
Abstract
Selenium (Se) is an essential micronutrient required by organisms of diverse lineage. Dietary Se is converted to hydrogen selenide either enzymatically or by endogenous antioxidant proteins. This convergent biochemical step crucially underlies the subsequent biological activity of Se and argues for inclusion of hydrogen selenide as the fourth endogenous gasotransmitter alongside nitric oxide, carbon monoxide and hydrogen sulfide.
Endogenously generated hydrogen selenide is incorporated into numerous ‘selenoprotein’ oxidoreductase enzymes, essential for maintaining redox-status homeostasis in health and disease. Direct effects of endogenous hydrogen selenide on cellular and molecular targets are currently unknown. Given exogenously, hydrogen selenide acts as a modulator of metabolism via transient inhibition of mitochondrial cytochrome C oxidase. Here we provide an overview of Se biology, its impact on several physiological systems (immune, endocrine, cardiovascular and metabolic) and its utility as a supplement in acute and critical illness states. We further explore the evidence base supporting its role as the fourth gasotransmitter and propose a strategic case towards generation of novel selenomimetic therapeutics.
Similar content being viewed by others


Introduction
Elemental selenium (Se; Greek [σελήνη] selene or ‘moon’) was discovered in 1817 by Swedish chemists Jöns Jakob Berzelius and Johan Gottlieb Gahn [1]. On communication of its discovery, Berzelius stated that ‘I have just examined it [selenium] more carefully and have found that what we took for tellurium is a new substance, endowed with interesting properties. This substance has the properties of a metal, combined with that of sulfur, to such a degree that one would say it is a new kind of sulfur’ [2].
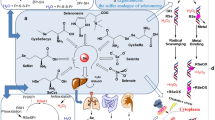
Selenium (alongside oxygen, sulfur and tellurium) belongs to the chalcogens—group 16 of the periodic table. It is an essential trace element required by organisms of diverse lineage (bacteria, archaea, eukaryotes) [3]. In addition to the elemental form, non-elemental forms and selenium-containing organic and inorganic molecules point towards a complex chemistry. Selenide is the reduced form of elemental selenium (oxidation state − 2), formed in biological systems or acidic environments from water-soluble selenium-containing compounds [4]. Endogenously generated hydrogen selenide is present as the small gaseous molecule, H2Se; analogous to sulfide, it is in equilibrium with the hydroselenide anion (HSe-) [5]. Similar to the other gaseous mediators reviewed in this issue, it also generates numerous oxidation products, the most abundant being selenite (SeO32-) and selenate (SeO42-), with oxidation states of + 4 and + 6, respectively [4, 6]. Crucially, conversion of all forms of intracellular Se-containing compounds to hydrogen selenide (either enzymatically or through redox reactions) represents a convergent and essential biochemical step (Fig. 1). This underpins its subsequent biological activity and, as we explore herein, adds support to its inclusion as the fourth endogenous ‘gasotransmitter’ alongside nitric oxide (NO), carbon monoxide (CO) and hydrogen sulfide (H2S/HS-).

Uptake and metabolism of Se-containing compounds. [CH3]2Se; dimethyl selenide, GSH; reduced glutathione, GSSeH; glutathioselenol, GSSeSG; selenodiglutathione, tRNA; transfer ribonucleic acid
In 2002, H2S was postulated to be the third member of a class of gaseous mediators, the gasotransmitters [7]. Five criteria were proposed to substantiate this claim, namely (i) they had to be small molecules of gas and (ii) freely permeable to cell membranes, (iii) they were endogenously and enzymatically generated albeit under regulatory controls, (iv) they had well-defined functions at physiologically relevant concentrations and (v) possessed cellular effects that may or may not be mediated by second messengers, but should have specific cellular and molecular targets [7]. To date, the importance of selenium in human biology has focussed on its incorporation into proteins [8, 9]. These ‘selenoproteins’, notable for their oxidoreductase activity and ability to impact on cellular redox status, have consequently drawn significant interest from the intensive care community and beyond. We propose that the physiological role(s) of endogenous hydrogen selenide extend beyond its incorporation into selenoproteins and, as a putative gasotransmitter, it is currently able to satisfy most of the above five criteria. Through a review of current knowledge and by using a hypothesis-driven approach, we here explore the evidence base arguing towards inclusion of hydrogen selenide as the fourth endogenous gasotransmitter. Necessarily, we provide an overview of selenium biology, including the functional roles of various proteins that constitute the ‘selenoproteome’. The significance of selenium and its derivatives in acute illness is considered, with a focus on immune, endocrine, circulatory and metabolic systems. Finally, we assess the utility of selenium supplementation and provide a strategic case towards the generation of new classes of putative selenomimetics.
