
Abstract
Background
Cell surface display system allows for endowing functional proteins expressed on bacterial surface by fusing different anchor proteins. Among PgsA, Blc, and Omp anchor, the antigen 43 (Ag43)-mediated surface display is a novel system in Escherichia coli. Here, we have demonstrated the red fluorescent protein (RFP) and cellulase (EC 3.2.1.4) on the cell surfaces at two different fusion sites in Ag43.
Results
We introduced two fusion sites which are unstructured domain (52–138 aa) and autochaperone domain (600–700 aa) at N-terminal for passenger proteins. As a result, the surface-displayed RFP expressed in plasmid pET28a, but the intracellular RFP expressed more than the surface-displayed RFP. Improved display efficiency of Ag43 was present when fusing at the site of the 138th amino acid (aa) compared to fusing at the site of the 700th aa. For endoglucanase, whole-cell surface-displayed Ag43-138-BsCel5 showed the highest specific activity which was 4.65-fold of BsCel5. Cell-displayed cellulase preserved residual activity ranging from 78% to 38% at temperatures from 55 °C to 80 °C, respectively.
Conclusions
This study is to demonstrate the novel surface display system of Ag43 in E. coli by targeting two different proteins RFP and BsCel5 that were successfully displayed on the cell surfaces at two different fusion sites. The Ag43 system displays surface heterologous proteins and is a potential whole-cell catalyst in the bioconversion of cellulose.

Similar content being viewed by others

Introduction
Cell surface display systems enable recombinant proteins to be displayed on the surface of microorganisms to bind external substrates and protect cells from membrane penetration of substrates. These functions allow cell surface display systems to be involved in applications such as high-throughput screening (Becker et al. 2007; Boder and Wittrup 1997), and whole-cell biocatalyst reactions (Muñoz-Gutiérrez et al. 2012, 2014; Tanaka et al. 2014; Zheng et al. 2012). Moreover, cell-surface engineering is a new era of combinatorial bioengineering for biotechnology which has been successfully used by yeast (Kondo and Ueda 2004). On the other hand, E. coli is the most widely used microorganism because of its easy genetic manipulation and rapid growth with high yield of heterologous proteins. To export passenger proteins to the cell surface, there are a variety of anchor proteins being developed for surface display, which include outer membrane proteins (e.g., OmpA, PgsA) (Verhoeven et al. 2009), surface appendages (e.g., Flagellin) (Westerlund-Wikström et al. 1997), ice nucleation protein (INP) (Yim et al. 2010), and auto-transporters (ATs), such as antigen 43 (Ag43) (Henderson and Owen 1999; Kranen et al. 2014; Lee et al. 2013).
The cell surface display based on ATs has been reported to be one of the most attractive systems for the large and complex proteins (van Bloois et al. 2011). ATs are a family of bacterial proteins composed of an N-terminal passenger domain and a C-terminal β-barrel-forming domain that is integrated into the outer membrane and therefore facilitates the surface display of the passenger domain (Jose 2006). Cell surface display based on ATs has been reported to be one of the most attractive systems for displaying large and complex proteins (van Bloois et al. 2011). For example, the IgE Fcε3 domain was integrated into Ag43 and displayed on the surface of E. coli Tan109, with regard to a potential model for asthma and other allergic diseases treatment (Huang et al. 2014). The β-glucosidase (BglC) was also displayed on the E. coli MS04 by Ag43 system, and successfully fermented cellulosic materials in the simultaneous saccharification and fermentation (SSF) process (Muñoz-Gutiérrez et al. 2014). AT systems are considered to have some striking advantages compared to other surface display systems: a higher number of recombinant proteins can be displayed at the cell surface without reducing cell viability; the β-barrel is freely mobile while serving as an anchor for the recombinant protein within the outer membrane; and an inorganic prosthetic group can be incorporated in an expressed apo-protein (Jose 2006).
Since 2016, there are 212 reports found from the Scifinder database with the keyword “Ag43”; however, only few reports are found with the keyword of “surface display.” Thus, Ag43 has a great potential as a protein display system (Woude and Henderson 2008). It has been shown that both two fusion types (i.e., 138th aa and 700th aa) can yield stable surface display, but perform with different efficiencies (Ramesh et al. 2012). Therefore, we constructed two types of plasmids for fusion of different target proteins [i.e., red fluorescent protein from Discosoma striata (RFP), and endoglucanase from Bacillus subtilis (BsCel5)] at both sites. Here, we adopted two different fusion sites and two different target proteins to study the function of Ag43 as a novel surface display system.
