
Abstract
Background
Saccade slowing has been proposed as endophenotype marker in Spinocerebellar Ataxia type 2 (SCA2), nevertheless the heritability of this trait has not been properly demonstrated. Thus the present paper was aimed to assess the heritability of different saccadic parameters in SCA2.
Methods
Forty-eight SCA2 patients, 25 preclinical carriers and 24 non-SCA2 mutation carriers underwent electronystagmographical assessments of saccadic eye movements as well as neurological examination and ataxia scoring. Estimates of heritability based on the intraclass correlation coefficients were calculated for saccade velocity, accuracy and latency as well as for age at disease onset from 36, 17 and 15 sibling pairs of SCA2 patients, preclinical carriers and controls, respectively.
Results
Saccade velocity was significantly reduced in SCA2 patients and preclinical carriers, whereas decreased saccade accuracy and increased saccade latency were only observed in the patients cohort. Intraclass correlation coefficient for saccade velocity was highly significant in SCA2 patients, estimating a heritability around 94%, whereas for the age at ataxia onset this estimate was around 68%.
Conclusions
Electronystagmographical measure of saccade velocity showed higher familial aggregation between SCA2 patients leading the suitability of this disease feature as endophenotype marker, with potential usefulness for the search of modifier genes and neurobiological underpinnings of the disease and as outcome measure in future neuroprotective clinical trials.
Similar content being viewed by others

Background
Endophenotypes are measurable components of a disease that have simpler associations to genetic underpinnings than the disease syndrome itself [1]. The term has been commonly applied in psychiatric genetics [2, 3] and recently in other diseases such as neurodegenerative disorders [4, 5].
Endophenotypes must to fit some criteria that define its validity and usefulness [1], such as: a) be associated with illness in the population, b) be heritable traits, c) be primarily state-independent (manifests in an individual whether or not illness is active), d) co-segregate within families, e) be found in non-affected family members at a higher rate than in the general population and f) be measured reliably. Moreover, considering that endophenotypes are biomarkers influenced by the same genetic factors conferring risk for a disease [1], its characterization offers several important advantages over clinical phenotypes in the understanding of gene pleiotropy, a well-established phenomenon of a single gene affecting multiple traits [6].
Among the hereditary ataxias, endophenotype term has been only applied in the Spinocerebellar Ataxia type 2 (SCA2) [7]. This autosomal dominant disease is caused by an unstable CAG repeat expansion on a coding region of ATXN2 gene, with the consequent expression of abnormally large polyglutamine tract in the ataxin-2 protein. SCA2 patients present a progressive cerebellar syndrome, usually accompanied by a combination of other non-cerebellar features such as slow saccades, peripheral neuropathy and others [8, 9]. Several of these non-cerebellar clinical characteristics appear before the ataxia onset and define the prodromal stage of the disease [10, 11]. SCA2 reaches the worldwide highest prevalence in Holguín, Cuba, as result of a founder effect [12, 13], identifying the Cuban SCA2 population as a homogeneous resource for genetic studies.
Electronystagmographical assessments have led to characterize the saccade slowing as SCA2 endophenotype due to this trait affects almost all patients [7, 14] and is present in a high proportion of preclinical carriers [15]. In addition, saccade slowing exhibits a significant correlation with the CAG expansion size [7], and can be reliably measured by more than a decade before the ataxia onset [11]. Nevertheless, the heritability of saccade slowing in SCA2 has not been assessed, which limits its value as disease endophenotype. Thus, the aim of this study was to determine the heritability of saccadic abnormalities in SCA2 through the intraclass correlation method in a cohort of patients and preclinical carriers.
Methods
Participants
Forty-eight patients and 25 preclinical carriers were admitted to the Centre for Research and Rehabilitation of Hereditary Ataxias in Holguín (CIRAH) for this study. Twenty-four non-SCA2 mutation carriers from Holguin province were admitted as control group. The main demographical, clinical and molecular data of each group is shown in the Table 1. All procedures were in accordance with the declaration of Helsinki and the standards of the institutional Ethics Committee for Scientific Research from the CIRAH. All participants gave their written informed consent prior to the experiments.
Clinical assessments
All subjects underwent standardized neurological examination [16] and the Scale for the Assessment and Rating of Ataxia (SARA) [17] to evaluate cerebellar signs.