Endogenous generation and metabolism of hydrogen selenide
Numerous published works have referred interchangeably to elemental Se and the oxidation products of hydrogen selenide. For simplicity, we too will herein refer to the above forms as ‘Se’, unless stated otherwise. It is notable that endogenous generation and metabolism of hydrogen selenide is analogous to many aspects of its chalcogenomimetic congener, hydrogen sulfide. Elemental selenium is obtained exclusively from dietary sources [10, 11], either as the oxidation products selenite or selenate, or following incorporation of Se into the amino acids cysteine and methionine (generating selenocysteine and selenomethionine, respectively). Entry of Se into cells has been studied in vitro, whereby its accumulation intracellularly was found to be both energy-dependent or independent [12, 13]. This likely reflects uptake of either gaseous (diffusion) or anionic (active transport) forms. The seleno-amino acids are substrates for an enzymatic intracellular metabolic pathway forming endogenous hydrogen selenide (Fig 1) [14]; selenocysteine, formed from selenomethionine via transsulfuration, is converted into hydrogen selenide by selenocysteine β-lyase (SCL; EC 4.4.1.16) [15, 16]. This enzyme, first isolated in 1982 [15], catalyses decomposition of selenocysteine to hydrogen selenide and alanine, utilising pyridoxal 5’-phosphate as a cofactor [15]. SCL has been found in the human liver, kidney, heart and adrenal and muscle tissue in decreasing order of specific activity [17], and can be regulated by hypoxia, oxidative stress, pro-inflammatory cytokines and glucocorticoids [18,19,20].
Intracellularly, the dietary oxidation product selenate is first reduced to selenite [21] and then to hydrogen selenide, either by thioredoxin reductase [22] or through a series of redox reactions coupled to reduced glutathione (GSH) [23]. Although essential, hydrogen selenide is intrinsically toxic at inappropriately high concentrations [24]. Consequently, in excess, Se is either excreted in faeces or urine (as oxidised forms, a trimethylselenonium ion or conjugated seleno-[hexose] sugars) or, at higher concentrations, exhaled following methylation (e.g. to dimethyl selenide) [25,26,27,28,29,30].
Selenoproteins and redox status
Selenoprotein synthesis involves incorporation of Se and this process mandates an uniquely adapted translational machinery [31, 32]. Insertion of Se at the catalytic site of selenoprotein enzymes greatly enhances their biological activity [33]. Their importance in biology is epitomised by inhibition of embryogenesis following genetic deletion of several selenoproteins [34,35,36,37]. The significance of this system has also been elegantly illustrated in vitro by showing that a reduction in selenoprotein abundance (through an antisense oligonucleotide) resulted in substantial increases in cellular reactive oxygen species (ROS) [38]. Cytochrome c- and caspase-dependent apoptosis was subsequently observed, yet incubation with the antioxidants glutathione, α-tocopherol and N-acetylcysteine, either alone or in combination, was unable to prevent cellular death [38].
This finding exemplifies the homeostasis of redox status as critical in health, and its dysregulation being a prominent factor across myriad pathologies. Many of these are encountered in critically ill patients (e.g. hypoxic or inflamed states), whereby excessive ROS production and/or inadequate detoxification generates significant oxidative stress with subsequent damage to lipids, proteins and DNA [39, 40]. Se plays an important role in maintaining redox homeostasis [41] following incorporation into the key antioxidant selenoenzymes, glutathione peroxidase (GPx), thioredoxin reductase (TrxR) and methionine sulfoxide reductase (Msr) [8]. Twenty-five selenoproteins have been characterised to date; the majority have been ascribed definitive and diverse functional roles and have been reviewed in detail elsewhere [42]. Cytosolic GPx-(isoform 1) was the first mammalian selenoprotein enzyme to be discovered [43,44,45]. Subsequently, a further four Se-containing isoforms have been identified (collectively, GPx; 1–4 and 6). The most abundant are GPx1, primarily involved in detoxifying intracellular hydrogen peroxide (H2O2) to water, and GPx4 with a greater affinity for lipid hydroperoxides [46]. Both prevent lipid peroxidation, thus limiting cellular damage in states of oxidative stress. TrxR consists of three isoenzymes involved in regulating the redox status of the antioxidant protein thioredoxin (Trx). Like GPx, Se-containing TrxR can catalyse reduction of H2O2 and organic hydroperoxides [47] and further functions to regulate DNA repair and intracellular redox signalling [48, 49]. Additional redox-active selenoproteins include (i) Msr, where the ratio of the reduced/oxidised form (of methionine) communicates cellular redox status [50], (ii) selenoprotein P that acts principally as a Se carrier but also undergoes redox reactions [51,52,53,54] and (iii) deiodinases that are oxidoreductase enzymes crucial for thyroid function [55].
Decreased plasma Se levels (up to 40%) are well recognised in heterogenous medical/surgical patient cohorts and ICU patients [56,57,58]. Of note, the level of deficiency is a prognosticator and correlates with disease severity [56,57,58]. Mechanism(s) underlying these deficiencies remain incompletely understood. Se has been described as a negative acute phase reactant; its deficiency could relate to either decreased Se intake or increased metabolism of Se-containing compounds. However, the most frequently cited cause is redistribution [57,58,59,60]. There is scant evidence that this deficiency, recorded in blood, translates to a reduction in selenoprotein synthesis in vital organs. However, in chronic deficiency states, a significant correlation is observed between plasma Se and erythrocyte GPx activity [61, 62]. Further sequelae of Se deficiency and its impact on several physiological systems relevant to critical illness are discussed below.