Materials and methods
Materials, strains, plasmids, and gene sources
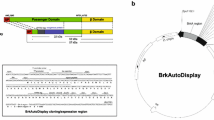
Bacterial strains, plasmids, and primers are shown in Table 1. Reagents used for plasmids construction including Ex-Taq polymerase, PCR-grade dNTPs, restriction enzymes, T4 ligase, and DNA ladder markers were purchased from Takara (Japan), while Phusion polymerase used for prolonged overlap extension PCR (POE-PCR) was obtained from NEB (USA). All DNA manipulations were performed according to standard protocols. PCR products and restriction-digested DNA were purified using PCR clean-up and gel-extraction kits (GeneDirex, USA). The plasmids were isolated using Plasmid Extraction Mini Kit (Favorgen, Taiwan). Genomic DNA was isolated from 5 mL cells (overnight culture at 37 °C in LB broth) using a bacteria genomic DNA miniprep Kit (Axygen, USA). Recombinant colonies grown on LB plates with appropriate antibiotics were confirmed by colony PCR. All the constructive plasmids were presented in Fig. 1. The gene sequence encoding Ag43 (GenBank: AAT48141.1) was amplified from E. coli MG1655 (ATCC: 47076). The genes encoding the mutant red fluorescent protein (RFP) from Discosoma striata (iGEM: BBa_E1010) and endoglucanase from Bacillus subtilis (BsCel5) (GenBank: CAA97610.1) were amplified using pSB1C3-RFP (iGEM: BBa_J04450) and the Bacillus subtilis genome (ATCC: 23857) as templates, respectively.

Maps of recombinant plasmids in this study. a pET28a-Ag43-138-RFP b pET28a-Ag43-138-BsCel5 c pET28a-Ag43-700-RFP d pET28a-Ag43-700-BsCel5 e pET28a-Ag43 f pET28a-BsCel5
Construction plasmids by ligating two digested fragments
Plasmids, pET28a-RFP and pET28a-Ag43, were constructed via DNA manipulation, double restriction-enzyme digestion, purification, ligation, and transformation into E. coli DH5α for plasmid propagation. The Ag43 fragment was digested with EcoRI and HindIII, while RFP fragment was digested with XhoI and BamHI, respectively. The two fragments were ligated into the pET28a which was digested with the same restriction enzymes to obtain plasmids, pET28a-Ag43 and pET28a-RFP, respectively.
Construction of plasmids by POE-PCR
We attempted to construct other plasmids using the method named prolonged overlap extension PCR (POE-PCR) (You and Zhang 2014). In detail, the insertion fragment and the linearized vector with two overlap ends were generated by high-fidelity PCR using NEB Phusion polymerase. Then the multimerization process of purifyng the above two PCR fragments with equimolar DNA was conducted using the method of POE-PCR without adding any primers. The resulting constructed plasmids were directly transformed into E. coli BL21 (DE3). A total of 4 vectors named pET28a-Ag43-138 or pET281-Ag43-700 were also constructed using the method of POE-PCR wherein passenger proteins RFP or BsCel5 are inserted within signal peptides of Ag43 and the C-terminal fragments 138–1039 aa of Ag43 (Fig. 1).
Cultivation conditions and induction
A 1% (v/v) precultured broth was transferred into fresh Luria–Bertani (LB) with 50 μg/mL kanamycin and grown at 37 °C with constant shaking at 200 rpm until OD600 nm is equal to 0.6–0.8. Afterward, cells harboring plasmids that used the T7 promoter were induced with 0.1 mM isopropyl β-d-thiogalactoside (IPTG), and the cultivation temperature was decreased to 22 °C for the subsequent 16 h.
