeloid differentiation factor 88 (MyD88)-dependent pathway and activate c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinases 1/2 (ERK1/2), p38 mitogen-activated protein kinase (MAPK) and the transcription factor nuclear factor (NF)-kB signaling pathways, which in turn induce the production of multiple proinflammatory cytokines, including interleukin (IL)-1, IL-6 and TNF-α [51, 52]. Several substances have been considered as myocardium-depressing factors, including TNF-α, IL-1, IL-6, complement anaphylatoxin (C5a) and LPS (Table 1) [12, 14]. For example, prolonged exposure of adult rat ventricular myocytes to a mixture of LPS, TNF-α, IL-1 and IL-6 inhibited cell contractility in vitro [53], and treatment with a monoclonal anti-TNF antibody in patients within 24 h of septic shock improved left ventricular function [54]. However, a recent clinical study investigated the relationship between serum cytokine concentrations (IL-1β, IL-6, IL-8, IL-10, IL-18, TNF-α and monocyte chemoattractant protein-1) and septic myocardial dysfunction. The authors defined reduced left ventricular EF of < 50 % or < 55 % as systolic dysfunction and e wave < 8 cm/s as diastolic dysfunction and found that none of these cytokines correlated with left ventricular EF and e’-wave velocities in septic patients. Similarly, there were no differences in cytokine concentrations between patients dichotomized to high and low left ventricular EF or e wave. Therefore, none of the measured circulating cytokines correlated with systolic or diastolic myocardial dysfunction in severe sepsis or septic shock in the clinical setting [55]. More recently, experimental studies found that LPS induced myocardial HMGB1 expression and increased plasma HMGB1 level in rats and mice and that HMGB1 stimulation produced a negative inotropic effect in the isolated rat heart [56, 57]. Similarly, another study demonstrated that increased circulating histone levels were significantly associated with new-onset left ventricular dysfunction and arrhythmias in septic patients with no previous cardiac dysfunction [58]. Nevertheless, the role of circulating HMGB1 and histones in SIMD deserves to be further investigated.
Indeed, it is likely that circulating myocardium-depressing factors are the initial stimuli and driving forces of septic myocardial dysfunction. It is well known that cardiomyocytes express Toll-like receptors such as TLR2 and TLR4 [59, 60]. LPS stimulates TLR4 on cardiomyocytes and leads to the phosphorylation of p38 MAPK and JNK and the activation of NF-kB, which induces cardiomyocyte TNF-α expression and decreases myocardial contractility [61]. Natural deletion of TLR4 [62] or MyD88 deletion in cardiomyocytes confers a profound protection with markedly improved cardiac function and survival in an LPS-induced shock model [63]. In addition, extracellular histones also stimulate cardiomyocyte TLR4 and induced myocardial dysfunction [64]. We recently observed that stimulation of cardiomyocyte β1-adrenoceptor promoted p38MAPK, JNK and NF-kB activation and subsequent TNF-α expression in LPS-treated cardiomyocytes [65]. Activation of cardiomyocyte α1-adrenoceptor can suppress LPS-induced cardiomyocyte TNF-α expression and improve cardiac dysfunction during endotoxemia [66]. We also found that blockade of the α2-adrenoceptor suppressed myocardial TNF-α and inducible nitric oxide synthase (iNOS) expression and cardiomyocyte apoptosis and cardiac dysfunction in endotoxemic animals [67]. Thus, it is necessary for regulating cardiomyocyte adrenergic signals to develop interventions for some myocardium-depressing factors and to provide therapeutic targets for SIMD.
In addition to cardiomyocytes, cardiac fibroblasts and endothelial cells are involved in SIMD. Cardiac fibroblasts make up 60 %–70 % of the total cell number in the heart. Tomita et al. demonstrated that LPS significantly increased the expression of TNF-α and matrix metalloproteinase (MMP)-9 in cultured cardiac fibroblasts. CLP induced cardiac MMP-9 expression, cardiac fibrosis and cardiac dysfunction in mice, and treatment with a broad-spectrum MMP inhibitor significantly alleviated these histological and functional changes during sepsis [68]. In addition, endothelial cell activation also plays a critical role in septic injury in multiple organs. It has been demonstrated that serum levels of sphingosine-1-phosphate, a potent regulator of endothelial integrity, are dramatically decreased and inversely associated with disease severity in septic patients [69]. Clinical evidence showed that systolic cardiac dysfunction was directly associated with markers of endothelial dysfunction in septic patients [70]. Some studies reported that circulating myocardium-depressing factors, such as TNF-α, increased the expression of intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) in coronary endothelial cells and cardiomyocytes [71, 72]. Blockade of VCAM-1 reduced myocardial neutrophil accumulation and abrogated LPS-induced cardiac dysfunction. The absence of ICAM-1 also reduced LPS-induced cardiac dysfunction, but without decreasing neutrophil accumulation. Moreover, the depletion of neutrophils failed to protect against LPS-induced myocardial dysfunction. These results indicate that endothelial and/or cardiomyocyte ICAM-1 and VCAM-1 mediate LPS-induced myocardial dysfunction independent of neutrophil infiltration [73]. Some investigators generated double transgenic mice that conditionally overexpress a degradation-resistant form of I-kB, an inhibitor of NF-kB, selectively on the endothelium. These authors demonstrated that endothelial-selective blockade of NF-kB activation markedly inhibited cardiac ICAM-1 and VCAM-1 expression and ameliorated myocardial injury in both LPS and CLP models of sepsis [74]. Therefore, circulating myocardium-depressing factors may activate cardiac fibroblasts and endothelial cells, which contribute to SIMD. Collectively, circulating PAMPs, DAMPs and cytokines can activate endothelial cells, cardiac fibroblasts and cardiomyocytes and increase the production of inflammatory mediators, which further stimulate iNOS expression and cause myocardial depression in sepsis [12, 50, 53, 64].