Immunity
Se intake influences its bioavailability to the immune system; selenium supplementation in healthy volunteers increased mRNA abundance of three selenoproteins (R, S and W) in peripheral blood mononuclear cells [63]. Classic innate immunity involves macrophages playing a vital role in controlling inflammatory status and phagocytosis of pathogens [64]. Se supplementation in mice conferred a switch in macrophage phenotype from the pro-inflammatory M1 to the anti-inflammatory M2 subtype following lipopolysaccharide (LPS) exposure [65]. Selenocysteine tRNA knockout mice exhibited reduced macrophage migration [66]. The transcription factor, nuclear factor (NF)-κB, is a significant component of innate immunity and a key mediator of inflammation. A murine macrophage cell line exposed to S. aureus had reduced NF-kB activation following Se treatment, with decreased expression of pro-inflammatory cytokines [67]. NF-kB inhibition was also observed following selenomethionine supplementation with decreased LPS-induced inflammation in chicken trachea [68].
While these reports indicate that Se acts an anti-inflammatory micronutrient, the impact of Se supplementation on adaptive immunity has yielded more variable results. CD4+ T helper cells can differentiate into various subtypes depending on the nature of stimulation. A high-Se diet administered to mice promoted differentiation of naive CD4+ T helper cells to a (pro-inflammatory) Th1 CD4+ phenotype [69]. By contrast, increased IL-4 mRNA concentration, an anti-inflammatory cytokine marker for the Th2 CD4+ T helper cell, was observed in mice administered a low-Se diet [69]. This suggests a restricted Se diet could result in a more favourable phenotypic switch in inflamed states, however, decreased Se abundance (and hence selenoprotein synthesis) also reduced T cell maturation, a necessary step for B cell signalling [70]. More globally, a U-shaped curve has been described for the relationship between cancer prevention and blood Se concentrations [71]. A relationship was reported between mortality (all-cause, cardiovascular and cancer-related) and Se status [72]. A significant increase in mortality in patients with selenoprotein activity outside the optimal range has been suggested [72]. This implies that for immunity and beyond, achieving an ‘adequate’ Se status is more likely to be favourable over supra- or under-supplementation.
Endocrine system
Se plays a key role in the endocrine system, particularly the thyroid gland where it is directly involved in thyroid hormone metabolism and protects against oxidative stress during thyroid hormone synthesis [73]. Adequate Se provision is necessary for the prevention of thyroid disease [74]. Indeed, relatively high concentrations of Se are present in the thyroid gland compared with other organs [75]. The most important selenoprotein classes involved in endocrine function are (i) deiodinases that convert thyroxine (T4) to the more biologically active triiodothyronine (T3), (ii) GPx that is responsible for glandular protection from ROS during generation of T4 and T3 and (iii) selenoprotein P (binding Se in plasma) that acts as a distributor to key organs (including the thyroid) when Se is restricted [74].
The importance of Se to thyroid function was demonstrated in rats administered a low-Se diet. GPx activity in the thyroid was reduced by 50% with concurrent though lesser falls in T4 and T3 synthesis [75]. This supports evidence that Se supply to deiodinases is prioritised over other selenoproteins such as GPx [74]. A fall in T4 and T3 upregulates synthesis and secretion of thyroid-stimulating hormone (TSH); this accelerates conversion of T4 to T3, generating significant quantities of hydrogen peroxide. Given the reduced activity of GPx in Se-deficient states, this can augment fibrosis of the thyroid and subsequent dysfunction [74]. Necrosis and fibrosis of the thyroid gland, particularly after iodide loading, is exacerbated by Se deficiency [76]. Given the aforementioned reduced availability of Se in critically ill patients, this could represent an important endocrine consideration for this cohort.
In addition to critically ill patients presenting with a low Se status on admission, they also typically show a combination of decreased T4 and T3 and an increase in reverse T3, with TSH within the reference range. This state is collectively known as the non-thyroidal illness syndrome (NTIS). Although considered to be protective in the first instance as an immediate, acute-phase response to nutrient restriction (and injury), NTIS is a prognosticator with likely complex implications for patients [77]. In a small cohort of trauma patients, a correlation was seen between Se and T3 deficiency, along with a parallel decrease in T4 deiodination [78]. At present, this relationship is associative rather than causal. However, given the integral function of selenoproteins in thyroid function, a direct role is plausible and warrants further investigation.