Electronystagmographical assessments
Horizontal saccades were recorded binocularly with a 2-channel electrooculography (EOG) device (Jaeger-Toennies, Germany), using Ag-AgCl electrodes over right and left outer canthus of the eyes. The EOG signal was amplified and bandpass filtered (0.2–70 Hz). The data were sampled at a frequency of 200 Hz with a time base of 1000 ms/division, sensitivity of 200 μV/division and a time constant of 8 s. To minimize head movements of participants during the recordings, a forehead and chin rest were used in all cases.
Subjects were asked to fixate the target in the central position and to redirect their gaze to the new location of the target as soon as it appeared in the periphery, and later back in the central position. Ten centrifugal saccades in both horizontal directions were registered at 60° predictable amplitudes of stimulus. EOG signals were calibrated for a horizontal angle of 30°. Each session of EOG lasted around only 6 min. Saccade latency, accuracy and maximal saccade velocity were analyzed.
Heritability analysis
The heritability estimates were obtained by analysis of variance components of the saccade parameters and age at onset among sibships and the error variance within the sibships, allowing for the computation of the sibling intraclass correlation (ICC) [18]. ICC values above zero indicate that error variance within the sibships is lower than variation between unrelated sibships. Then, assuming that the contribution of shared environmental factors to within-sibship variability is negligible, doubling the value of the sibling ICC provides an estimate of the heritability, which upper bound is the familiarity [19]. As expanded CAG repeats have a significant influence on the saccade velocity [7, 14, 15], we used an standard linear regression model to adjust the effects of this genetic parameter on the saccade variables before the analyses.
Heritability analyses in SCA2 patients were conducted in 21 sibling groups, 17 of them had two members, three had three members and only one had five members, which yielded a total of 36 sibling pairs. Among preclinical carriers, a total of 17 sibling pairs were obtained from 11 sibling groups, eight of them with two members and three with three members. Finally, 15 sibling pairs of non-SCA2 mutation carriers were obtained from nine siblings groups with two members and two siblings groups with three members.
Statistical analyses
Intergroup differences of frequencies for clinical features were computed by Chi-square (χ2) tests. One-way ANOVAs, followed by Fisher-LSD post hoc tests, were carried up to compare the saccade variables between groups. Correlation analyses (followed by Bonferroni correction) were performed by the Pearson correlation test. All analyses were conducted using the STATISTICA software package (StatSoft, Inc., 2003, v6).
Results
Clinical characterization
The frequency comparisons of main clinical features between the groups are showed in the Fig. 1A. All SCA2 patients exhibited a cerebellar syndrome, characterized by gait ataxia, postural instability, dysarthria, dysmetria and dysdiadochocinesia. Consistent with previous studies [8, 9, 12], muscle cramps and sensory abnormalities were significantly reported by patients and preclinical carriers, as compared to controls. Hyperreflexia was only significantly detected in preclinical carriers, but not in patients due to the effect of the peripheral sensory neuropathy affecting more notably the patients than preclinical carriers [20]. It is known that SCA2 mutation carriers commonly show a conversion from deep tendon hyperreflexia to hyporeflexia even since late prodromal stage which is associated to the progression of sensory neuropathy [10]. Slowed horizontal saccades at bedside examination were highly frequent in SCA2 patients. Age at disease onset (r = 0.55; p < 0.0001) and SARA score (r = 0.42; p = 0.003) were significantly correlated to expanded CAG repeats in the patients’s cohort.

Intergroup comparisons of clinical (a) and electronystagmographical abnormalities (b). Bars represent the mean, and error bars the standard error of the means. The findings of X2 test and the Fisher LSD post-hoc comparisons are showed by letters (a,b,c) code. The same letter above distinct bars represents no significant differences (p > 0.05), whereas different letter means significant differences (p < 0.05)
Characterization of saccade abnormalities
Similar to preceding works [7, 14, 15], saccade velocity was significantly reduced in SCA2 patients and preclinical carriers, whereas decreased saccade accuracy and increased saccade latency were only observed in SCA2 patients (Fig. 1b).
Correlation analyses only disclosed significant associations of saccade velocities with expanded CAG repeats (r = 0.60; p = 0.0001), age at onset (r = −0.45; p = 0.002), aging (r = 0.59; p = 0.0001) and SARA score (r = −0.37; p = 0.012) in SCA2 patients.
Heritability estimates.
Analyses of variance components of residual age at onset in SCA2 patients showed an ICC of 0.34, suggesting upper bounds of heritability or familiarity of ~68%. For saccade parameters, the data of heritability analyses are presented in Table 2.