Autonomic dysregulation
The autonomic nervous system plays an important role in sepsis. Some evidence indicates that sepsis induces autonomic dysregulation, including neuronal and glial apoptosis within the autonomic centers of the heart, high plasma levels of catecholamines, reduced heart rate variability and decreased cardiac responsiveness to intrinsic catecholamines, which may contribute to SIMD. A number of studies showed decreased densities of β1-adrenoceptors, reduced levels of stimulatory G-proteins and increased expression of inhibitory G-proteins in cardiomyocytes during sepsis. These results indicate that impaired myocardial responsiveness to catecholamines in sepsis can be attributed to the downregulation of adrenergic receptors and/or post-receptor signaling [6, 75]. In addition, β3-adrenoceptors, which mediate an increased negative inotropic response to agonists, were found to be upregulated during sepsis, suggesting that activation of β3-adrenoceptors by catecholamines may contribute to SIMD [76].
Dysfunction of intracellular Ca2+ transporters in cardiomyocytes
In addition to a decrease in myofilament Ca2+ sensitivity due to a sustained increase in cardiac troponin I phosphorylation at Ser23/24 [77], a dysfunction of intracellular Ca2+ transporters in cardiomyocytes underlies SIMD. During sepsis, downregulated L-type calcium channels and a suppressed sarcoplasmic reticulum (SR) pump lead to a decrease in the amplitude of cellular Ca2+ transients and SR calcium load in cardiomyocytes [78]. CLP increased myocardial angiotensin II content, which may be associated with the disturbance of Ca2+ transport in the cardiac SR [79]. LPS also specifically impaired sarcolemmal diastolic Ca2+ extrusion pathways by depressing the function of the Na+/Ca2+exchanger and the plasmalemmal Ca2+ ATPase, which in turn resulted in intracellular diastolic Ca2+ overload [80]. This disruption of cellular Ca2+ homeostasis in cardiomyocytes may contribute to SIMD. However, the mechanism underlying the reduction in transient systolic Ca2+ is not well established. A recent study demonstrated that sepsis induced a decrease in sodium current in cardiomyocytes, which reduced cardiac excitability. This reduction in the density of Na+ channels might lessen the transient Ca2+ action potential in cardiomyocytes by decreasing the number of Ca2+ channels that open during the action potential due to the reduction in peak depolarization or/and by reducing Ca2+ entry due to a shortening of the action potential [81].
Energetic starvation of cardiomyocytes
Although oxygenation of the myocardium does not appear to be altered during sepsis, accumulating evidence indicates that impaired metabolism and reduced energy production in cardiomyocytes play a critical role in SIMD. Under normal conditions, approximately 70 % of adenosine triphosphate (ATP) in cardiomyocytes is produced via fatty acid oxidation, and the remainder is produced via glucose oxidation. A small amount of ATP is also derived from the catabolism of lactate and ketone bodies [82]. During sepsis, inflammatory cytokines, such as IL-1β, can downregulate very low-density lipoprotein receptor expression in cardiomyocytes [83]. Decreased expression of the very low-density lipoprotein receptor and the fatty acid transporter CD36 inhibits lipid uptake by cardiomyocytes [82]. Importantly, Toll-like receptor-mediated inflammatory signaling reprograms cardiac energy metabolism, leading to a reduced expression of fatty-acid-binding protein, acyl-CoA synthetase, and fatty acid oxidation-associated transcriptional factors, including peroxisome proliferator activated receptors (PPARs) and PPARγ-coactivator-1 [82, 84]. Recently, Drosatos et al. discovered that cardiomyocyte Krüppel-like factor five upregulated PPARγ expression through direct promoter binding, which was blocked in sepsis. Depletion of cardiac myocyte-specific Krüppel-like factor five not only reduced myocardial PPARγ expression, fatty acid oxidation and ATP levels, but also increased myocardial triglyceride accumulation and induced myocardial dysfunction [85]. These data indicate that sepsis inhibits intracellular fatty acid oxidation and could eventually reduce cardiomyocyte ATP production and myocardial function. Restoration of myocardial fatty acid oxidation improves SIMD. Drosatos et al. demonstrated that both cardiomyocyte-specific expression of PPARγ and activation of PPARγ by rosiglitazone increased myocardial fatty acid oxidation and prevented LPS-induced cardiac dysfunction, but without affecting the expression of myocardial inflammatory cytokines [86]. Thus, although inflammation is an important component of mechanisms that mediate SIMD, the decrease in myocardial fatty acid oxidation constitutes another critical mechanism responsible for this disorder.
