
Abstract
Poly (ADP-ribose) polymerases (PARPs) are an important family of nucleoproteins highly implicated in DNA damage repair. Among the PARP families, the most studied are PARP1, PARP2 and PARP 3. PARP1 is found to be the most abundant nuclear enzyme under the PARP series. These enzymes are primarily involved in base excision repair as one of the major single strand break (SSB) repair mechanisms. Being double stranded, DNA engages itself in reparation of a sub-lethal SSB with the aid of PARP. Moreover, by having a sister chromatid, DNA can also repair double strand breaks with either error-free homologous recombination or error-prone non-homologous end-joining. For effective homologous recombination repair, DNA requires functional heterozygous breast cancer genes (BRCA) which encode BRCA1/2. Currently, the development of PARP inhibitors has been one of the promising breakthroughs for cancer chemotherapy. In March 2017, the United States Food and Drug Administration (FDA) approved niraparib for maintenance therapy of recurrent gynecologic cancers (epithelial ovarian, primary peritoneal and fallopian tube carcinomas) which are sensitive to previous platinum based chemotherapy irrespective of BRCA mutation and homologous recombination deficiency status. It is the third drug in this class to receive FDA approval, following olaparib and rucaparib and is the first global approval for maintenance therapy of the aforementioned cancers. Niraparib preferentially blocks both PARP1 and PARP2 enzymes. The daily tolerated dose of niraparib is 300 mg, above which dose limiting grade 3 and 4 toxicities were observed. In combination with humanized antibody, pembrolizumab, it is also under investigation for those patients who have triple negative breast cancer. By and large, there are several clinical trials that are underway investigating clinical efficacy and safety, as well as other pharmacokinetic and pharmacodynamic profiles of this drug for various malignancies.
Similar content being viewed by others


Introduction
General principles of DNA repair
The human genome is constantly under stress due to insults from both endogenous (free radicals or reactive oxygen species derived from metabolic processes) and exogenous (irradiation, chemicals, clinical drugs, and viruses, among others) sources. This results in routine DNA damage that may in turn lead to a serious genetic instability and cell death if it is left unrepaired. Being double stranded and having a sister chromatid, DNA repairs itself prior to cell division in any one of the following repair mechanisms (Fig. 1) [1,2,3].

Illustrated diagram describing DNA repair pathways. (Note: BER, base excision repair; DSB, double strand break; HRR, homologous recombination repair; NER, nucleoside excision repair; NHEJ, non-homologous end-joining; SSB, single strand break)
Direct reversal is highly efficient, applicable when there is a single lesion, and is essentially error-free. The lesion may be tolerated or bypassed if it does not have a significant risk on the ongoing DNA replication and the genetic stability in general. Coming to the single strand break (SSB) repair, since the damage involves one strand of replicating DNA, it can be repaired by undergoing either excision of damaged site (base or nucleotide) or correcting the mismatch bases complementary to the anti-sense (template) strand [2, 4, 5].When a SSB is left unrepaired due to genetic and/or epigenetic factors, it will progress to double strand break (DSB) during DNA replication. By having homologous chromosomes, DNA has a back up to undergo DSB repair either by error-free homologous recombination (HR) or error-prone non-homologous end-joining (NHEJ). If the DSB is left unrepaired, it leads to a breakdown of the chromosome into smaller fragments, genomic instability, cell cycle arrest and apoptosis [1, 4, 6, 7]. The more faithful process of HR repair of DSBs involves localization of BRCA-1 and BRCA-2 proteins encoded from breast cancer gene to sites of DNA damage, resection of the DSB, and gap-filling DNA synthesis using the homologous sister chromatid as a template [8]. Before the DNA enters the repair process, cellular response depends upon the magnitude of the damage, resulting in induction of cell-cycle checkpoint pathways and DNA repair mechanisms. G2/M check point is a critical point where DNA must be repaired before the cell enters cell division/mitosis. If the damage is extensive and irreparable, induction of cell death occurs [3, 7].
The role of poly (ADP-ribose) polymerases (PARPs) in DNA repair
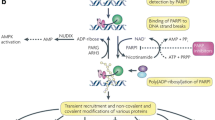
PARPs are a member of nuclear protein enzymes highly implicated in DNA damage repair. During SSB, PARP detects the damaged site and undergoes post translational modification of targeted proteins by the process known as ADP-ribosylation. This process creates a conducive environment for recruiting several DNA repair proteins including topoisomerases, DNA ligase III, DNA polymerase β, and scaffolding proteins such as X-ray cross complementing protein 1 (XRCC1), among others. The ribosylation process also leads in relaxation of tightened chromatins and histones and results in unwinding of DNA to make it accessible for repair processes. What is more, PARP facilitates HR by recruiting factors such as ataxia telangiectasia-mutated kinase (ATM), mitotic recombination 11 (Mre11), and Nijmegen breakage syndrome 1 (Nbs1) to sites of DSBs (Fig. 2) [9,10,11]. When PARP activity is compromised, these SSBs cannot be repaired and progress to DSBs at DNA replication forks. In a normal cell, there is a cellular backup by which DSBs are repaired with the aid of HR, a mechanism different from base excision repair (BER) and hence, even in the absence of PARP activity and loss of BER, DNA repair can be effectively taken place by this pathway. However, cells can have a double-hit whereby both BER and HR are compromised. These cells rely on error-prone NHEJ for damage repair, which results in DNA instability and chromosomal aberrations, eventually resulting in apoptosis. The dual-insult of HR and BER defects results in “synthetic lethality” justifying the potent and lethal synergy between these two otherwise non-lethal event when they occur alone [1, 11]. Therefore, this review aims to address the role of common PARP inhibitors on cancer chemotherapy with special focus on niraparib and its first global approval for maintenance therapy of gynecologic cancers.