The sibling ICCs were only significant for saccade velocity in the SCA2 patients, indicating that the within-siblings variability is significantly less than the between-siblings variability. Therefore, the upper bound of heritability for saccade velocity was ~94%. No significant ICCs were observed for the remaining saccade parameters in SCA2 patients as well as for all saccade parameters in preclinical carriers and controls.
Discussion
This paper is the first study assessing the heritability of saccades in SCA2 or any hereditary ataxia. According to our results, 94% of the residual variance of saccade velocity is explained by genetic factors, regardless the expanded CAG repeats, and/or environmental factors shared by families, supporting the classification of this oculomotor trait as disease endophenotype.
Our findings contradict a previous paper [21] reporting significant differences in saccade velocity between two SCA2 monozygotic twin patients. This discrepancy could be explained by the reduced number of twin pairs enrolled and the existence of non-germline factors that differentially affect each twin, such as somatic mosaicism, epigenetic factors and differential heteroplasmy of disease-modifying mitochondrial mutations.
It is known that somatic mutations continuously occur along the life and twins may show different somatic mutation rates depending on environmental influence [22]. Indeed, previous evidences have suggested that somatic mosaicism of expanded CAG repeats is an important modifying factor of SCA2 disease progression [23,24,25] and its prevention by CAA-interruptions in the ATXN2 gene’s CAG tract seems to be related to the SCA2 parkinsonian phenotype [25].
Moreover, differential DNA methylation levels in the ataxin-2 gene promoter might also have modifying effects on the SCA2 phenotype [26,27,28]. For example, a significant disease onset anticipation was observed in one SCA2 family without intergenerational CAG repeat instability but distinct patterns of DNA-methylation in the ATNX2 gene [26]. Interestingly, epigenetic differences in discordant monozygotic twins have been reported for amyotrophic lateral sclerosis, a devastating late-onset neurodegenerative condition in which the ATXN2 gene can be also implicated, but not differential methylation within its specific promoter was observed among the discordant twins [29]. Likewise, epigenetic differences in monozygotic twins explain discordant phenotypes for Alzheimer disease [30], major depressive disorder [31] and schizophrenia [32]. Nevertheless, none of these works have the effects of the differential epigenetics factors in specific disease endophenotypes.
In contrast to the abovementioned work, a large study enrolling 112 normal twins showed significant heritability estimates for saccade velocity and other oculomotor traits [33]. Nevertheless, longitudinal genetic studies in twins are needed to assess more robustly the genetic factors underlying the individual variability of these parameters.
The lack of familial aggregation for saccade accuracy and latency in SCA2 patients could be explained by the higher heterogeneity of these measures, which are more susceptible to additional influences such as the visual feedbacks that modify the accuracy of slowed saccades at flight [34], as well as attentional levels [35] and rehabilitation effects [36] on the saccade latency.
The heritability of saccade velocity was higher compared with the age at onset’s estimates obtained in the present study and in previously studied cohort [37, 38]. These results suggest that saccade velocity may be more appropriate for genetic studies than age at onset in SCA2 and so, it’s use could result in more sensitive approaches to detect disease modifier genes and the neurobiological bases of SCA2.
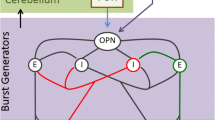
The link between the genetic basis of SCA2 and the saccade slowing consists in the preferential vulnerability of saccadic excitatory burst neurons (EBN) to expanded ataxin-2 [39]. Within the saccadic circuitry, these neurons receive excitatory inputs from the caudal pole of the contralateral cerebellar fastigial nucleus, which is controlled by inhibitory inputs from the Purkinje cells and in turns, they drive abducens motoneurons to produce saccades [40]. Then, this susceptibility could be explained by the excitotoxic effects caused the deliberated cerebellar outputs as result of the loss of inhibitory control from the Purkinje cells [8, 9]. Additionally, expanded ataxin-2 could impair the calcium signaling pathways, mitochondrial function and bioenergetic sensoring in EBN cells [41,42,43,44], three key functions for these high-energy consuming neurons.
Therefore, the differences observed in the saccade velocity between SCA2 patients with relatively similar mutation sizes could result from differential effects of modifier genes on the PolyQ toxicity in the EBN cells. Hence, the study of patients who have saccade velocities out of the mean range adjusted on CAG repeats offers important advantages for the identification of these modifying genes and the understanding of its mechanisms of actions.