Circulatory system
A role for Se in the cardiovascular system is particularly notable with regard to its dietary provision, a factor that shows significant geographical variation. For example, populations with Se deficiency reside in areas of low-selenium soil concentration, most notably in regions of China. By contrast, areas of Venezuela are considered ‘seleniferous’ due to high soil levels and therefore agricultural produce content [79, 80]. Keshan disease, named after a region of Heilongjiang province in China, is characterised by congestive cardiomyopathy caused by a combination of dietary Se deficiency and the presence of a mutated strain of Coxsackie virus [81]. Although it still exists in rural areas where treatment is not readily available, Se administration offers a preventative measure and is reputed to protect the infected myocardium against oxidative stress [82]. In Se-deficient individuals, a lack of GPx activity resulted in increased peroxide levels and decreased prostacyclin synthetase activity [83]. Decreased prostacyclin, increased thromboxane levels and a reduction in prostacyclin/thromboxane ratio can result in a platelet pro-aggregatory state and systemic vasoconstriction [84], potentially increasing the risk of thromboembolic events and coronary disease. An extensive literature reviewed in detail elsewhere [85] has focussed on Se deficiency contributing to the oxidation of low-density lipoproteins. These modified fats are subject to uncontrolled uptake by M2-macrophage foam cells and contribute to atherosclerotic plaques. Accordingly, numerous meta-analyses have reported on Se-status (mostly serum Se levels) and the epidemiology of cardiovascular disease [86,87,88,89]. These studies unequivocally show that a low-Se status is associated with an increased incidence of cardiovascular disease, comprising both coronary heart disease and stroke. However, as the adjusted risk between studies shows considerable variation, there is insufficient evidence to recommend Se treatment as a preventive therapy [89].
Other gasotransmitters (most notably NO and hydrogen sulfide) exert significant effects on vascular tone through well-defined cellular and molecular mechanisms [90, 91]. The administration of these molecules (or their derivatives) are antihypertensive while their inhibition (for example using nitric oxide synthase inhibitors) can increase blood pressure in states of circulatory shock. Direct vasoactive mechanisms of Se derivatives have yet to be explored, however, we postulate that they may show similar actions to the accepted members of the gasotransmitter class. Elsewhere, a role for a selenomimetic group of compounds, the phenylaminoalkyl selenides, has been reported [92]. Although their mechanism of action likely excludes a direct role for hydrogen selenide, the redox chemistry of the selenium moiety allows these molecules to propagate an ascorbate redox cycle in adrenergic nerve terminals, regulating a key enzyme of catecholamine metabolism, dopamine-β-monooxygenase. This limits conversion of dopamine to norepinephrine with resulting blood pressure–lowering effects [93].
Metabolism
A landmark study in 2005 showed that mice breathing hydrogen sulfide gas entered a profound and reversible ‘suspended animation-like state’ [94]. Ten years later, the same group reported on the ability of exogenous hydrogen selenide gas to also induce significant metabolic effects [95]. A four-fold decrease in oxygen consumption and carbon dioxide production was observed in H2Se-treated mice, as well as a fall in core temperature [95]. The inhibitory metabolic role of exogenous hydrogen sulfide is however contentious as endogenous sources support basal bioenergetic function [96]. Nonetheless, when considering pharmacology alone (and a putative role for metabolism-modifying therapeutics), similarities in the effects of exogenous sulfide and selenide gases on aerobic respiration were noted; this prompted a subsequent commentary citing potential inclusion for hydrogen selenide as the fourth endogenous gasotransmitter [97].
Hydrogen selenide is likely to modulate aerobic metabolism, especially given the proximity of selenium to oxygen and sulfur in the periodic table, with both being notable for their interaction with mitochondria, and the known metabolic activity of NO, CO and hydrogen sulfide. While precise molecular mechanism(s) of metabolic inhibition in mice breathing (exogenous) H2Se were not elucidated [95], we propose mitochondrial cytochrome C oxidase (Complex IV) inhibition as a major pathway. We discovered in preliminary experiments using rat liver homogenate that the basic salt, sodium hydrogen selenide (NaHSe), could inhibit oxygen consumption in respiring tissue and, specifically, mitochondrial cytochrome C oxidase activity [98]. A notable difference between hydrogen selenide and hydrogen sulfide with regard to cytochrome C oxidase inhibition was the duration of effect. Hydrogen selenide acted in a transient, reversible manner, with a duration of action more than 3-fold shorter than hydrogen sulfide.
We have long held an interest in the use of mitochondrial inhibitors as adjunct therapies during revascularisation of ischemic organs. We have argued that short-term inhibitors of cytochrome C oxidase can prevent reperfusion-induced overproduction of mitochondria-derived ROS, while still allowing a degree of metabolism that supports cellular function [99, 100]. The transient nature of inhibition with hydrogen selenide could reveal it to be an attractive therapeutic target. Moreover, NaHSe given to mice at reperfusion protected against myocardial ischemia/reperfusion injury [95]. Selenide (but not the oxidation product, selenite) could target injured tissues with (radioactive) Se accumulation in the heart directly correlating to injury severity [95].