DNA repair processes with the aids of poly (ADP-ribose) polymerase. (Notes: XRCC1, X-ray cross complementing protein 1; ATM, ataxia telangiectasia-mutated kinase; MRE11, mitotic recombination 11. Others include: - Nijmegen breakage syndrome 1 (Nbs1), DNA ligase III, and DNA polymerase β)
Methods
A total of 945 articles were retrieved from various legitimate data bases and indexing services (Directory of open access journals, PubMed, PubMed Central, MEDLINE, Scopus and ProQuest), as well as other supplemental sources and search engines (CrosRef, WorldCat, and Google Scholar) with the aid of key terms: “PARP”, “PARP inhibitors”, “DNA repair”, “cancer”, “malignant tumors”, “Niraparib”, “MK-4827”, “Zejula”, “maintenance therapy” and “companion diagnostic*”. Boolean operators (AND, OR, NOT) were appropriately used for increasing the chance of obtaining relevant literature for this topic. Moreover, truncation was also applied to expand the literature searches and increase the number of related articles for inclusion. Following database searches and in-depth screening of each article by authors, majority of articles were removed from this study including duplicate articles from various databases and search engines; unrelated titles and abstracts; abstracts without full texts and full texts with insufficient information for data extraction. Finally, 68 references were included for the study from which, 22 articles were critically reviewed to summarize the current therapeutic profile of niraparib. Coming to the data extraction process, general background information concerning the role of PARP in DNA repair, cancer therapeutics and earlier inhibitors of this enzyme (s) were highlighted. Coming to the drug of interest, niraparib, data regarding the chemistry, pharmacology, primary outcomes of preclinical studies as well as completed clinical trials were extracted from respective individual studies. Furthermore, important data about ongoing clinical trials were also retrieved upon visiting https://clinicaltrials.gov/ web site. Data were collected from June to August, 2017.
Review
The role of PARP inhibitors in cancer chemotherapy
Ovarian cancer is the 5th leading cause of cancer-related deaths in women in the United States. It is estimated that in 2017, more than 22,440 women will be diagnosed with ovarian cancer leading to more than 14,080 deaths. However, cancer chemotherapy has shown little improvement over time [12, 13]. So far, recurrent ovarian cancer has been dichotomized into two categories based on the sensitivity of platinum based therapy as ‘platinum sensitive’ and ‘platinum resistant’. This classification considers the number of months within which the patient can be freed from platinum based therapy from the last time of infusion to recorded recurrence. From molecular perspective, there is no clear cut demarcation to divide cancers based on sensitivity to platinum chemotherapy. With advancement in science and technology, targeted therapies like PARP inhibitors have led to a more holistic approach to the treatment of disease recurrence [14,15,16]. It is estimated that approximately 50% of high-grade serous ovarian cancer (HGSOC) show alterations in the Fanconi anemia–BRCA pathway [17]. Mutations in this pathway, including genes involved in HR repair such as RAD51C/D, and BRIP1 have been associated with homologous recombination deficiency (HRD) and hereditary ovarian cancer [18]. Epigenetic mechanisms can also contribute to the development of HRD. For example, silencing of BRCA1 in HGSOC has been shown to occur via epigenetic changes such as hypermethylation of BRCA1 promoter [17]. PARP inhibitors have been developed in the recurrence and maintenance treatment settings in epithelial ovarian cancer. As they inhibit SSB repair, inducing synthetic lethality in cells with underlying HRD as seen in BRCA1/2 mutant tumors (Fig. 3). Marked responses have been observed in ovarian cancers with BRCA1/2 mutation, even if up to 50% of HGSOC having HRD may also be better treated compared to cancers with HRD negative (HR proficient) genotypes [19, 20].

The influence of PARP inhibitors and BRCA mutation status in DNA repair and apoptosis of cancer cell. (Note: SSB, single strand break; BER, base excision repair; DSB, double strand break; HRR, homologous recombination repair; NHEJ, Non-homologous end-joining)
Among the newly diagnosed ovarian cancers, around 25% of them carry BRCA1/2 mutations from which majority (18%) are germline mutations whereas the remaining (7%) cancers are associated with somatic mutations [21]. In the absence of either germline or somatic mutations of BRCA1/2, HRD can occur in a variety of mechanisms as studied in several serious malignancies. The HR defects that occur due to aberration in genes other than BRCA exhibit closely resembling phenotypic characteristics secondary to PARP inhibitors and the condition is referred to as ‘BRCA-ness. This has been clearly demonstrated in either genetic mutations of ATM, RAD51C/D, check point kinases 2 (CHK2), phosphatase and tensin homolog (PTEN) or epigenetic silencing of BRCA1/2 promoter as having difficulty of effectively repairing DSBs by HR [22,23,24,25].
Overview of common PARP inhibitors that got FDA approval
Beginning from the first efficacious in vitro study of PARP inhibitors, several agents have been studied in ovarian cancer [26, 27]. The best studied include olaparib, veliparib, talazoparib, rucaparib and niraparib (Table 1). Each PARP inhibitor possesses subtly different targets of PARP isoenzymes [28, 29]. PARP-1 is the most abundant and founding member of poly ADP-ribosylating proteins (a family of around 17 proteins) known as the ADP-ribosyltransferase diphtheria toxin-like proteins [16, 30]. In addition to the canonical targets of niraparib, PARP1 and PARP2, subsequent functional validation suggested that inhibition of deoxycytidine kinase by it could have detrimental effects when combined with nucleoside analogs used for the treatment of various diseases [31].