Mitochondrial dysfunction and oxidative-nitrosative stress
Although early myocardial dysfunction during sepsis is associated with myocardial inflammation rather than mitochondrial injury [87], enzyme activities of nicotinamide-adenine dinucleotid cytochrome c reductase, succinate cytochrome c reductase and cytochrome c oxidase were found to be significantly suppressed during sepsis. Mitochondrial complex II and complex IV were also downregulated, and the myocardial ATP content markedly declined during the late stage of sepsis [88]. These results indicate that mitochondrial dysfunction associated with a decrease in myocardial ATP content is likely correlated with the deterioration of myocardial function during the late stage of sepsis. Furthermore, pharmacological inhibition of mitochondrial permeability transition by cyclosporine derivatives was found to improve myocardial dysfunction and survival in animal models of CLP-induced sepsis [89]. Similarly, the administration of mitochondria-targeted vitamin E also protected myocardial mitochondrial structure and function, inhibited mitochondrial oxidative stress, and improved myocardial function in septic rats [90]. Thus, it is likely that mitochondrial dysfunction is causative rather than epiphenomenal and is relevant to SIMD. However, the underlying mechanisms responsible for sepsis-induced mitochondrial dysfunction are still not completely elucidated. Oxidative-nitrosative stress due to excessive production of mitochondrial reactive oxygen species and nitric oxide, increased mitochondrial permeability transition pore opening and increased mitochondrial uncoupling may contribute to this type of mitochondrial dysfunction [91, 92].
Cardiomyocyte apoptosis
In addition to leading to DNA fragmentation, activated caspases can directly induce the breakdown of myofibrillar proteins, decrease ATPase activity and force development in cardiomyocytes [93]. Although cardiomyocyte apoptosis has not been observed in human autopsy specimens, there is increasing evidence that caspase-3 activation and cardiomyocyte apoptosis contribute to SIMD [94–96]. It is generally accepted that the over-production of inflammatory mediators and reactive oxygen species plays a critical role in capase-3 activation and cardiomyocyte apoptosis during sepsis [95, 97–99]. However, we recently found that reduced cardiac endogenous norepinephrine or blockade of β1-adrenoceptors almost completely abolished cardiomyocyte apoptosis in LPS-challenged mice [67]. We further demonstrated that β1-adrenoceptor activation promotes LPS-induced cardiomyocyte apoptosis [65]. Therefore, β1-adrenoceptor activation appears to be more important than cytokines in LPS-induced cardiomyocyte apoptosis. In fact, a randomized clinical trial observed that therapy with the β-blocker esmolol reduced heart rate, increased left ventricular stroke work index and decreased 28-day mortality in septic shock patients [100]. The reader can refer to the excellent systematic review on β-blockers in septic patients [101].
Conclusions
SIMD refers to the intrinsic myocardial systolic and diastolic dysfunction of both the left and right sides of the heart during sepsis. Early recognition of intrinsic myocardial dysfunction is critical for the administration of the most appropriate therapy for septic patients. However, traditional insensitive parameters, such as EF, cannot accurately assess sepsis-induced intrinsic myocardial dysfunction. It is now suggested that the routine use of speckle tracking and tissue Doppler echocardiography may be valuable in the identification of SIMD in septic patients. The circulating myocardium-depressing factors are only initial stimulators that induce cardiac structure and function damage, in which cardiomyocytes, cardiac endothelial cells and even fibroblasts are involved. A deeper understanding of the effects of immuno-metabolic and neuroendocrine factors on cardiomyocytes, endothelial cells and fibroblasts at molecular and subcellular levels will expand our knowledge of the mechanisms contributing to SIMD. The elucidation of these mechanisms should help identify new cardiac-specific therapeutic targets and improve the prognosis of septic patients.