Then, with the higher heritability estimates observed in this paper for saccade slowing, we satisfy all criteria defining endophenotypes, which were shown in Introduction and thus we offer new evidences about the conceptualization of this oculomotor trait as endophenotype marker in SCA2 (Fig. 2). As endophenotype, the saccade slowing confers neurobiological insights beyond the traditional symptomatic approaches of the disease and its assessment could be help to stratify SCA2 patients and preclinical carriers into more homogeneous cohorts, leading to more objective physiopathological approaches and meaningful clinical trials.

Criteria defining saccade slowing as endophenotypical marker in SCA2. White drawings denote subjects with normal saccade velocities, whereas blue drawings denote subjects with saccade slowing. Drawings with different blue shades denote different degrees of saccade slowing
A limitation of this paper was the inclusion of a relative small number of subjects. Nevertheless, even in this small cohort, we obtained significant heritability estimates for saccade slowing, suggesting the robustness of this heritable measure.
Conclusions
As conclusion, saccade slowing, as assessed by electronystagmography, showed higher familial aggregation between SCA2 patients, supporting the suitability of this oculomotor trait as disease endophenotype, a sensitive tool for the search of modifier genes and neurobiological underpinnings of the disease, as well as to evaluate the efficacy of future neuroprotective clinical trials.
Abbreviations
- CAG:
-
Cytosine-adenine-guanine
- EBN:
-
Saccadic excitatory burst neurons
- ICC:
-
Intraclass correlation
- PolyQ:
-
Polyglutamine
- SARA:
-
Scale for the Assessment and Rating of Ataxia
- SCA2:
-
Spinocerebellar Ataxia type 2
References
Gottesman II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry. 2003;160:636–45.
Cannon TD, Keller MC. Endophenotypes in the genetic analyses of mental disorders. Annu Rev Clin Psychol. 2006;2:267–90.
Ferrarelli F. Endophenotypes and biological markers of schizophrenia: from biological signs of illness to novel treatment targets. Curr Pharm Des. 2013;19(36):6462–79.
Louwersheimer E, Wolfsgruber S, Espinosa A, et al. Alzheimer's disease risk variants modulate endophenotypes in mild cognitive impairment. Alzheimers Dement. 2016;12(8):872–81.
Racette BA1, Good L, Antenor JA, McGee-Minnich L, Moerlein SM, Videen TO, et al. [18F] FDOPA PET as an endophenotype for Parkinson's disease linkage studies. Am J Med Genet B Neuropsychiatr Genet2006;141B(3):245–249.
Paaby AB, Rockman MV. The many faces of pleiotropy. Trends Genet. 2013;29(2):66–73.
Velázquez-Pérez L, Seifried C, Santos-Falcón N, Abele M, Ziemann U, Almaguer LE, et al. Saccade velocity is controlled by polyglutamine size in spinocerebellar ataxia 2. Ann Neurol. 2004;56(3):444–7.
Magaña JJ, Velázquez-Pérez L, Cisneros B. Spinocerebellar ataxia type 2: clinical presentation, molecular mechanisms, and therapeutic perspectives. Mol Neurobiol. 2013;47(1):90–104.
Auburger GW. Spinocerebellar ataxia type 2. Handb Clin Neurol. 2012;103:423–36.
Velázquez-Pérez L, Rodríguez-Labrada R, Canales-Ochoa N, Montero JM, Sánchez-Cruz G, Aguilera-Rodríguez R, et al. Progression of early features of spinocerebellar ataxia type 2 in individuals at risk: a longitudinal study. Lancet Neurol. 2014;13(5):482–9.
Velázquez-Pérez L, Rodríguez-Labrada R, Laffita-Mesa JM. Prodromal spinocerebellar ataxia type 2: Prospects for early interventions and ethical challenges. Mov Disord. 2017;32(5):708–18.
Velázquez Pérez L, Cruz GS, Santos Falcón N, Enrique Almaguer Mederos L, Escalona Batallan K, Rodríguez Labrada R, et al. Molecular epidemiology of spinocerebellar ataxias in Cuba: insights into SCA2 founder effect in Holguin. Neurosci Lett. 2009;454(2):157–60.
Auburger G, Diaz GO, Capote RF, Sanchez SG, Perez MP, del Cueto ME, et al. Autosomal dominant ataxia: genetic evidence for locus heterogeneity from a Cuban founder-effect population. Am J Hum Genet. 1990;46(6):1163–77.