Olaparib
The PARP inhibitor olaparib (Lynparza®) was the first to be approved in advanced ovarian cancer therapy for those with gBRCA1/2 mutations. Following phase I safety and efficacy studies, a multicenter phase II study demonstrated response to olaparib in patients with gBRCA1/2 mutations in recurrent ovarian cancer and breast cancer with at least 3 prior chemotherapy regimens. A subgroup analysis of patients with advanced ovarian cancer patients revealed an overall response rate (ORR) of 34% [32, 33]. These findings led to the fast track approval of olaparib capsules in the USA in December, 2014 as fourth line therapy. The approval of olaparib for advanced ovarian cancer patients with BRCA 1/2 mutation and who are intensively pretreated with chemotherapy becomes a major therapeutic breakthrough for this lethal and difficult to treat disease. Even if it is the first agent in its class to get fast track approval by FDA, rucaparib and niraparib received recent approval in slightly different settings. Several PARP inhibitors are also under clinical development either alone or in combination with other treatment modalities including radiation therapies, cytotoxic agents and antiangiogenic agents [6] (Table 1). On August 17, 2017, FDA granted regular approval to olaparib tablets (Lynparza, AstraZeneca) for the maintenance treatment settings of adult patients with recurrent gynecologic cancers, who are in a complete or partial response to platinum-based chemotherapy [34]. The recent approval of olaparib in the maintenance setting was based on two randomized, placebo-controlled, double-blind, multicenter trials in patients with recurrent gynecologic cancers who were in response to platinum-based therapy.
Study 19 (NCT00753545), a phase II trial engaged 265 patients with platinum sensitive HGSOC regardless of BRCA status (1:1) to receive olaparib capsules 400 mg orally twice daily or placebo. Study 19 demonstrated a statistically significant improvement in investigator-assessed progression-free survival (PFS) in patients treated with olaparib compared to placebo (Hazard Ratio (HR') = 0.35; 95% CI: 0.25, 0.49; p < 0.0001). The estimated median PFS was 8.4 months and 4.8 months in the olaparib and placebo arms, respectively. Clinical outcomes between placebo- and olaparib-treated patients with somatic BRCA1/2 mutations were similar to those with germline BRCA1/2 mutations, indicating that patients with somatic BRCA1/2 mutations benefit from treatment with olaparib in maintenance setting [35]. SOLO-2/ENGOT-Ov21 (NCT01874353), a phase III clinical trial, randomized 295 patients with recurrent germline BRCA-mutated gynecologic cancers (2:1) to receive olaparib tablets 300 mg orally twice daily or placebo. SOLO-2 demonstrated a statistically significant improvement in investigator-assessed PFS in patients randomized to olaparib compared with those who received placebo (HR' = 0.30; 95% CI: 0.22, 0.41; p < 0.0001). The estimated median PFS was 19.1 and 5.5 months in the olaparib and placebo arms, respectively [36].
Rucaparib
Coming to rucaparib (Rubraca®), it is a potent inhibitor of PARP1, PARP2 and PARP 3 with the greatest affinity towards PARP1. Both phase I and II clinical trials demonstrated that it has a promising efficacy in ovarian cancers with both BRCA mutation (including germline and somatic subtypes) and tumors with HRD positive. In December, 2016, US FDA approved rucaparib for the treatment of advanced ovarian cancer patients with general BRCA1/2 mutation and who took at least 2 lines of previous platinum based therapy. The accelerated approval was based upon ORR (54%) [37]. In ARIEL2 part 1, patients with recurrent, platinum-sensitive, high-grade ovarian carcinomas were classified into one of three predefined HRD subgroups on the basis of tumor mutational analysis: BRCA mutant (deleterious germline or somatic), BRCA wild-type and loss of heterozygosity (LOH) high (LOH high group), or BRCA wild-type and LOH low (LOH low group). Median PFS after rucaparib treatment was 12·8 months (95% CI 9·0–14·7) in the BRCA mutant subgroup, 5·7 months (5·3–7·6) in the LOH high subgroup, and 5·2 months (3·6–5·5) in the LOH low subgroup. PFS was significantly longer in the BRCA mutant (HR' = 0·27, 95% CI 0·16–0·44, p < 0·0001) and LOH high (0·62, 0·42–0·90, p = 0·011) subgroups compared with the LOH low subgroup. Part 2 of the ARIEL2 trial is ongoing, and it will prospectively evaluate rucaparib responsiveness in patient sub-groups defined by LOH scores [38].
ARIEL3 (NCT01968213), a randomised, double-blind, placebo-controlled, phase 3 trial, demonstrated improved PFS by investigator review for rucaparib compared with placebo in all three primary efficacy analyses: BRCA mutation (16.6 months vs. 5.4 months; HR': 0.23, P < 0.001); HRD-positive (13.6 months vs. 5.4 months; HR': 0.32, P < 0.001); overall intent-to-treat populations (10.8 months vs. 5.4 months; HR': 0.36, P < 0.001) [39]. On October 10, 2017, FDA approval was sought for maintenance therapy of rucaparib in ovarian cancer following promising findings from ARIEL3 clinical trial (Table 1) [40].
Niraparib
Among the PARP inhibitor series, niraparib (Zejula) is the third drug in this class, to receive FDA approval for cancer chemotherapy. However, the previous PARP inhibitors, olaparib and rucaparib have been approved for simple treatment rather than maintenance for those patients who are responsive to previous chemotherapy. Niraparib has become the first drug that got global approval for maintenance therapy of patients with recurrent gynecologic cancers regardless of their BRCA mutation and HRD status [41, 42] (Table 1). Hereafter, this review focuses on summarizing the chemistry, pharmacology, preclinical studies, completed and ongoing clinical trials as well as toxicological concerns of niraparib.