Abbreviations
- BNP:
-
B-type natriuretic peptide
- CLP:
-
Cecal ligation and puncture
- CO:
-
Cardiac output
- cTn:
-
cardiac-specific troponins
- DAMPs:
-
Damage-associated molecular patterns
- EF:
-
Ejection fraction
- ERK1/2:
-
Extracellular signal-regulated kinases 1/2
- HMGB1:
-
High mobility group box 1
- ICAM-1:
-
Intercellular adhesion molecule-1
- iNOS:
-
inducible nitric oxide synthase
- JNK:
-
c-Jun N-terminal kinase
- LPS:
-
Lipopolysaccharide
- MAPK:
-
Mitogen-activated protein kinase
- MMP:
-
Matrix metalloproteinase
- MyD88:
-
Myeloid differentiation factor 88
- NF-kB:
-
Nuclear factor-kB
- PAMPs:
-
Pathogen-associated molecular patterns
- PPARs:
-
Peroxisome proliferator activated receptors
- SIMD:
-
Sepsis-induced myocardial dysfunction
- SR:
-
Sarcoplasmic reticulum
- SVR:
-
Systemic vascular resistance
- TNF-α:
-
Tumor necrosis factor-α
- TLRs:
-
Toll-like receptors
- VCAM-1:
-
Vascular cell adhesion molecule-1
References
Dellinger RP, Levy MM, Rhodes A, Annane D, Gerlach H, Opal SM, et al. Surviving Sepsis Campaign Guidelines Committee including The Pediatric Subgroup. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock, 2012. Intensive Care Med. 2013;39:165–228.
Deutschman CS, Tracey KJ. Sepsis: current dogma and new perspectives. Immunity. 2014;40:463–75.
Levy MM, Dellinger RP, Townsend SR, Linde-Zwirble WT, Marshall JC, Bion J, et al. The Surviving Sepsis Campaign: results of an international guideline-based performance improvement program targeting severe sepsis. Intensive Care Med. 2010;36:222–31.
Jawad I, Lukšić I, Rafnsson SB. Assessing available information on the burden of sepsis: global estimates of incidence, prevalence and mortality. J Glob Health. 2012;2:010404.
Charpentier J, Luyt CE, Fulla Y, Vinsonneau C, Cariou A, Grabar S, et al. Brain natriuretic peptide: A marker of myocardial dysfunction and prognosis during severe sepsis. Crit Care Med. 2004;32:660–5.
Hochstadt A, Meroz Y, Landesberg G. Myocardial dysfunction in severe sepsis and septic shock: more questions than answers? J Cardiothorac Vasc Anesth. 2011;25:526–35.
Landesberg G, Gilon D, Meroz Y, Georgieva M, Levin PD, Goodman S, et al. Diastolic dysfunction and mortality in severe sepsis and septic shock. Eur Heart J. 2012;33:895–903.
Sanfilippo F, Corredor C, Fletcher N, Landesberg G, Benedetto U, Foex P, et al. Diastolic dysfunction and mortality in septic patients: a systematic review and meta-analysis. Intensive Care Med. 2015;41:1004–13.
Palmieri V, Innocenti F, Guzzo A, Guerrini E, Vignaroli D, Pini R. Left ventricular systolic longitudinal function as predictor of outcome in patients with sepsis. Circ Cardiovasc Imaging. 2015;8:e003865.
Rudiger A, Dyson A, Felsmann K, Carré JE, Taylor V, Hughes S, et al. Early functional and transcriptomic changes in the myocardium predict outcome in a long-term rat model of sepsis. Clin Sci (Lond). 2013;124:391–401.
Li C, Hua F, Ha T, Singh K, Lu C, Kalbfleisch J, et al. Activation of myocardial phosphoinositide-3-kinase p110α ameliorates cardiac dysfunction and improves survival in polymicrobial sepsis. PLoS One. 2012;7:e44712.
Antonucci E, Fiaccadori E, Donadello K, Taccone FS, Franchi F, Scolletta S. Myocardial depression in sepsis: from pathogenesis to clinical manifestations and treatment. J Crit Care. 2014;29:500–11.
Parker MM, Shelhamer JH, Bacharach SL, Green MV, Natanson C, Frederick TM, et al. Profound but reversible myocardial depression in patients with septic shock. Ann Intern Med. 1984;100:483–90.
Romero-Bermejo FJ, Ruiz-Bailen M, Gil-Cebrian J, Huertos-Ranchal MJ. Sepsis-induced cardiomyopathy. Curr Cardiol Rev. 2011;7:163–83.
Repessé X, Charron C, Vieillard-Baron A. Evaluation of left ventricular systolic function revisited in septic shock. Crit Care. 2013;17:164.
Zaky A, Deem S, Bendjelid K, Treggiari MM. Characterization of cardiac dysfunction in sepsis: an ongoing challenge. Shock. 2014;41:12–24.
Weil MH, Maclean LD, Visscher MB, Spink WW. Studies on the circulatory changes in the dog produced by endotoxin from gram-negative microorganisms. J Clin Invest. 1956;35:1191–8.
Clowes Jr GH, Vucinic M, Weidner MG. Circulatory and metabolic alterations associated with survival or death in peritonitis: clinical analysis of 25 cases. Ann Surg. 1966;163:866–85.
McLean LD, Mulligan WG, McLean AP, Duff JH. Patterns of septic shock in man–a detailed study of 56 patients. Ann Surg. 1967;166:543–62.
Rabuel C, Mebazaa A. Septic shock: a heart story since the 1960s. Intensive Care Med. 2006;32:799–807.
Parker MM, McCarthy KE, Ognibene FP, Parrillo JE. Right ventricular dysfunction and dilatation, similar to left ventricular changes, characterize the cardiac depression of septic shock in humans. Chest. 1990;97:126–31.