The whole-cell protease treatment and SDS-PAGE analysis
The whole-cell protease treatment was originally described by Maurer et al. (1997). Two milliliters of cell culture (OD600 nm of 4) was concentrated in a 1.5 mL microcentrifuge tube by centrifugation at 10,000×g for 10 min, and the cell pellet was washed twice with 1 ml phosphate-buffered saline (PBS, pH = 7.4). The cell pellet was resuspended in 950 μL PBS, and 50 μL of 10 g/L trypsin was added. After 60 min of incubation at 37 °C, the reaction was stopped and was followed by washing the cells twice with 1 mL phosphate salt buffer (PBS). The cell pellet was then resuspended in 1 mL PBS. Finally, samples were stored for subsequent SDS-PAGE analysis with 10% (w/v) and 4% (w/v) concentrations as separating and stacking gels, respectively. Tris–glycine buffer (pH 8.3) containing 0.1% SDS was used as electrode buffer. Samples with the same concentration were suspended in protein buffer containing 1% SDS and 5 mM mercaptoethanol, and heated at 100 °C for 10 min before loading onto the gel. Electrophoresis was conducted at a constant current of 20 mA per slab at room temperature in a Biorad mini gel electrophoresis unit. Separated proteins on SDS-PAGE were visualized by staining with Coomassie blue R-250, and then using Image scanner.
Detection of BsCel5 activity on plates using congo red staining
The activity of E. coli strain BL21 (DE3) carrying recombinant BsCel5 can be detected on LB plates containing 10 g/L carboxymethyl cellulose (CMC), 50 μg/mL kanamycin resistance, and 0.1 mM IPTG (Ng et al. 2009). A 2 μL drop of overnight culture was added onto the surface of the CMC-LB plate described above and incubated at 37 °C for 12 h followed by incubation at 55 °C for another 2 h. Plates were then immersed in 2 g/L congo red for 30 min and de-stained by washing twice with 1 M NaCl for total 30 min.
The whole-cell activity assay for BsCel5
The whole-cell activity was done as reported previously with some modifications (Ng et al. 2013). A 300-μL reaction mixture (270 μL of 50 mM sodium acetate buffer (pH 5.5) containing 10 g/L CMC and 30 μL of cell culture (OD600 nm of 2) was incubated at 55 °C for 15 min. For thermostability, enzymes were preheated at 55 °C to 80 °C for 1 h. The hydrolysis activity was determined by the reducing sugars released from the reaction, which was measured using the dinitrosalicylic acid (DNS) method (Miller 1959). In brief, the supernatant of the reaction mixtures (200 μL) was mixed with the same volume of DNS reagent and maintained in a boiling water bath for 10 min. The solutions were cooled at room temperature immediately, and color intensity measurement at 540 nm was then performed.
Results and discussion
The target proteins fused at the 138th aa and the 700th aa of Ag43
Ag43 is composed of a signal peptide (1–52 aa), a N-terminal unstructured passenger (53–137 aa), a linker (138–600 aa), a putative auto-chaperone (600–700 aa), and a C-terminal β-barrel pore (701–1039 aa) (Fig. 2a) (Ramesh et al. 2012). The signal peptide, which is cleaved and left in the inner membrane, enables Ag43 to reach and fold in the periplasm. Subsequently, the β-barrel domain integrates into the outer membrane where it forms a pore; the passenger domain translocates through this pore to the cell exterior (Kjaergaard et al. 2000). The signal peptide and C-terminal β-barrel pore are necessary in this translocation process, while the other domains would not be replaced. Thus, we constructed two types of plasmids for the fusion of target proteins at two different sites (i.e., the 138th aa and the 700th aa). The plasmid named Ag43-138 inserted a passenger protein through genetic replacement of 53–137 aa and kept the other domains intact, whereas the plasmid named Ag43-700 inserted a passenger protein through genetic replacement of 53–699 aa (Fig. 2b). All the transformants are successful which presented on the surfaces of LB plates (Fig. 3a), as confirmed by PCR and shown in Fig. 3b.