Chemistry, pharmacology and preclinical data of niraparib
Niraparib (Zejula, MK-4827; (2-[4-[(3S)-piperidin-3-yl], phenyl]indazole-7-carboxamide) is a potent and selective inhibitor of PARP-1 and PARP-2 enzymes. Its molecular formula is C19H20N4O and has a molar mass of 320.396 g/mol (Fig. 4) [43].

Chemical structure of niraparib
Niraparib is administered orally on once daily basis and can be taken without consideration to meals since food does not significantly affect the absorption and/or the metabolism of niraparib [44]. It is readily absorbed from the oral route and its bioavailability is approximately 73% in humans as per the phase III clinical trials revealed. There is no significant difference in pharmacokinetic parameters between the feeding and fasting states. For example, the mean ratios of maximum plasma concentration (Cmax) and area under the curve (AUC0-∞) in the fed to fasted state were 0.83 and 1.08, respectively. In both cases, niraparib possesses long terminal half-life (t1/2) of greater than 2 days (57 and 59 h for feeding and fasting states, respectively). This is consistent with once daily dosing of niraparib for cancer chemotherapy [45]. Coming to the metabolic profile of niraparib, it had been shown that niraparib is moderately metabolized in humans primarily via hydrolytic (phase I) and conjugative (Phase II) pathways in the liver. The hepatic phase I metabolism is via carboxylesterase-catalyzed amide hydrolysis, leading to the formation of inactive metabolite (carboxylic acid derivative) which in turn undergoes a phase II conjugation reaction called glucuronidation for ease of biliary and renal excretion [46]. Unlike rucaparib and olaparib, studies indicated that cytochrome P-450 enzymes (CYP) including CYP 1A2 play a negligible role in the metabolism of niraparib in humans [46,47,48]. Moreover, from the total administered dose, 31.6% and 40.0% are recovered in feces and urine, respectively, whereas 29.9% of the dose is excreted unchanged in the urine and feces [46].
In preclinical trial of rodent’s model, it was also indicated that almost similar concentration-time profile of niraparib was obtained from both brain and plasma samples, and the mean brain-to-plasma concentration ratios following a single oral dose ranged from 0.85–0.99 of the brain Tmax (Table 2) [49]. Moreover, different preclinical and clinical studies indicated that niraparib induces chemo- and radio-sentiziation and hence facilitates cell death in cancer cells. Combinational therapy of niraparib with topoisomerase inhibitors such as irinotecan chemosensitize cancer cells as observed both in in-vivo and in vitro studies. In several breast and lung cancer models, niraparib enhances the therapeutic effects of radiation therapy independent of the tumor suppressor P-53 function (Table 3). Having impaired the BER function by niraparib, exposure of radiation converts the sub-lethal SSBs into lethal DSBs leading to synthetic lethality of cancer cells [50,51,52].
The activity of niraparib in sporadic prostate cancer provides a strong clinical evidence for developing PARP inhibitor based therapies for metastatic castration resistant prostate cancer (CRPC). Erythroblast transformation-specific (ETS) gene rearrangement and loss of PTEN are among the common genetic alterations in prostate cancer and have been linked to increased sensitivity to PARP inhibitors in preclinical models [4]. Moreover, compared to other PARP inhibitors, niraparib was found to be effective as a monotherapeutic agent in several cell lines tested in pediatric diffuse intrinsic pontine glioma and pediatric high-grade astrocytoma [53].
Evidences from clinical trials of niraparib
Niraparib is approved for the maintenance therapy of adult patients with recurrent gynecologic cancers that are in a complete or partial response to previous platinum-based chemotherapy. In Europe, it is under review by European Medicine Agency for maintenance therapy of recurrent ovarian cancer patients who are sensitive to earlier platinum chemotherapy [42].
In a phase 1 dose-escalation study, 100 patients with advanced solid tumors were enrolled in two parts. In part A, cohorts of three to six patients, enriched for BRCA1 and BRCA2 mutation carriers, received niraparib daily at ten escalating doses from 30 mg to 400 mg in a 21-day cycle to establish the maximum tolerated dose. In part B, further investigation was conducted to determine the maximum tolerated dose in patients with sporadic platinum-resistant high-grade serous ovarian cancer and sporadic prostate cancer. Considering various side effects associated with dose escalation, 300 mg/day was established as the maximum tolerated dose (Table 4). Niraparib was found to inhibit tumor growth in models with loss of BRCA activity and loss of function mutation of tumor suppressor PTEN proteins. Sandhu et al. administered niraparib to a small cohort of patients enriched for BRCA-deficient and sporadic cancers associated with defects in HR repair. Thirty-nine patients were treated, 11 of whom had gBRCA1/2 mutations. Eight of the BRCA1/2 mutation carriers with ovarian cancer had a partial response. What is more, antitumor activity was also found in sporadic HGSOC [54].
While only a minority of prostate cancer patients carries germline mutations, many sporadic CRPCs harbor epigenetic and genetic disruption of genes that are crucial for the HR pathway including BRCA1, BRCA2, FANC, ATM, CHEK1/2, MRE11A and RAD51. Some of these aberrations have been associated with responsiveness to PARP inhibitors and platinum, justifying a synthetic lethality between platinum or PARP inhibitors and these sporadic DNA repair gene defects [55] (Table 4).