Burns JR, Menapace FJ. Acute reversible cardiomyopathy complicating toxic shock syndrome. Arch Intern Med. 1982;142:1032–4.
Ozier Y, Guéret P, Jardin F, Farcot JC, Bourdarias JP, Margairaz A. Two-dimensional echocardiographic demonstration of acute myocardial depression in septic shock. Crit Care Med. 1984;12:596–9.
Vieillard Baron A, Schmitt JM, Beauchet A, Augarde R, Prin S, Page B, et al. Early preload adaptation in septic shock? A transesophageal echocardiographic study. Anesthesiology. 2001;94:400–6.
Huang SJ, Nalos M, McLean AS. Is early ventricular dysfunction or dilatation associated with lower mortality rate in adult severe sepsis and septic shock? A meta-analysis. Crit Care. 2013;17:R96.
Dalla K, Hallman C, Bech-Hanssen O, Haney M, Ricksten SE. Strain echocardiography identifies impaired longitudinal systolic function in patients with septic shock and preserved ejection fraction. Cardiovasc Ultrasound. 2015;13:30.
Y-Hassan S, Settergren M, Henareh L. Sepsis-induced myocardial depression and takotsubo syndrome. Acute Card Care. 2014;16:102–9.
Kasner M, Westermann D, Steendijk P, Gaub R, Wilkenshoff U, Weitmann K, et al. Utility of Doppler echocardiography and tissue Doppler imaging in the estimation of diastolic function in heart failure with normal ejection fraction: a comparative Doppler-conductance catheterization study. Circulation. 2007;116:37–47.
Mousavi N, Czarnecki A, Ahmadie R, Tielan F, Kumar K, Lytwyn M, et al. The utility of tissue Doppler imaging for the noninvasive determination of left ventricular filling pressures in patients with septic shock. J Intensive Care Med. 2010;25:163–7.
Sturgess DJ, Marwick TH, Joyce C, Jenkins C, Jones M, Masci P, et al. Prediction of hospital outcome in septic shock: a prospective comparison of tissue Doppler and cardiac biomarkers. Crit Care. 2010;14:R44.
Brown SM, Pittman JE, Hirshberg EL, Jones JP, Lanspa MJ, Kuttler KG, et al. Diastolic dysfunction and mortality in early severe sepsis and septic shock: a prospective, observational echocardiography study. Crit Ultrasound J. 2012;4:8.
Terradellas JB, Bellot JF, Sarís AB, Gil CL, Torrallardona AT, Garriga JR. Acute and transient ST segment elevation during bacterial shock in seven patients without apparent heart disease. Chest. 1982;81:444–8.
Tariq M, Ageel AM, Parmar NS. Decrease by naloxone of some electrocardiographic and biochemical changes following endotoxin induced shock in rats. Toxicon. 1986;24:101–3.
Martinez JD, Babu RV, Sharma G. Escherichia coli septic shock masquerading as ST-segment elevation myocardial infarction. Postgrad Med. 2009;121:102–5.
Guenancia C, Binquet C, Laurent G, Vinault S, Bruyère R, Prin S, et al. Incidence and Predictors of New-Onset Atrial Fibrillation in Septic Shock Patients in a Medical ICU: Data from 7-Day Holter ECG Monitoring. PLoS One. 2015;10:e0127168.
Clerico A, Iervasi G, Mariani G. Pathophysiologic relevance of measuring the plasma levels of cardiac natriuretic peptide hormones in humans. Horm Metab Res. 1999;31:487–98.
Chua G, Kang-Hoe L. Marked elevations in N-terminal brain natriuretic peptide levels in septic shock. Crit Care. 2004;8:R248–50.
Potz BA, Sellke FW, Abid MR. Endothelial ROS and impaired myocardial oxygen consumption in sepsis-induced cardiac dysfunction. J Intensive Crit Care. 2016;2.
Roch A, Allardet-Servent J, Michelet P, Oddoze C, Forel JM, Barrau K, et al. NH2 terminal pro-brain natriuretic peptide plasma level as an early marker of prognosis and cardiac dysfunction in septic shock patients. Crit Care Med. 2005;33:1001–7.
Turner KL, Moore LJ, Todd SR, Sucher JF, Jones SA, McKinley BA, et al. Identification of cardiac dysfunction in sepsis with B-type natriuretic peptide. J Am Coll Surg. 2011;213:139–46.
Maeder M, Fehr T, Rickli H, Ammann P. Sepsis-associated myocardial dysfunction: diagnostic and prognostic impact of cardiac troponins and natriuretic peptides. Chest. 2006;129:1349–66.
ver Elst KM, Spapen HD, Nguyen DN, Garbar C, Huyghens LP, Gorus FK. Cardiac troponins I and T are biological markers of left ventricular dysfunction in septic shock. Clin Chem. 2000;46:650–7.