Rodríguez-Labrada R, Velázquez-Pérez L, Auburger G, Ziemann U, Canales-Ochoa N, Medrano-Montero J, et al. Spinocerebellar ataxia type 2: measures of saccade changes improve power for clinical trials. Mov Disord. 2016;31(4):570–8.
Velázquez-Pérez L, Seifried C, Abele M, Rodríguez-Labrada R, Santos-Falcón N, Sánchez-Cruz g, et al. Saccade velocity is reduced in presymptomatic spinocerebellar ataxia type 2. Clin Neurophysiol. 2009;120(3):632–5.
Denny-Brown D, Dawson DM, Tyler HR. Handbook of neurological examination and case recording. 3rd ed. Cambridge, Mass: Harvard University Press; 1982.
Schmitz-Hubsch T, du Montcel ST, Baliko L, Berciano J, Boesch S, Depondt C, et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology. 2006;66(11):1717–20.
Schork NJ. The design and use of variance component models in the analysis of human quantitative pedigree data. Biom J. 1993;35(4):387–405.
Falconer D. Introduction to quantitative genetics. New York: Wiley; 1989.
Velázquez-Perez L, Rodríguez-Labrada R, Canales-Ochoa N, Sanchez-Cruz G, Fernandez-Ruiz J, Montero JM, et al. Progression markers of Spinocerebellar ataxia 2. A twenty years neurophysiological follow up study. J Neurol Sci. 2010;290(1–2):22–6.
Anderson JH, Christova PS, Xie TD, Schott KS, Ward K, Gomez CM. Spinocerebellar ataxia in monozygotic twins. Arch Neurol. 2002;59(12):1945–51.
Castillo-Fernandez JE, Spector TD, Bell JT. Epigenetics of discordant monozygotic twins: implications for disease. Genome Med. 2014;31;6(7):60.
Laffita-Mesa, JM, Vazquez-Mojena, Y, Cuello-Almarales, DA, Velazquez-Perez LC. The contribution of the self PolyQ load somatic mosaicism in the CNS to the onset, disease duration and progression rate of SCA2 and phenotypic delineation. Mov Disord 2012;27;Suppl1:1487.
Matsuura T, Sasaki H, Yabe I, Hamada K, Hamada T, Shitara M, Tashiro K. Mosaicism of unstable CAG repeats in the brain of spinocerebellar ataxia type 2. J Neurol. 1999;246(9):835–9.
Charles P, Camuzat A, Benammar N, et al. Are interrupted SCA2 CAG repeat expansions responsible for parkinsonism? Neurology. 2007;20;69(21):1970–5.
Laffita-Mesa J, Bauer P, Kourí V, Peña Serrano L, Roskams J, Almaguer Gotay D, et al. Epigenetics DNA methylation in the core ataxin-2 gene promoter: novel physiological and pathological implications. Hum Genet. 2012;131(4):625–38.
Bauer PO, Zumrova A, Matoska V, Mitsui K, Goetz P. Can ataxin-2 be down-regulated by allele-specific de novo DNA methylation in SCA2 patients? Med Hypotheses. 2004;63(6):1018–23.
Laffita-Mesa JM, Bauer P. Epigenetic heredity (deoxyribonucleic acid methylation): clinical context in neurodegenerative disorders and ATXN2 gene. Med Clin (Barc). 2014;143(8):360–5.
Young PE, Kum Jew S, Buckland ME, Pamphlett R, Suter CM. Epigenetic differences between monozygotic twins discordant for amyotrophic lateral sclerosis (ALS) provide clues to disease pathogenesis. PLoS One. 2017;12(8):e0182638.
Mastroeni D, McKee A, Grover A, Rogers J, Coleman PD. Epigenetic differences in cortical neurons from a pair of monozygotic twins discordant for Alzheimer’s disease. PLoS One. 2009;4:e6617.
Malki K, Koritskaya E, Harris F, Bryson K, Herbster M, Tosto MG. Epigenetic differences in monozygotic twins discordant for major depressive disorder. Transl Psychiatry. 2016;6(6):e839. https://doi.org/10.1038/tp.2016.101.
Dempster EL, Pidsley R, Schalkwyk LC, Owens S, Georgiades A, Kane F, Kalidindi S, et al. Disease-associated epigenetic changes in monozygotic twins discordant for schizophrenia and bipolar disorder. Hum Mol Genet. 2011;20:4786–96.