In the multinational, randomized, double blind, phase III clinical trial (ENGOT-OV16/NOVA trial), adult patients were dichotomized in two cohorts, each containing two arms, based on the status of gBRCA mutation (gBRCA cohort and non-gBRCA cohort). The categorization was based on BRCAnalysis CDxBRCA testing (Myriad Genetics, Salt Lake City, USA). Patients were randomly assigned in 2:1 ratio to receive 300 mg niraparib or placebo in each cohort. In this study, the primary end point (outcome) measured was the PFS. To further fine tune the efficacy of niraparib on different ovarian cancer histological subtypes, the non gBRCA cohort was further classified in to HRD positive (HR deficient) and HRD negative (HR proficient) based on myChoice HRD™ test (Myriad Genetics). In this trial, the total number of patients enrolled was 553 and from those 203 were assigned to gBRCA cohort (138 to treatment arm (niraparib) and 65 to placebo) while the remaining 350 patients were assigned to non gBRCA cohort (234:116 for niraparib to placebo group). In gBRCA cohort, patient in the niraparib arm had significantly longer median PFS period (21 months) compared to placebo group (5.5 months) [HR', 0.27; 95% CI: 0.17–0.4. Coming to the non gBRCA cohort with HRD positive patients, the median PFS was found to be 12.9 months and 3.8 months for niraparib and placebo groups, respectively [HR', 0.38; (95% CI: 0.24–0.59]. The overall PFS in non-gBRCA cohort regardless of HRD status was 9.3 months vs 3.9 months [HR', 0.45; 95% CI: 0.34–0.61]. Generally, from this clinical trial, we can conclude that niraparib can be given to recurrent ovarian cancer patients regardless of gBRCA and HRD status (Table 4) [56].
The successful phase 3 niraparib ENGOT-OV16/NOVA trial also included a substantial number of patients with platinum resistant ovarian cancer. In this study, 181 patients were assigned to placebo (65 gBRCAmuts and 116 non-gBRCAmuts). The prevalence of platinum resistance estimated for the gBRCAmut, non-gBRCAmut, and pooled cohorts were 42%, 53%, and 49%, respectively (Table 4). Approximately half of the patients in the NOVA study, where niraparib treatment met its primary endpoint by prolonging PFS following a response to platinum, had developed resistance at last line of chemotherapy (Table 4) [57].
Toxicological concerns of niraparib
In phase I dose escalation trial, common treatment-related toxic effects were anemia (48%), nausea (42%), fatigue (42%), thrombocytopenia (35%), anorexia (26%), neutropenia (24%), constipation (23%), and vomiting (20%), and were predominantly grade 1 or 2. The most common grade 3 or 4 adverse events that were reported in the niraparib group were thrombocytopenia (33.8%), anemia (25.3%), and neutropenia (19.6%), which were managed with dose modifications [54]. Niraparib is also associated with serious risks, such as hypertension, hypertensive crisis, myelodysplastic syndrome, acute myeloid leukemia, and bone marrow suppression. Women who are pregnant or breastfeeding should not take niraparib because it may cause harm to a developing conceptus or a newborn baby [41, 56].
Companion diagnostic tests
Companion diagnostic tests are very critical to identify cancer patients who are best treated by PARP inhibitors. Myriad’s BRCA analysis CDx™ is the only FDA-approved test to determine olaparib treatment eligibility. Rucaparib’s companion diagnostic test (FoundationFocus™CDxBRCA test that detects germline and somatic BRCA1/2 mut) is the first FDA-approved next-generation sequencing (NGS)-based test designed to identify patients likely to respond to rucaparib [58,59,60]. Coming to niraparib, the eligibility is determined with myChoice HRD™ test (Myriad Genetics). While BRCA analysis CDx™, as the name explains, evaluates only BRCA, myChoiceHRD™, developed by the same company, evaluates LOH beyond BRCA and can be considered an enhancement of BRCA analysis CDx™. It is an NGS-based assay that assesses BRCA1/2 sequences, and genomic scarring (HRD score), composed by LOH, telomeric allelic balance and large-scale transitions [61].
Ongoing clinical trials
There are several clinical trials that are underway for invesigating safety, tolerability, efficacy, and other pharmacokinetic and pharmacodynamic profiles of niraparib in different treatment modalities (single and/or combination therapies) and cancers of diverse histological origin. Several phase I clinical trials are underway investigating the maximum tolerated dose of niraparib when used in combiation with different treatment modalitites: with enzalutamide in CRPC, with everolimus in ovarian cancer, as well as with temozolomide and irinotecan in case of ewing sarcoma, among others. In phase II clinical trials, the safety and efficacy of niraparib alone is on the way to be investigated. Coming to the phase III clinical trials, the primary outcome measures of niraparib alone and in comparison with physician’s choice have been under evaluation for maintenance therapy of advanced ovarian cancer patients following a response to front line platinum based therapy and human epidermal growth factor receptor 2 negative (HER2-), gBRCA mut-positive breast cancer patients, respectively(Table 5) [62].