Sheyin O, Davies O, Duan W, Perez X. The prognostic significance of troponin elevation in patients with sepsis: a meta-analysis. Heart Lung. 2015;44:75–81.
Yan GT, Lin J, Hao XH, Xue H, Zhang K, Wang LH. Heart-type fatty acid-binding protein is a useful marker for organ dysfunction and leptin alleviates sepsis-induced organ injuries by restraining its tissue levels. Eur J Pharmacol. 2009;616:244–50.
Jo YH, Kim K, Lee JH, Rhee JE, Lee JH, Kang KW, et al. Heart-type fatty acid-binding protein as a prognostic factor in patients with severe sepsis and septic shock. Am J Emerg Med. 2012;30:1749–55.
Zhang ZC, Dai HW, Yu YH, Yang JD, Hu CB. Usefulness of heart-type fatty acid-binding protein in patients with severe sepsis. J Crit Care. 2012;27:415.e13–8.
Celes MR, Prado CM, Rossi MA. Sepsis: going to the heart of the matter. Pathobiology. 2013;80:70–86.
Schmittinger CA, Dünser MW, Torgersen C, Luckner G, Lorenz I, Schmid S, et al. Histologic pathologies of the myocardium in septic shock: a prospective observational study. Shock. 2013;39:329–35.
Soriano FG, Guido MC, Barbeiro HV, Caldini EG, Lorigados CB, Nogueira AC. Endotoxemic myocardial dysfunction: subendocardial collagen deposition related to coronary driving pressure. Shock. 2014;42:472–9.
Kakihana Y, Ito T, Nakahara M, Yamaguchi K, Yasuda T. Sepsis-induced myocardial dysfunction: pathophysiology and management. J Intensive Care. 2016;4:22.
Denk S, Perl M, Huber-Lang M. Damage- and pathogen-associated molecular patterns and alarmins: keys to sepsis? Eur Surg Res. 2012;48:171–9.
Vénéreau E, Ceriotti C, Bianchi ME. DAMPs from cell death to new life. Front Immunol. 2015;6:422.
Hobai IA, Morse JC, Siwik DA, Colucci WS. Lipopolysaccharide and cytokines inhibit rat cardiomyocyte contractility in vitro. J Surg Res. 2015;193:888–901.
Vincent JL, Bakker J, Marécaux G, Schandene L, Kahn RJ, Dupont E. Administration of anti-TNF antibody improves left ventricular function in septic shock patients. Results of a pilot study. Chest. 1992;101:810–5.
Landesberg G, Levin PD, Gilon D, Goodman S, Georgieva M, Weissman C, et al. Myocardial Dysfunction in Severe Sepsis and Septic Shock: No Correlation With Inflammatory Cytokines in Real-life Clinical Setting. Chest. 2015;148:93–102.
Hagiwara S, Iwasaka H, Uchino T, Noguchi T. High mobility group box 1 induces a negative inotropic effect on the left ventricle in an isolated rat heart model of septic shock: a pilot study. Circ J. 2008;72:1012–7.
Xu H, Su Z, Wu J, Yang M, Penninger JM, Martin CM, et al. The alarmin cytokine, high mobility group box 1, is produced by viable cardiomyocytes and mediates the lipopolysaccharide-induced myocardial dysfunction via a TLR4/phosphatidylinositol 3-kinase gamma pathway. J Immunol. 2010;184:1492–8.
Alhamdi Y, Abrams ST, Cheng Z, Jing S, Su D, Liu Z, et al. Circulating histones are major mediators of cardiac injury in patients with sepsis. Crit Care Med. 2015;43:2094–103.
Nemoto S, Vallejo JG, Knuefermann P, Misra A, Defreitas G, Carabello BA, et al. Escherichia coli LPS-induced LV dysfunction: role of toll-like receptor-4 in the adult heart. Am J Physiol Heart Circ Physiol. 2002;282:H2316–23.
Boyd JH, Mathur S, Wang Y, Bateman RM, Walley KR. Toll-like receptor stimulation in cardiomyoctes decreases contractility and initiates an NF-kappaB dependent inflammatory response. Cardiovasc Res. 2006;72:384–93.
Zhang T, Feng Q. Nitric oxide and calcium signaling regulate myocardial tumor necrosis factor-α expression and cardiac function in sepsis. Can J Physiol Pharmacol. 2010;88:92–104.
Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science. 1998;282:2085–8.
Feng Y, Zou L, Chen C, Li D, Chao W. Role of cardiac- and myeloid-MyD88 signaling in endotoxin shock: a study with tissue-specific deletion models. Anesthesiology. 2014;121:1258–69.
Kalbitz M, Grailer JJ, Fattahi F, Jajou L, Herron TJ, Campbell KF, et al. Role of extracellular histones in the cardiomyopathy of sepsis. FASEB J. 2015;29:2185–93.
Wang Y, Wang Y, Yang D, Yu X, Li H, Lv X, et al. B1-adrenoceptor stimulation promotes LPS-induced cardiomyocyte apoptosis through activating PKA and enhancing CaMKII and IkBα phosphorylation. Crit Care. 2015;19:76.