Katsanis J, Taylor J, Iacono WG, Hammer MA. Heritability of different measures of smooth pursuit eye tracking dysfunction: a study of normal twins. Psychophysiology. 2000;37:724–30.
Federighi P, Cevenini G, Dotti MT, Rosini F, Pretegiani E, Federico A, et al. Differences in saccade dynamics between spinocerebellar ataxia 2 and late-onset cerebellar ataxias. Brain. 2011;134:879–91.
Darrien JH, Herd K, Starling LJ, Rosenberg JR, Morrison JD. An analysis of the dependence of saccadic latency on target position and target characteristics in human subjects. BMC Neurosci. 2001;2:13.
Rodríguez JC, Velázquez L, Sánchez G, Almaguer Mederos L, Almaguer Gotay D, García-Rodriguez JC, et al. Evaluación de la restauración neurológica en pacientes con ataxia SCA2 cubana. Plast & Rest Neurol. 2008;7(1):13–8.
Figueroa KP, Coon H, Santos N, Velazquez L, Mederos LA, Pulst SM. Genetic analysis of age at onset variation in spinocerebellar ataxia type 2, 2017. Neurol Genet. 15;3(3):e155.
Almaguer-Mederos. Genetic and physiological modifierfactors of clinical phenotype in cuban families with Spinocerebellar Ataxia type 2. [Doctoral thesis]. In: Repository of doctoral thesis. 2010. http://tesis.repo.sld.cu/297/. Accessed 17 Jan 2017.
Geiner S, Horn AK, Wadia NH, Sakai H, Buttner-Ennever JA. The neuroanatomical basis of slow saccades in spinocerebellar ataxia type 2 (Wadia-subtype). Prog Brain Res. 2008;171:575–81.
Ramat S, Leigh RJ, Zee DS, Optican LM. What clinical disorders tell us about the neural control of saccadic eye movements. Brain. 2007;130(Pt 1):10–35.
Liu J, Tang T-S, Tu H, Nelson O, Herndon E, Huynh DP, et al. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 2. J Neurosci. 2009;29:9148–62.
Simon DK, Zheng K, Velazquez L, Santos N, Almaguer L, Figueroa KP, et al. Mitochondrial complex I gene variant associated with early age at onset in spinocerebellar ataxia type 2. Arch Neurol. 2007;64:1042–4.
Lastres-Becker I, Nonis D, Eich F, Klinkenberg M, Gorospe M, Kötter P, et al. Mammalian ataxin-2 modulates translation control at the pre-initiation complex via PI3K/mTOR and is induced by starvation. Biochim Biophys Acta. 2016;1862(9):1558–69.
Auburger G, Sen NE, Meierhofer D, Başak AN, Gitler AD. Efficient prevention of neurodegenerative diseases by depletion of starvation response factor Ataxin-2. Trends Neurosci. 2017;40(8):507–16.
Acknowledgements
This research was supported by Cuban Ministry of Public Health. We wish to thank to SCA2 patients, preclinical carriers and controls for their participation in this work.
Funding
This work was funded by the Cuban Ministry of Public Health.
Availability of data and materials
The datasets generated and/or analyzed during the current study are not publically available due to institutional regulations of the intellectual property policy. These data are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
RRL and LVP participated in the study design and coordination. RRL, NCO and JMM performed the electronystagmographical assessments. LVP, RRL and JMM performed the neurological assessments and ataxia scoring, YVM performed the molecular assessments. RRL and YVM performed the statistical analyses and drafted the manuscript. LVP performed the critical revision of the final version and all authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Authors’ information
LVP was recipient of the Georg Foster award from the Alexander Von Humboldt foundation in 2013. LVP and RRL are members of the Cuban Academy of Sciences.
Ethics approval and consent to participate
All procedures were in accordance with the declaration of Helsinki and the standards of the institutional Ethics Committee for Scientific Research from the CIRAH. All participants gave their written informed consent prior to the experiments.
Consent for publication
Not required for this paper.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Rodríguez-Labrada, R., Vázquez-Mojena, Y., Canales-Ochoa, N. et al. Heritability of saccadic eye movements in spinocerebellar ataxia type 2: insights into an endophenotype marker. cerebellum ataxias 4, 19 (2017). https://doi.org/10.1186/s40673-017-0078-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40673-017-0078-2