Conclusion and future prospects
The role of PARP family enzymes in DNA repair and cancer therapeutics was well emphasized in this review article. The development of PARP inhibitors has become one of the promising breakthroughs and hot spots in the area of experimental oncology. As documented in various histological subtypes of cancer, there are several germline and/or somatic mutations, as well as epigenetic alterations compromising effective reparation of DSBs by HR repair. This will create medically important and selective situation whereby cancer cells will be subjected to dual insult of PARP inhibitors and mutations of HR genes including BRCA1/2. Normal cells are less likely to be affected by PARP inhibitors since they have functional HR for DSB. Based on this evidence, scientists are striving to discover PARP inhibitors which have superior safety and efficacy profiles than the existing medications for cancer chemotherapy. Even if niraparib is the third drug to get FDA approval from its class, it is the first one to receive global approval for maintenance therapy of patients with recurrent gynecologic cancers regardless of BRCA and HRD status. Maintenance therapy is an important part of cancer chemotherapy for patients who have responded positively to a primary treatment. Niraparib offers patients a new treatment option that may help delay the future growth of these cancers, regardless of whether they have a specific genetic mutation. Niraparib has also several important pharmacokinetic features including negligible interaction with food; once daily dosing regimen; less likely to interact with other coadministered drugs since it is primarily metabolized by hydrolytic and conjugative pathways, and lower dosage requirement than previously approved PARP inhibitors (olaparib and rucaparib). It is also a potent inhibitor of PARP 1 and PARP 2 enzymes. Evidence from randomized phase III clinical trials indicated that niraparib can be given to any ovarian cancer patients who are responsive to previous therapy. Additional feature here is that this drug can also be given to patients irrespective of HRD status: HRD negative (HR proficient) and HRD positive (HR deficient) cells as statistically significant median PFS was observed in niraparib arms of both cohorts compared to placebo. Generally, niraparib is under investigation either alone or in combination with other treatment modalities for several cancer types. Among them, niraparib alone is under study in phase III clinical trial for maintenance treatment of patients with advanced ovarian cancer following a response on front line platinum based therapy. In combination with pembrolizumab, it is at the transition of phase I/II trials investigating the dose limiting toxicity in triple negative breast cancer patients. Moreover, the efficacy (PFS) of niraparib in comparison with physicians’ choice is also under consideration in phase III (BRAVO) trial for HER2- breast cancer patients.
Abbreviations
- BER:
-
Base Excision Repair
- BRCA:
-
Breast Cancer gene
- CHK2:
-
Check point Kinase 2
- CRPC:
-
Castration Resistant Prostate Cancer
- DSB:
-
Double Strand Break
- ETS:
-
Erythroblast Transformation-Specific gene
- FDA:
-
Food and Drug Administration
- gBRCA:
-
Germline mutation of Breast Cancer gene
- HER2-:
-
Human Epidermal growth factor Receptor 2 negative
- HGSOC:
-
High Grade Serious Ovarian Cancer
- HR:
-
Homologous Recombination
- HR':
-
Hazard Ratio
- HRD:
-
Homologous Recombination Deficiency
- LOH:
-
Loss of Heterozygosity
- NHEJ:
-
Non Homologous End Joining
- PARP:
-
Poly (ADP-Ribose) Ploymerase
- PFS:
-
Progression Free Survival
- PTEN:
-
Phosphatase and Tensin homolog
- SSB:
-
Single Strand Break
- TNBC:
-
Triple Negative Breast Cancer
References
Liu JF, Matulonis UA. What is the place of PARP inhibitors in ovarian cancer treatment? Curr Oncol Rep. 2016;18(5):29.
Rabenau K, Hofstatter E. DNA damage repair and the emerging role of poly (ADP-ribose) polymerase inhibition in cancer therapeutics. Clin Ther. 2016;38(7):1577–88.
Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071.
Zhang J. Poly (ADP-ribose) polymerase inhibitor: an evolving paradigm in the treatment of prostate cancer. Asian J Androl. 2014;16(3):401–6.
Dantzer F, de la Rubia G, Ménissier-de Murcia J, Hostomsky Z, de Murcia G, Schreiber V. Base excision repair is impaired in mammalian cells lacking poly (ADP-ribose) polymerase-1. Biochemistry. 2000;39(25):7559–69.
Coyne GOS, Chen A, Kummar S. Delivering on the promise: PARP inhibition as targeted anti-cancer therapy. Curr Opin Oncol. 2015;27(6):475–81.
Frey MK, Pothuri B. Targeting DNA repair: poly (ADP-ribose) polymerase inhibitors. Trans Cancer Res. 2015;4(1):84–96.
Gudmundsdottir K, Ashworth A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene. 2006;25(43):5864–74.
Shen Y, Aoyagi-Scharber M, Wang B. Trapping poly(ADP-ribose) polymerase. J Pharmacol Exp Ther. 2015;353(3):446–57.
Do K, Chen AP. Molecular pathways: targeting PARP in cancer treatment. Clin Cancer Res. 2013;19(5):977–84.
Fortini P, Pascucci B, Parlanti E, D’errico M, Simonelli V, Dogliotti E. The base excision repair: mechanisms and its relevance for cancer susceptibility. Biochimie. 2003;85(11):1053–71.
American Cancer Society. Cancer facts and figures 2017. Atlanta: American Cancer Society; 2017.
Miller RE, Ledermann JA. The status of poly (adenosine diphosphate-ribose) polymerase (PARP) inhibitors in ovarian cancer, part 2: extending the scope beyond olaparib and BRCA1/2 mutations. Clin Adv Hematol Oncol. 2016;14(9):704–11.
Alvarez RD, Matulonis UA, Herzog TJ, Coleman RL, Monk BJ, Markman M. Moving beyond the platinum sensitive/resistant paradigm for patients with recurrent ovarian cancer. Gynecol Oncol. 2016;141(3):405–9.
Pujade-Lauraine E, Combe P. Recurrent ovarian cancer. Ann Oncol. 2016;27(suppl_1):i63–5.
Coyne GOS, Chen AP, Meehan R, Doroshow JH. PARP inhibitors in reproductive system cancers: current use and developments. Drugs. 2017;77(2):113–30.
Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D'Andrea AD. Homologous recombination deficiency: exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov. 2015;5(11):1137–54.
Pennington KP, Swisher EM. Hereditary ovarian cancer: beyond the usual suspects. Gynecol Oncol. 2012;124(2):347–53.