Yu X, Jia B, Wang F, Lv X, Peng X, Wang Y, et al. α1 adrenoceptor activation by norepinephrine inhibits LPS-induced cardiomyocyte TNF-α production via modulating ERK1/2 and NF-kB pathway. J Cell Mol Med. 2014;18:263–73.
Wang Y, Yu X, Wang F, Wang Y, Wang Y, Li H, et al. Yohimbine promotes cardiac NE release and prevents LPS-induced cardiac dysfunction via blockade of presynaptic α2A-adrenergic receptor. PLoS One. 2013;8:e63622.
Tomita K, Takashina M, Mizuno N, Sakata K, Hattori K, Imura J, et al. Cardiac fibroblasts: contributory role in septic cardiac dysfunction. J Surg Res. 2015;193:874–87.
Winkler MS, Nierhaus A, Holzmann M, Mudersbach E, Bauer A, Robbe L, et al. Decreased serum concentrations of sphingosine-1-phosphate in sepsis. Crit Care. 2015;19:372.
Furian T, Aguiar C, Prado K, Ribeiro RV, Becker L, Martinelli N, et al. Ventricular dysfunction and dilation in severe sepsis and septic shock: relation to endothelial function and mortality. J Crit Care. 2012;27:319.e9–15.
Kacimi R, Karliner JS, Koudssi F, Long CS. Expression and regulation of adhesion molecules in cardiac cells by cytokines: response to acute hypoxia. Circ Res. 1998;82:576–86.
Henninger DD, Panés J, Eppihimer M, Russell J, Gerritsen M, Anderson DC, et al. Cytokine-induced VCAM-1 and ICAM-1 expression in different organs of the mouse. J Immunol. 1997;158:1825–32.
Raeburn CD, Calkins CM, Zimmerman MA, Song Y, Ao L, Banerjee A, et al. ICAM-1 and VCAM-1 mediate endotoxemic myocardial dysfunction independent of neutrophil accumulation. Am J Physiol Regul Integr Comp Physiol. 2002;283:R477–86.
Ye X, Ding J, Zhou X, Chen G, Liu SF. Divergent roles of endothelial NF-kappaB in multiple organ injury and bacterial clearance in mouse models of sepsis. J Exp Med. 2008;205:1303–15.
Hoover DB, Ozment TR, Wondergem R, Li C, Williams DL. Impaired heart rate regulation and depression of cardiac chronotropic and dromotropic function in polymicrobial sepsis. Shock. 2015;43:185–91.
Moniotte S, Belge C, Sekkali B, Massion PB, Rozec B, Dessy C, et al. Sepsis is associated with an upregulation of functional beta3 adrenoceptors in the myocardium. Eur J Heart Fail. 2007;9:1163–71.
Marshall M, Anilkumar N, Layland J, Walker SJ, Kentish JC, Shah AM, et al. Protein phosphatase 2A contributes to the cardiac dysfunction induced by endotoxemia. Cardiovasc Res. 2009;82:67–76.
Hobai IA, Edgecomb J, LaBarge K, Colucci WS. Dysregulation of intracellular calcium transporters in animal models of sepsis-induced cardiomyopathy. Shock. 2015;43:3–15.
Ji Y, Ren X, Zhao Y, Dong L, Wu L, Su J. Role of intracardiac angiotensin II in cardiac dysfunction of rat during septic shock. Chin Med J (Engl). 1996;109:864–7.
Wagner S, Schürmann S, Hein S, Schüttler J, Friedrich O. Septic cardiomyopathy in rat LPS-induced endotoxemia: relative contribution of cellular diastolic Ca(2+) removal pathways, myofibrillar biomechanics properties and action of the cardiotonic drug levosimendan. Basic Res Cardiol. 2015;110:507.
Koesters A, Engisch KL, Rich MM. Decreased cardiac excitability secondary to reduction of sodium current may be a significant contributor to reduced contractility in a rat model of sepsis. Crit Care. 2014;18:R54.
Drosatos K, Lymperopoulos A, Kennel PJ, Pollak N, Schulze PC, Goldberg IJ. Pathophysiology of sepsis-related cardiac dysfunction: driven by inflammation, energy mismanagement, or both? Curr Heart Fail Rep. 2015;12:130–40.
Jia L, Takahashi M, Morimoto H, Takahashi S, Izawa A, Ise H, et al. Changes in cardiac lipid metabolism during sepsis: the essential role of very low-density lipoprotein receptors. Cardiovasc Res. 2006;69:545–55.
Schilling J, Lai L, Sambandam N, Dey CE, Leone TC, Kelly DP. Toll-like receptor-mediated inflammatory signaling reprograms cardiac energy metabolism by repressing peroxisome proliferator-activated receptor γ coactivator-1 signaling. Circ Heart Fail. 2011;4:474–82.