Schematic representation of the composition of Ag43 and the different fusion sites. a Signal peptide, passenger, linker, autochaperone, and β-barrel domain are indicated in yellow, red, green, purple, and blue, respectively. b Presentation of two different fusion positions (the 138th aa and the 700th aa) of Ag43 where the passenger proteins fused

Photographs of transformants on LB plates and confirmation by PCR. a CFU on plate of different genetic E. coli: (1) pET28a-Ag43-138-RFP, (2) pET28a-Ag43-700-RFP, (3) pET28a-Ag43-138-BsCel5 and (4) pET28a-Ag43-700-BsCel5. b PCR confirm of pET28a-Ag43 (3120 bp) on left, pET28a-Ag43-700-RFP and pET28a-Ag43-138-RFP (RFP is 675 bp) in the middle, and pET28a-Ag43-138-BsCel5 and pET28a-Ag43-700-BsCel5 (BsCel5 is 1413 bp) on right
Surface display confirmation by whole-cell trypsin treatment
Trypsin cleaves proteins on the C-terminal side of lysine and arginine amino acid residues. Therefore, by taking advantage of this mechanism, proteins attached to the cell service with extracellular exposure can be removed using trypsin (Maurer et al. 1997). Figure 4a shows the SDS-PAGE analysis for trypsin-treated and untreated whole cells expressing Ag43-138-RFP and Ag43-700-RFP. In contrast to the bands of untreated cells, the SDS-PAGE bands of trypsin-treated cells were much thinner. These data confirmed that cells carrying pET28a-Ag43-138-RFP and pET28a-Ag43-700-RFP both produced surface-displayed RFP. Moreover, cells carrying pET28a-Ag43-138-RFP produced more protein exposed on the cell surface than that of cells carrying pET28a-Ag43-700-RFP.

Whole-cell protease treatment analysis. a The whole-cell SDS-PAGE analysis of protease treatment (+) and non-treatment (−) for RFP. b The whole-cell SDS-PAGE analysis of protease treatment (+) and non-treatment (−) for BsCel5. M indicates a protein marker. The red arrows and boxes indicate the recombinant proteins
Whole cells with intracellular BsCel5 (pET28a-BsCel5) showed no differences between trypsin-treated and untreated groups. However, there was an appreciably thinner band in trypsin-treated cells carrying pET28a-Ag43-138-BsCel5 compared with untreated cells; trypsin treatment had no effect on cells carrying pET28a-Ag43-700-BsCel5 (Fig. 4b). Therefore, pET28a-Ag43-138-BsCel5 produced more protein exposed on the cell surface than that of pET28a-Ag43-700-BsCel5, which is in agreement with our RFP results. Thus, the fusion site at the 138th aa was superior to the 700th aa in terms of cell surface display. This finding may be explained by the fact that protein translocation and folding may be enhanced when the proteins are fused at the 138th aa (Ag43-138), as the fragments of 138–1039 aa are unchanged and contain intact linker and auto-chaperone domains.
Can the cell turn red by displaying RFP fused to Ag43?
Fluorescent proteins are used to quickly estimate the efficiency of a display system because they are easily expressed and can be observed from the color of the cell (Kan et al. 2015). As shown in Fig. 3a, RFP expressions are ranking in the order of pET281-Ag43-700-RFP (i.e., 67 kDa) > pET28a-RFP (i.e., 32 kDa) > pET28a-Ag43-138-RFP (i.e., 124 kDa). The protein is soluble (S) in the expression via used pET28a. By surface display in Ag43, proteins are dominating in the pellet (P) or on cell surface which has already proofed by trypsin digestion (Fig. 4). The proteins on the cell surface showed a decreased fluorescence compared to that in the soluble protein. Thus, cell color followed the order of pET28a-RFP > pET28a-700-RFP > pET28a-Ag43-138-RFP (Fig. 3b). RFP expression of pET28a-700-RFP is more than that of pET28a-Ag43-138-RFP, thus cells expressing RFP display deeper red color (Fig. 5). Because the fluorescent protein is easily reduced by the other materials in the extracellular environment and, as a result, loses its fluorescence on the cell surface (Yang et al. 1996).

a SDS-PAGE analysis and cell pellets of recombinant RFP. pET28a-Ag43-138-RFP (lanes 1–4), pET28a-Ag43-700-RFP (lanes 5–8), and pET28a-RFP (lanes 9–11) were analyzed by SDS-PAGE. The red arrows indicate the recombinant proteins. M indicates a protein marker. W, PP, S, and P indicate whole cell, periplasm, supernatant, and pellet, respectively. b The performance of cell surface display for RFP. The cells carrying recombinant plasmids pET28a-Ag43-138-RFP, pET28a-Ag43-700-RFP, and pET28a-RFP could turn red
The cellulase activity of BsCel5 fused to Ag43
BsCel5 was used to estimate the efficiency of our Ag43-mediated surface display system. The obvious hydrolytic zones on CMC-congo red plates are shown for the fusion plasmids (pET28a-Ag43-138-BsCel5 and pET28a-Ag43-700-BsCel5) and for intracellular BsCel5 (pET28a-BsCel5) (Fig. 6A). The hydrolytic zones of intracellular BsCel5 were smaller than those of the surface-displayed BsCel5 on agar plates. Thus, Ag43 is a promising cell display system for cellulases, as BsCel5 on the cell surface that more readily contacted the external environment was more direct than BsCel5 not displayed on the surface. The contact area of an enzyme and its substrate is fixed when the former is displayed on the cell surface, thereby accelerating the interaction between substrates and displayed enzymes.