Selenomimetics and selenium supplementation
Given the unique and intriguing biochemistry of Se, and its impact on physiology in health and disease, several selenomimetics have been trialled across diverse pathologies. The most frequently studied is ebselen, an organoselenium compound cited in over 1000 reports [101]. Ebselen catalyses the reduction of hydroperoxides by thiol compounds (e.g. glutathione), thereby mimicking the enzymatic activity of GPx [93]. In preclinical studies, it has proven efficacy in numerous models ranging from cardiovascular and neurodegenerative diseases, to alcoholic liver disease and cancer [101]. In Europe, it did not continue beyond Phase I human studies owing to toxicity concerns of the Se moiety [101]. Clinical trials in Japan however assessed the efficacy of ebselen against oxidative tissue damage following acute ischemic stroke; although showing initial promise [102], it was later discontinued due to lack of (long-term) efficacy [103]. That notwithstanding, a recent review cites a remaining appetite for the development of this molecule [104]. Other notable selenomimetics include selenazofurin, an anti-neoplastic and anti-viral agent; selenotifen, an histaminergic anti-allergic drug; and selenium sulfide, an anti-viral compound used for treating seborrhea and tinea versicolor [93].
The use of selenomimetics, either as pharmacological tools or nutritional supplements, has yet to reach fruition in acute medicine and critical care. To date, 19 clinical trials have assessed Se supplementation in critically ill patients [105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123]. A meta-analysis found a significant but modest reduction in overall mortality and length of hospital stay; however, other endpoints showed no significant difference, including 28-day all-cause mortality, length of ICU stay, incidence of new infections and duration of mechanical ventilation [124]. The cohorts analysed were heterogenous, comprising elective (cardiac) surgical patients, traumatic brain injury, sepsis and acute pancreatitis. Furthermore, the quantity of Se administered also varied considerably, both in the amount given as a loading dose (500–4000 μg), and the presence and quantity of subsequent doses or continuous infusions. This represents the prototypical conundrum of who, when and how to treat, how to assess the efficacy of the therapy and when to discontinue treatment. The likely corollary is that some patients may benefit while others may be harmed or unaffected, and the resulting meta-analysis, perhaps predictably, revealed only modest improvement or no overall effect.
The oxidation status of Se supplements could also be of importance and, to date, overlooked. The aforementioned clinical trials all utilised intravenous sodium selenite and this oxidation product would require reduction by endogenous antioxidants (e.g. glutathione, thioredoxins) to bioactive selenide. It is feasible that this may not be achievable in some critically ill patients in whom antioxidant defences are already strained. Furthermore, the aforesaid study in mice [95] found that hydrogen selenide but not selenite was effective in mitigating reperfusion injury, thus suggesting the oxidation status of the therapy does impact upon its efficacy. Additionally, lessons should be learned from initial studies using basic sulfur salts where non-targeted release of sulfide had resulting implications for pharmacokinetics, safety and efficacy. Thereafter, intelligent drug design yielded complex molecules that enabled more controlled sulfide delivery [125,126,127], improved targeting to its intended site of action (e.g. the mitochondrion) and at concentrations that better reflect those derived from endogenous sources [128, 129]. We postulate that the same may hold true for Se-based compounds, whereby using bespoke complex molecules that target the intended matrix, therapeutic concentrations could be delivered in a controllable manner yielding more favourable results.
Conclusion
The indispensable micronutrient Se undergoes intracellular conversion to endogenous and bioactive hydrogen selenide. This makes a case for its inclusion as the fourth endogenous gasotransmitter alongside CO, NO and H2S. Hydrogen selenide currently satisfies most but not all of the criteria required for its full inclusion as a gasotransmitter. It is present physiologically as a small gaseous molecule and capable of passive transmembrane transport. It is generated enzymatically and non-enzymatically in pathways similar to hydrogen sulfide, and regulation of the enzymatic process can be modified by physical stressors and endogenous signalling molecules. Thereafter, selenoprotein synthesis is essential for the preservation of redox-balance over numerous physiological systems. Hydrogen selenide has yet to be ascribed further functional roles and is currently not associated with other well-defined cellular and molecular targets. However, we have recently demonstrated that exogenous administration can modulate aerobic respiration via inhibition of mitochondrial complex IV, and its role as an endogenous signalling molecule is under further investigation. We propose that intelligent selenomimetic drug design and delivery may generate more favorable pharmacological (and nutritional) tools over those currently used, providing an array of novel therapeutics that can confer protection against redox-based pathologies encountered in acute medicine and critical illness.
Availability of data and materials
Available from the corresponding author on reasonable request.