Kanjanapan Y, Lheureux S, Oza AM. Niraparib for the treatment of ovarian cancer. Expert Opin Pharmacother. 2017;18(6):631–40.
Sabatucci I, Lorusso D. Il ruolo di niraparib nel trattamento del carcinoma ovarico: attualita e prospettive. Recenti Prog Med. 2017;108(6):265–8.
Pennington KP, Walsh T, Harrell MI, Lee MK, Pennil CC, Rendi MH, et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res. 2014;20(3):764–75.
McCabe N, Turner NC, Lord CJ, Kluzek K, Białkowska A, Swift S, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly (ADP-ribose) polymerase inhibition. Cancer Res. 2006;66(16):8109–15.
Anwar M, Aslam HM, Anwar S. PARP inhibitors. Hered Cancer Clin Prac. 2015;13(1):4.
Konstantinopoulos P, Spentzos D, Karlan B, Taniguchi T, Fountzilas E, Francoeur N, et al. A gene expression profile of BRCAness that correlates with responsiveness to platinum and PARP inhibitors. J Clin Oncol. 2010;28(15_suppl):5004.
Dedes KJ, Wetterskog D, Mendes-Pereira AM, Natrajan R, Lambros MB, Geyer FC, et al. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci Transl Med. 2010;2(53):53ra75.
Bryant HE, Schultz N, Thomas HD. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–7.
Farmer H, McCabe N, Lord CJ. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–21.
Scott C, Swisher E, Kaufmann S. Poly (ADP-ribose) polymerase inhibitors: recent advances and future development. J Clin Oncol. 2015;33(12):1397–406.
Thorsell AG, Ekblad T, Karlberg T, Low M, Pinto AF, Tresaugues L, et al. Structural basis for potency and promiscuity in poly(ADP-ribose) polymerase (PARP) and Tankyrase inhibitors. J Med Chem. 2017;60(4):1262–71.
Hassa PO, Hottiger MO. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front Biosci:J virtual library. 2008;13(13):3046–82.
Knezevic CE, Wright G, Remsing Rix LL, Kim W, Kuenzi BM, Luo Y, et al. Proteome-wide profiling of clinical PARP inhibitors reveals compound-specific secondary targets. Cell Chem Biol. 2016;23(12):1490–503.
Kaufman B, Shapira-Frommer R, Schmutzler RK, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33(3):244–50.
Domchek SM, Aghajanian C, Shapira-Frommer R, et al. Efficacy and safety of olaparib monotherapy in germline BRCA1/2 mutation carriers with advanced ovarian cancer and three or more lines of prior therapy. Gynecol Oncol. 2016;140(2):199–203.
US FDA. FDA approves olaparib tablets for maintenance treatment in ovarian cancer. Available at:- https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm572143.htm. Accessed 10 Oct 2017.
Dougherty BA, Lai Z, Hodgson DR, Orr MCM, Hawryluk M, Sun J, et al. Biological and clinical evidence for somatic mutations in BRCA1 and BRCA2 as predictive markers for olaparib response in high-grade serous ovarian cancers in the maintenance setting. Oncotarget. 2017;8(27):43653–61.
Pujade-Lauraine E, Ledermann JA, Selle F, Gebski V, Penson RT, Oza AM, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 18(9):1274–1284.
Dockery L, Gunderson C, Moore K. Rucaparib: the past, present, and future of a newly approved PARP inhibitor for ovarian cancer. OncoTargets Ther. 2017;10:3029–37.
Swisher EM, Lin KK, Oza AM, Scott CL, Giordano H, Sun J, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017;18(1):75–87.
Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390(10106):1949–61.
Broderick JM: FDA Approval Sought for Maintenance Rucaparib in Ovarian Cancer. Available at: http://www.onclive.com/web-exclusives/fda-approval-sought-for-maintenance-rucaparib-in-ovarian-cancer. Accessed 20 Oct 2017.
Tarapchak P. FDA Approves Maintenance Treatment for Recurrent Epithelial Ovarian, Fallopian Tube, or Primary Peritoneal Cancers. Oncology Times 2017: http://journals.lww.com/oncology-times/blog/fdaactionsandupdates/pages/post.aspx?PostID=231.
Scott LJ. Niraparib: first global approval. drugs; 2017. p. 1–6.
National Center for Biotechnology Information. PubChem Compound Database; CID=24958200. Available at: https://pubchem.ncbi.nlm.nih.gov/compound/1038915-60-4#section=Top. Accessed 15 Oct 2017.
Gonzalez-Martin A, Backes FJ, Baumann KH, Chase DM, Fehr MK, Coleman RL, et al. A randomized, double-blind phase III trial of niraparib maintenance treatment in patients with HRD+ advanced ovarian cancer after response to front-line platinum-based chemotherapy. J Clin Oncol. 2016;34(15 suppl):TPS5606-TPS5606.
Moore KN, Zhang Z-Y, Agarwal S, Patel MR, Burris HA, Martell RE, et al. Food effect substudy of a phase 3 randomized double-blind trial of maintenance with niraparib (MK4827), a poly(ADP)ribose polymerase (PARP) inhibitor versus placebo in patients with platinum-sensitive ovarian cancer. J Clin Oncol. 2014;32(15_suppl):e16531.
van Andel L, Zhang Z, Lu S, Kansra V, Agarwal S, Hughes L, et al. Human mass balance study and metabolite profiling of 14C–niraparib, a novel poly(ADP-Ribose) polymerase (PARP)-1 and PARP-2 inhibitor, in patients with advanced cancer. Investig New Drugs. 2017.
United States Product Insert. 2017. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2016/209115s000lbl.pdf. Accessed 3 Aug 2017.