The performance of cell surface display for BsCel5. A Congo red tests for cells harboring plasmids pET28a (a), pET28a-BsCel5 (b), pET28a-Ag43-138-BsCel5 (c), and pET28a-Ag43-700-BsCel5 (d). Left and right images represent before and after congo red staining, respectively. B Whole-cell activity assay for cells harboring pET28a-BsCel5, pET28a-Ag43-138-BsCel5, and pET28a-Ag43-700-BsCel5. The activity was measured by the amount of unit possessed per milliliter culture (OD600 nm of 2). One unit of activity was defined as the amount of glucose generated per minute. The activity of pET28a-BsCel5 was 0.27 U/mL and defined as 1. C Thermal stability of BsCel5 in the different recombinants. The activities of BsCel5 at 55, 60, 70, and 80 °C are indicated with stacked bars in green, pink, gray, and yellow (from top to down), respectively
Whole-cell activity was also measured and is shown in Fig. 6B. The sample of Ag43-138-BsCel5 obtained the highest specific activity, which was 4.65 times of cells containing intracellular BsCel5. On the other hand, the activity of cells containing Ag43-700-BsCel5 was 1.77 times of cells containing intracellular BsCel5. In addition, all the recombinant BsCel5 are thermal stable. At temperatures ranging from 55 to 60 °C, surface-displayed cellulase (pET28a-Ag43-138-BsCel5) shows a higher activity than that of intracellular cellulase (BsCel5). At higher temperatures (up to 79.5 °C), the displayed BsCel5 decreased to 38% residual activity, while BsCel5 preserves 55.2% residual activity (Fig. 6C). It is because the BsCel5 on the cell surface contacted the external environment more directly and became more unstable.
The whole-cell catalyst has been developed by expressing the endoglucanase (Cel5A), exoglucanase (Cel9E), and β-glucosidase (BGL) from Clostridium cellulolyticum on the cell surface of E. coli via the co-displaying by three cellulases fusing with PgsA separately or positional assembly by outer membrane vesicles (OMV) (Ryu and Karim 2011; Park et al. 2012). Moreover, the cellulase was also displayed in another strain, Zymobacter palmae (Kojima et al. 2012). Cellulase-mediated biodegradation of cellulose is one promising approach in global carbon recycling (Ng et al. 2010, 2011), and this biodegradation is also a potential consolidated bioprocessing (CBP) platform for effective production of biocommodities (Arora et al. 2015; Liao et al. 2011; Obeng et al. 2017). Therefore, BsCel5 surface display by Ag43 may be a new approach for whole-cell catalysis and CBP in the future.
Conclusion
We successfully expressed targeted passenger proteins displayed on the cell surface of E. coli by means of Ag43. The fusion site at the 138th aa was better for surface display of target proteins than the site at the 700th aa. Thus, the Ag43 fragment of 138–700 aa may be critically important for the translocation process. Moreover, this is the first attempt to explore the bioconversion of cellulose by surface display of cellulase (BsCel5) via an Ag43-mediated system which is a simple and cost-effective strategy for cellulosic biorefinery in the future.