Abbreviations
- CO:
-
Carbon monoxide
- DNA:
-
Deoxyribonucleic acid
- GPx:
-
Glutathione peroxidase
- GSH:
-
Reduced glutathione
- GSSeH:
-
Glutathioselenol
- GSSeSG:
-
Selenodiglutathione
- H2O2 :
-
Hydrogen peroxide
- H2S:
-
Hydrogen sulfide (gas)
- H2Se:
-
Hydrogen selenide (gas)
- HS- :
-
Hydrosulfide anion
- HSe- :
-
Hydroselenide anion
- IL:
-
Interleukin
- LDL:
-
Low-density lipoprotein
- LPS:
-
Lipopolysaccharide
- mRNA:
-
Messenger ribonucleic acid
- Msr:
-
Methionine sulfoxide reductase
- NaHSe:
-
Sodium hydrogen selenide
- NF-κB:
-
Nuclear factor kappa B
- NO:
-
Nitric oxide
- NTIS:
-
Non-thyroidal illness syndrome
- ROS:
-
Reactive oxygen species
- Se:
-
Selenium
- SCL:
-
Selenocysteine β-lyase
- SeO3 2- :
-
Selenite
- SeO4 2- :
-
Selenate
- T3:
-
Triiodothyronine
- T4:
-
Thyroxine
- TNF-α:
-
Tumour necrosis factor alpha
- tRNA:
-
Transfer ribonucleic acid
- Trx:
-
Thioredoxin
- TrxR:
-
Thioredoxin reductase
- TSH:
-
Thyroid stimulating hormone
References
National Research Council (1983) Selenium in nutrition: Revised Edition. The National Academies Press, Washington, DC
Weeks ME (1932) The discovery of the elements. VI. Tellurium and selenium. J Chem Educ 9:474
Gladyshev VN, Hatfield DL (1999) Selenocysteine-containing proteins in mammals. J Biomed Sci 6:151–160
Barceloux DG (1999) Selenium. J Toxicol Clin Toxicol 37:145–172
Cupp-Sutton K, Ashby M (2016) Biological chemistry of hydrogen selenide. Antioxidants 5:42
Fordyce F (2007) Selenium geochemistry and health. Ambio 36:94–97
Wang R (2002) Two’s company, three’s a crowd: can H2S be the third endogenous gaseous transmitter? FASEB J 16:1792–1798
Brown KM, Arthur JR (2001) Selenium, selenoproteins and human health: a review. Public Health Nutr 4:593–599
Labunskyy VM, Hatfield DL, Gladyshev VN (2014) Selenoproteins: molecular pathways and physiological roles. Physiol Rev 94:739–777
Ross AC, Caballero BH, Cousins RJ, et al (2012) Modern nutrition in health and disease: Eleventh edition. Wolters Kluwer Health Adis (ESP)
Chun OK, Floegel A, Chung S-J et al (2010) Estimation of antioxidant intakes from diet and supplements in U.S. adults. J Nutr 140:317–324
Ganyc D, Self WT (2008) High affinity selenium uptake in a keratinocyte model. FEBS Lett 582:299–304
Misra S, Kwong RWM, Niyogi S (2012) Transport of selenium across the plasma membrane of primary hepatocytes and enterocytes of rainbow trout. J Exp Biol 215:1491–1501
Esaki N, Nakamura T, Tanaka H et al (1981) Enzymic synthesis of selenocysteine in rat liver. Biochemistry 20:4492–4496
Esaki N, Nakamura T, Tanaka H, Soda K (1982) Selenocysteine lyase, a novel enzyme that specifically acts on selenocysteine. Mammalian distribution and purification and properties of pig liver enzyme. J Biol Chem 257:4386–4391
Yudkoff M (2012) Chapter 42 - Disorders of amino acid metabolism. In: Brady ST, Siegel GJ, Albers RW, Price DL (eds) Basic Neurochemistry (Eighth Edition). Academic Press, New York, pp 737–754
Daher R, Van Lente F (1992) Characterization of selenocysteine lyase in human tissues and its relationship to tissue selenium concentrations. J Trace Elem Electrolytes Health Dis 6:189–194
Wray JR, Davies A, Sefton C et al (2019) Global transcriptomic analysis of the arcuate nucleus following chronic glucocorticoid treatment. Mol Metab 26:5–17
Seale LA (2019) Selenocysteine β-Lyase: biochemistry, regulation and physiological role of the selenocysteine decomposition enzyme. Antioxidants 8:357
Becker N-P, Martitz J, Renko K et al (2014) Hypoxia reduces and redirects selenoprotein biosynthesis. Met Integr Biometal Sci 6:1079–1086
Burk RF, Hill KE (2015) Regulation of selenium metabolism and transport. Annu Rev Nutr 35:109–134
Kumar S, Björnstedt M, Holmgren A (1992) Selenite is a substrate for calf thymus thioredoxin reductase and thioredoxin and elicits a large non-stoichiometric oxidation of NADPH in the presence of oxygen. Eur J Biochem 207:435–439
Ganther HE (1971) Reduction of the selenotrisulfide derivative of glutathione to a persulfide analog by gluthathione reductase. Biochemistry 10:4089–4098
Spallholz JE (1994) On the nature of selenium toxicity and carcinostatic activity. Free Radic Biol Med 17:45–64
Byard JL (1969) Trimethyl selenide. A urinary metabolite of selenite. Arch Biochem Biophys 130:556–560
Kobayashi Y, Ogra Y, Ishiwata K et al (2002) Selenosugars are key and urinary metabolites for selenium excretion within the required to low-toxic range. Proc Natl Acad Sci U S A 99:15932–15936