Drosatos K, Pollak NM, Pol CJ, Ntziachristos P, Willecke F, Valenti MC, et al. Cardiac Myocyte KLF5 Regulates Ppara Expression and Cardiac Function. Circ Res. 2016;118:241–53.
Drosatos K, Khan RS, Trent CM, Jiang H, Son NH, Blaner WS, et al. Peroxisome proliferator-activated receptor-γ activation prevents sepsis-related cardiac dysfunction and mortality in mice. Circ Heart Fail. 2013;6:550–62.
Smeding L, van der Laarse WJ, van Veelen TA, Lamberts RR, Niessen HW, Kneyber MC, et al. Early myocardial dysfunction is not caused by mitochondrial abnormalities in a rat model of peritonitis. J Surg Res. 2012;176:178–84.
Chen HW, Hsu C, Lu TS, Wang SJ, Yang RC. Heat shock pretreatment prevents cardiac mitochondrial dysfunction during sepsis. Shock. 2003;20:274–9.
Larche J, Lancel S, Hassoun SM, Favory R, Decoster B, Marchetti P, et al. Inhibition of mitochondrial permeability transition prevents sepsis-induced myocardial dysfunction and mortality. J Am Coll Cardiol. 2006;48:377–85.
Zang QS, Sadek H, Maass DL, Martinez B, Ma L, Kilgore JA, et al. Specific inhibition of mitochondrial oxidative stress suppresses inflammation and improves cardiac function in a rat pneumonia-related sepsis model. Am J Physiol Heart Circ Physiol. 2012;302:H1847–59.
Cimolai MC, Alvarez S, Bode C, Bugger H. Mitochondrial mechanisms in septic cardiomyopathy. Int J Mol Sci. 2015;16:17763–78.
Neri M, Riezzo I, Pomara C, Schiavone S, Turillazzi E. Oxidative-nitrosative stress and myocardial dysfunctions in sepsis: Evidence from the literature and postmortem observations. Mediators Inflamm. 2016;2016:3423450.
Communal C, Sumandea M, de Tombe P, Narula J, Solaro RJ, Hajjar RJ. Functional consequences of caspase activation in cardiac myocytes. Proc Natl Acad Sci U S A. 2002;99:6252–6.
Nevière R, Fauvel H, Chopin C, Formstecher P, Marchetti P. Caspase inhibition prevents cardiac dysfunction and heart apoptosis in a rat model of sepsis. Am J Respir Crit Care Med. 2001;163:218–25.
Kumar A, Kumar A, Michael P, Brabant D, Parissenti AM, Ramana CV, et al. Human serum from patients with septic shock activates transcription factors STAT1, IRF1, and NF-kappaB and induces apoptosis in human cardiac myocytes. J Biol Chem. 2005;280:42619–26.
Neviere R, Hassoun SM, Decoster B, Bouazza Y, Montaigne D, Maréchal X, et al. Caspase-dependent protein phosphatase 2A activation contributes to endotoxin-induced cardiomyocyte contractile dysfunction. Crit Care Med. 2010;38:2031–6.
Carlson DL, Willis MS, White DJ, Horton JW, Giroir BP. Tumor necrosis factor-alpha-induced caspase activation mediates endotoxin-related cardiac dysfunction. Crit Care Med. 2005;33:1021–8.
Niu J, Azfer A, Kolattukudy PE. Protection against lipopolysaccharide-induced myocardial dysfunction in mice by cardiac-specific expression of soluble Fas. J Mol Cell Cardiol. 2008;44:160–9.
Matsuno K, Iwata K, Matsumoto M, Katsuyama M, Cui W, Murata A, et al. NOX1/NADPH oxidase is involved in endotoxin-induced cardiomyocyte apoptosis. Free Radic Biol Med. 2012;53:1718–28.
Morelli A, Ertmer C, Westphal M, Rehberg S, Kampmeier T, Ligges S, et al. Effect of heart rate control with esmolol on hemodynamic and clinical outcomes in patients with septic shock: a randomized clinical trial. JAMA. 2013;310:1683–91.
Sanfilippo F, Santonocito C, Morelli A, Foex P. Beta-blocker use in severe sepsis and septic shock: a systematic review. Curr Med Res Opin. 2015;31:1817–25.
Acknowledgments
The authors’ work cited in this review was supported by grants from the National Natural Science Foundation of China (81372028, 81170222), the Guangzhou Science and Technology Projects (201508020005) and the Project of the Department of Education of Guangdong Province (No.2013KJCX0019). The funding agencies had no role in the study design, data collection and analysis, preparation of the manuscript or the decision to publish. All aspects of the experimental research on animals were approved by the Experimental Animal Care and Use Committee at Jinan University, which conform to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Authors’ contributions
XL conducted the literature review and drafted the manuscript. HW contributed to the review and the revised manuscript. Both authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lv, X., Wang, H. Pathophysiology of sepsis-induced myocardial dysfunction. Military Med Res 3, 30 (2016). https://doi.org/10.1186/s40779-016-0099-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40779-016-0099-9