Abbreviations
- Ag43:
-
antigen 43
- ATs:
-
auto-transporters
- BsCel5:
-
Bacillus subtilis cellulase
- IPTG:
-
β-d-thiogalactoside
- CMC:
-
carboxymethyl cellulose
- Omp:
-
outer membrane proteins
- POE-PCR:
-
prolonged overlap extension PCR
- RFP:
-
red fluorescent protein
- SDS-PAGE:
-
sodium dodecyl sulfate polyacrylamide gel electrophoresis
References
Arora R, Behera S, Sharma NK, Kumar S (2015) Bioprospecting thermostable cellulosomes for efficient biofuel production from lignocellulosic biomass. Bioresour Bioprocess 2:38
Becker S, Michalczyk A, Wilhelm S, Jaeger KE, Kolmar H (2007) Ultrahigh-throughput screening to identify E. coli cells expressing functionally active enzymes on their surface. ChemBioChem 8:943–949
Boder ET, Wittrup KD (1997) Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol 15:553–557
Henderson IR, Owen P (1999) The major phase-variable outer membrane protein of Escherichia coli structurally resembles the immunoglobulin A1 protease class of exported protein and is regulated by a novel mechanism involving Dam and OxyR. Biotechnol J 181:2132–2141
Huang FY, Wang CC, Huang YH, Zhao HG, Guo JL, Zhou SL, Wang H, Lin YY, Tan GH (2014) Antigen 43/Fcε3 chimeric protein expressed by a novel bacterial surface expression system as an effective asthma vaccine. Immunology 143:230–240
Jose J (2006) Autodisplay: efficient bacterial surface display of recombinant proteins. Appl Microbiol Biotechnol 69:607–614
Kan SC, Chen CM, Lin CC, Wu JY, Shieh CJ, Liu YC (2015) Deciphering EGFP production via surface display and self-cleavage intein system in different hosts. J Taiwan Inst Chem Eng 55:1–6
Kjaergaard K, Schembri MA, Hasman H, Klemm P (2000) Antigen 43 from Escherichia coli induces inter- and intraspecies cell aggregation and changes in colony morphology of Pseudomonas fluorescens. J Bacteriol 182:4789–4796
Kojima M, Akahoshi T, Okamoto K, Yanase H (2012) Expression and surface display of Cellulomonas endoglucanase in the ethanologenic bacterium Zymobacter palmae. Appl Microbiol Biotechnol 96:1093–1104
Kondo A, Ueda M (2004) Yeast cell-surface display: applications of molecular display. Appl Microbiol Biotechnol 64:28–40
Kranen E, Detzel C, Weber T, Jose J (2014) Autodisplay for the co-expression of lipase and foldase on the surface of E. coli: washing with designer bugs. Microb Cell Fact. 13:19
Lee H, Park SJ, Han MJ, Eom GT, Choi MJ, Kim SH, Oh YH, Song BK, Lee SH (2013) Expression of a lipase on the cell-surface of Escherichia coli using the OmpW anchoring motif and its application to enantioselective reactions. Biotechnol Lett 35:1677–1683
Liao H, Zhang XZ, Rollin JA, Zhang YH (2011) A minimal set of bacterial cellulases for consolidated bioprocessing of lignocellulose. Biotechnol J 6:1409–1418
Maurer J, Jose J, Meyer TF (1997) Autodisplay: one-component system for efficient surface display and release of soluble recombinant proteins from Escherichia coli. J Bacteriol 179:794–804
Miller GL (1959) Use of dinitrosalicylic as reagent for the determination of reducing sugars. Anal Chem 31:426–428
Muñoz-Gutiérrez I, Oropeza R, Gosset G, Martinez A (2012) Cell surface display of a β-glucosidase employing the type V secretion system on ethanologenic Escherichia coli for the fermentation of cellobiose to ethanol. J Ind Microbiol Biotechnol 39:1141–1152
Muñoz-Gutiérrez I, Moss-Acosta C, Trujillo-Martinez B, Gosset G, Martinez A (2014) Ag43-mediated display of a thermostable β-glucosidase in Escherichia coli and its use for simultaneous saccharification and fermentation at high temperatures. Microb Cell Fact 13:106
Ng IS, Li CW, Yeh YF, Chen PT, Chir JL, Ma CH, Yu SM, Ho THD, Tong CG (2009) A novel endoglucanase from the thermophilic bacterium Geobacillus sp. 70PC53 with high activity and stability over a broad range of temperatures. Extremophiles 13:425–435
Ng IS, Li CW, Chan SP, Chir JL, Chen PT, Tong CG, Yu SM, Ho THD (2010) High-level production of a thermoacidophilic β-glucosidase from Penicillium citrinum YS40-5 by solid-state fermentation with rice bran. Bioresour Technol 101:1310–1317