Palmer IS, Fischer DD, Halverson AW, Olson OE (1969) Identification of a major selenium excretory product in rat urine. Biochim Biophys Acta BBA 177:336–342
Suzuki Y, Hashiura Y, Matsumura K et al (2010) Dynamic pathways of selenium metabolism and excretion in mice under different selenium nutritional statuses. Metallomics 2:126–132
McConnell KP, Portman OW (1952) Excretion of dimethyl selenide by the rat. J Biol Chem 195:277–282
McConnell KP, Roth DM (1966) Respiratory excretion of selenium. Proc Soc Exp Biol Med 123:919–921
Hoffmann PR, Berry MJ (2005) Selenoprotein synthesis: a unique translational mechanism used by a diverse family of proteins. Thyroid 15:769–775
Papp LV, Holmgren A, Khanna KK (2010) Selenium and selenoproteins in health and disease. Antioxid Redox Signal 12:793–795
Kim H-Y, Gladyshev VN (2005) Different catalytic mechanisms in mammalian selenocysteine- and cysteine-containing methionine-R-sulfoxide reductases. PLoS Biol 3:e375
Seiler A, Schneider M, Förster H et al (2008) Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab 8:237–248
Imai H, Hirao F, Sakamoto T et al (2003) Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem Biophys Res Commun 305:278–286
Jakupoglu C, Przemeck GKH, Schneider M et al (2005) Cytoplasmic thioredoxin reductase is essential for embryogenesis but dispensable for cardiac development. Mol Cell Biol 25:1980–1988
Conrad M, Jakupoglu C, Moreno SG et al (2004) Essential role for mitochondrial thioredoxin reductase in hematopoiesis, heart development, and heart function. Mol Cell Biol 24:9414–9423
Papp LV, Lu J, Bolderson E et al (2010) SECIS-binding protein 2 promotes cell survival by protecting against oxidative stress. Antioxid Redox Signal 12:797–808
Goodyear-Bruch C, Pierce JD (2002) Oxidative stress in critically ill patients. Am J Crit Care 11:543–551
Halliwell B, Gutteridge JMC (1999) Free radicals in biology and medicine, 3rd ed. Clarendon Press ; Oxford University Press, Oxford : New York
Burk RF (2002) Selenium, an antioxidant nutrient. Nutr Clin Care Off Publ Tufts Univ 5:75–79
Rose AH, Hoffmann P (2015) Selenoproteins and cardiovascular stress. Thromb Haemost 113:494–504
Papp LV, Lu J, Holmgren A, Khanna KK (2007) From selenium to selenoproteins: synthesis, identity, and their role in human health. Antioxid Redox Signal 9:775–806
Steinbrenner H, Sies H (2009) Protection against reactive oxygen species by selenoproteins. Biochim Biophys Acta BBA 1790:1478–1485
Flohe L, Günzler WA, Schock HH (1973) Glutathione peroxidase: a selenoenzyme. FEBS Lett 32:132–134
Yant LJ, Ran Q, Rao L et al (2003) The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic Biol Med 34:496–502
Björnstedt M, Hamberg M, Kumar S et al (1995) Human thioredoxin reductase directly reduces lipid hydroperoxides by NADPH and selenocystine strongly stimulates the reaction via catalytically generated selenols. J Biol Chem 270:11761–11764
Arnér ES, Holmgren A (2000) Physiological functions of thioredoxin and thioredoxin reductase. Eur J Biochem 267:6102–6109
Maulik N, Das DK (2008) Emerging potential of thioredoxin and thioredoxin interacting proteins in various disease conditions. Biochim Biophys Acta BBA 1780:1368–1382
Kryukov GV, Kumar RA, Koc A et al (2002) Selenoprotein R is a zinc-containing stereo-specific methionine sulfoxide reductase. Proc Natl Acad Sci U S A 99:4245–4250
Steinbrenner H, Alili L, Bilgic E et al (2006) Involvement of selenoprotein P in protection of human astrocytes from oxidative damage. Free Radic Biol Med 40:1513–1523
Steinbrenner H, Steinbrenner H, Bilgic E et al (2006) Selenoprotein P protects endothelial cells from oxidative damage by stimulation of glutathione peroxidase expression and activity. Free Radic Res 40:936–943
Atkinson JB, Hill KE, Burk RF (2001) Centrilobular endothelial cell injury by diquat in the selenium-deficient rat liver. Lab Invest 81:193–200
Traulsen H, Steinbrenner H, Buchczyk DP et al (2004) Selenoprotein P protects low-density lipoprotein against oxidation. Free Radic Res 38:123–128
Aaseth J, Frey H, Glattre E et al (1990) Selenium concentrations in the human thyroid gland. Biol Trace Elem Res 24:147–152
Sakr Y, Reinhart K, Bloos F et al (2007) Time course and relationship between plasma selenium concentrations, systemic inflammatory response, sepsis, and multiorgan failure. Br J Anaesth 98:775–784
Hawker FH, Stewart PM, Snitch PJ (1990) Effects of acute illness on selenium homeostasis. Crit Care Med 18:442–446