Zhang Z-Y, Kansra V, van Andel L, Tibben M, Rosing H, Lu S, et al. Characterization of absorption, metabolism, and elimination of Niraparib, an investigational poly (ADP-ribose) polymerase inhibitor, in cancer patients. Clin Ther. 2017;39(8):e7–8.
Mikule K, Wilcoxen K. Abstract B168: the PARP inhibitor, niraparib, crosses the blood brain barrier in rodents and is efficacious in a BRCA2-mutant intracranial tumor model. AACR; 2015.
Bridges KA, Toniatti C, Buser CA, Liu H, Buchholz TA, Meyn RE. Niraparib (MK-4827), a novel poly(ADP-ribose) polymerase inhibitor, radiosensitizes human lung and breast cancer cells. Oncotarget. 2014;5(13):5076–86.
Wang L, Mason KA, Ang KK, Buchholz T, Valdecanas D, Mathur A, et al. MK-4827, a PARP-1/−2 inhibitor, strongly enhances response of human lung and breast cancer xenografts to radiation. Investig New Drugs. 2012;30(6):2113–20.
Williams SMG, Kuznicki AM, Andrade P, Dolinski BM, Elbi C, O’Hagan RC, et al. Treatment with the PARP inhibitor, niraparib, sensitizes colorectal cancer cell lines to irinotecan regardless of MSI/MSS status. Cancer Cell Int. 2015;15(1):14.
Chornenkyy Y, Agnihotri S, Yu M, Buczkowicz P, Rakopoulos P, Golbourn B, et al. Poly-ADP-ribose polymerase as a therapeutic target in pediatric diffuse intrinsic Pontine Glioma and pediatric high-grade Astrocytoma. Mol Cancer Ther. 2015;14(11):2560–8.
Sandhu SK, Schelman WR, Wilding G, Moreno V, Baird RD, Miranda S, et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2013;14(9):882–92.
Nappi L, Gleave ME. PARP inhibition in castration-resistant prostate cancer. Future Med. 2016;12(5). https://doi.org/10.2217/fon.2216.2211.
Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375(22):2154–64.
Campo JMD, Mirza MR, Berek JS, Provencher DM, Emons G, Fabbro M, et al. The successful phase 3 niraparib ENGOT-OV16/NOVA trial included a substantial number of patients with platinum resistant ovarian cancer (OC). J Clin Oncol. 2017;35(15_suppl):5560.
Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31(11):1023–31.
Lin K, Sun J, Maloney L, Goble S, Oza A, Coleman R, et al. 2701 quantification of genomic loss of heterozygosity enables prospective selection of ovarian cancer patients who may derive benefit from the PARP inhibitor rucaparib. Eur J Cancer. 2015;51:S531–2.
Jenner ZB, Sood AK, Coleman RL. Evaluation of rucaparib and companion diagnostics in the PARP inhibitor landscape for recurrent ovarian cancer therapy. Future Oncol. 2016;12(12):1439–56.
Wilcoxen KM, Becker M, Neff C, Abkevich V, Jones JT, Hou X, et al. Use of homologous recombination deficiency (HRD) score to enrich for niraparib sensitive high grade ovarian tumors. J Clin Oncol. 2015;33
US National Library of Medicine. clinicaltrials.gov. Available online at: https://clinicaltrials.gov/ct2/results?cond=Cancer&term=Niraparib&cntry1=&state1=&recrs. Accessed 10 Aug 2017.
FDA: FDA grants orphan drug designation to veliparib for advanced NSCLC. HemOnc Today 2016.
US National Library of Medicine. clinicaltrials.gov. Available at: https://clinicaltrials.gov/ct2/show/NCT03075462?term=fluzoparib&rank=2. Accessed 16 Oct 2017.
Litton JK, Scoggins M, Whitman GJ, Barcenas CH, Moulder SL, Murthy RK, et al. A feasibility study of neoadjuvant talazoparib for early-stage breast cancer patients with a germline BRCA pathogenic variant: NCT02282345. Am Soc Clin Oncol. 2017.
Deas O, Cairo S, Wilcoxen K, Mikule K, Tran T-A, Timms K, et al: Preclinical evaluation of the PARP inhibitor niraparib and cytotoxic chemotherapy alone or in combination in a panel of 25 triple-negative breast cancer PDX models: relevance of BRCA mutations, HRD status and other biomarkers. AACR; 2016.
Wang Y, Cairo S, Nicolle D, Cristescu R, Loboda A, Nebozhyn M, et al. The PARP inhibitor niraparib demonstrates robust activity in a subset of patient-derived triple-negative breast cancer xenograft models. AACR; 2014.
Mueller S, Bhargava S, Molinaro AM, Yang X, Kolkowitz I, Olow A, et al. Poly (ADP-ribose) polymerase inhibitor MK-4827 together with radiation as a novel therapy for metastatic neuroblastoma. Anticancer Res. 2013;33(3):755–62.
Acknowledgments
We would like to thank Haramaya University School of Pharmacy staffs who gave us invaluable intellectual feedback for the realization of this article.
Funding
We didn’t receive any specific grant for this review article.
Availability of data and materials
Not applicable
Author information
Authors and Affiliations
Contributions
Both authors designed the study, collected scientific literatures, critically appraised individual articles for inclusion, wrote the review article and drafted the manuscript. MS also prepared the final manuscript for publication. Both authors read and approved the final version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sisay, M., Edessa, D. PARP inhibitors as potential therapeutic agents for various cancers: focus on niraparib and its first global approval for maintenance therapy of gynecologic cancers. gynaecol oncol res pract 4, 18 (2017). https://doi.org/10.1186/s40661-017-0055-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40661-017-0055-8