Ng IS, Tsai SW, Ju YM, Yu SM, Ho THD (2011) Dynamic synergistic effect on Trichoderma reesei cellulases by novel beta-glucosidases from Taiwanese fungi. Bioresour Technol 102:6073–6081
Ng IS, Chi XQ, Wu XM, Bao ZW, Lu YH, Chang JS, Ling XP (2013) Cloning and expression of Cel8A from Klebsiella pneumoniae in Escherichia coli and comparison to cel gene of Cellulomonas uda. Biochem Eng J 78:53–58
Obeng EM, Adam SNN, Budiman C, Ongkudon CM, Maas R, Jose J (2017) Lignocellulases: a review of emerging and developing enzymes, systems, and practices. Bioresour Bioprocess 4:16
Park M, Sun Q, Liu F, DeLisa MP, Chen W (2012) Positional assembly of enzymes on bacterial outer membrane vesicles for cascade reactions. PLoS ONE 9:e97103
Ramesh B, Sendra VG, Cirino PC, Varadarajan N (2012) Single-cell characterization of autotransporter-mediated Escherichia coli surface display of disulfide bond-containing proteins. J Biol Chem 287:38580–38589
Ryu S, Karim MN (2011) A whole cell biocatalyst for cellulosic ethanol production from dilute acid-pretreated corn stover hydrolyzates. Appl Microbiol Biotechnol 91:529–542
Tanaka T, Hirata Y, Nakano M, Kawabata H, Kondo A (2014) Creation of cellobiose and xylooligosaccharides-coutilizing Escherichia coli displaying both β-glucosidase and β-xylosidase on its cell surface. ACS Synth Biol 3:446–453
van Bloois E, Winter RT, Kolmar H, Fraaije MW (2011) Decorating microbes: surface display of proteins on Escherichia coli. Trends Biotechnol 29:79–86
Verhoeven GS, Alexeeva S, Dogterom M, Blaauwen TD (2009) Differential bacterial surface display of peptides by the transmembrane domain of OmpA. PLoS ONE 4:e6739
Westerlund-Wikström B, Tanskanen J, Virkola R, Hacker J, Lindberg M, Skurnik M, Korhonen TK (1997) Functional expression of adhesive peptides as fusions to Escherichia coli flagellin. Protein Eng 10:1319–1326
Woude MWV, Henderson IR (2008) Regulation and function of Ag43 (Flu). Annu Rev Microbiol 62:153–169
Yang F, Moss LG, Phillips GN Jr (1996) The molecular structure of green fluorescent protein. Nat Biotechnol 14:1246–1251
Yim SK, Kim DH, Jung HC, Pan JG, Kang HS, Ahn T, Yun CH (2010) Surface display of heme- and diflavin-containing cytochrome P450 BM3 in Escherichia coli: a whole-cell biocatalyst for oxidation. J Microbiol Biotechnol 20:712–717
You C, Zhang YHP (2014) Simple cloning and DNA assembly in Escherichia coli by prolonged overlap extension PCR. Methods Mol Biol 1116:183–192
Zheng Z, Chen T, Zhao M, Wang Z, Zhao X (2012) Engineering Escherichia coli for succinate production from hemicellulose via consolidated bioprocessing. Microb Cell Fact 11:37
Authors’ contributions
KJJ and ISN designed the experiment and analyzed the data, YLG performed all of experiments. ISN and YLG wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors are grateful for the financial support received for this project from the Ministry of Science and Technology (MOST 105-2221-E-006-225-MY3 and MOST 105-2621-M-006-012-MY3) in Taiwan. This work was also supported by the National Natural Science Foundation of China (No. 21776232). The authors also sincerely appreciate the academic connection between the Xiamen University (China) and the National Cheng Kung University (Taiwan).
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The authors approved the availability of data and materials for publishing the manuscript.
Consent for publication
The authors approved the consent for publishing the manuscript.
Ethics approval and consent to participate
All the authors have read and agreed the ethics for publishing the manuscript.
Funding
This research is support by funding from the Ministry of Science and Technology (MOST 105-2221-E-006-225-MY3 and MOST 105-2621-M-006-012-MY3) in Taiwan, and the National Natural Science Foundation of China (No. 21776232).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Jing, K., Guo, Y. & Ng, IS. Antigen-43-mediated surface display revealed in Escherichia coli by different fusion sites and proteins. Bioresour. Bioprocess. 6, 14 (2019). https://doi.org/10.1186/s40643-019-0248-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40643-019-0248-6