Forceville X, Vitoux D, Gauzit R et al (1998) Selenium, systemic immune response syndrome, sepsis, and outcome in critically ill patients. Crit Care Med 26:1536–1544
de Oliveira Iglesias SB, Leite HP, Paes ÂT et al (2014) Low plasma selenium concentrations in critically ill children: the interaction effect between inflammation and selenium deficiency. Crit Care 18:R101
Strachan S, Wyncoll D (2009) Selenium in critically ill patients. J Intensive Care Soc 10:38–43
Neve J, Vertongen F, Peretz A, Carpentier YA (1989) Usual values of selenium and glutathione peroxidase in a Belgian population. Ann Biol Clin 47:138–143
Nève J (1991) Methods in determination of selenium states. J Trace Elem Electrolytes Health Dis 5:1–17
Goldson AJ, Fairweather-Tait SJ, Armah CN et al (2011) Effects of selenium supplementation on selenoprotein gene expression and response to influenza vaccine challenge: a randomised controlled trial. PLoS ONE 6:e14771
Fairweather D, Cihakova D (2009) Alternatively activated macrophages in infection and autoimmunity. J Autoimmun 33:222–230
Nelson SM, Lei X, Prabhu KS (2011) Selenium levels affect the IL-4–induced expression of alternative activation markers in murine macrophages. J Nutr 141:1754–1761
Carlson BA, Yoo M-H, Sano Y et al (2009) Selenoproteins regulate macrophage invasiveness and extracellular matrix-related gene expression. BMC Immunol 10:57
Bi C-L, Wang H, Wang Y-J et al (2016) Selenium inhibits Staphylococcus aureus-induced inflammation by suppressing the activation of the NF-κB and MAPK signalling pathways in RAW264.7 macrophages. Eur J Pharmacol 780:159–165
Shi X, Wang W, Zheng S et al (2019) Selenomethionine relieves inflammation in the chicken trachea caused by LPS though inhibiting the NF-κB pathway. Biol Trace Elem Res. https://doi.org/10.1007/s12011-019-01789-1 [Epub ahead of print]
Hoffmann FW, Hashimoto AC, Shafer LA et al (2010) Dietary selenium modulates activation and differentiation of CD4+ T cells in mice through a mechanism involving cellular free thiols. J Nutr 140:1155–1161
Carlson BA, Yoo M-H, Shrimali RK et al (2010) Role of selenium-containing proteins in T-cell and macrophage function. Proc Nutr Soc 69:300–310
Rayman MP (2012) Selenium and human health. Lancet 379:1256–1268
Bleys J (2008) Serum selenium levels and all-cause, cancer, and cardiovascular mortality among US adults. Arch Intern Med 168:404–410
Fairweather-Tait SJ, Bao Y, Broadley MR et al (2011) Selenium in human health and disease. Antioxid Redox Signal 14:1337–1383
Ventura M, Melo M, Carrilho F (2017) Selenium and thyroid disease: from pathophysiology to treatment. Int J Endocrinol 2017:1–9
Bermano G, Nicol F, Dyer JA et al (1995) Tissue-specific regulation of selenoenzyme gene expression during selenium deficiency in rats. Biochem J 311:425–430
Arthur JR, Nicol F, Beckett GJ (1992) The role of selenium in thyroid hormone metabolism and effects of selenium deficiency on thyroid hormone and iodine metabolism. Biol Trace Elem Res 34:321–325
Fliers E, Bianco AC, Langouche L, Boelen A (2015) Thyroid function in critically ill patients. Lancet Diabetes Endocrinol 3:816–825
Berger MM, Lemarchand-Béraud T, Cavadini C, Chioléro R (1996) Relations between the selenium status and the low T3 syndrome after major trauma. Intensive Care Med 22:575–581
Reilly C (2006) Selenium in food and health, 2nd ed. Springer US
Underwood EJ (1977) Selenium. In: Underwood EJ (ed) Trace elements in human and animal nutrition (Fourth Edition). Academic Press, pp 302–346
Beck MA, Levander OA, Handy J (2003) Selenium deficiency and viral infection. J Nutr 133:1463S–1467S
Chen J (2012) An original discovery: selenium deficiency and Keshan disease (an endemic heart disease). Asia Pac J Clin Nutr 21:320–326
McCarty MF (1986) An antithrombotic role for nutritional antioxidants: implications for tumor metastasis and other pathologies. Med Hypotheses 19:345–357
Schiavon R, Freeman GE, Guidi GC et al (1984) Selenium enhances prostacyclin production by cultured endothelial cells: possible explanation for increased bleeding times in volunteers taking selenium as a dietary supplement. Thromb Res 34:389–396
Gharipour M, Sadeghi M, Behmanesh M et al (2017) Selenium homeostasis and clustering of cardiovascular risk factors: a systematic review. Acta Bio-Medica Atenei Parm 88:263–270
Hu XF, Stranges S, Chan LHM (2019) Circulating selenium concentration is inversely associated with the prevalence of stroke: results from the Canadian Health Measures Survey and the National Health and Nutrition Examination Survey. J Am Heart Assoc 8:e012290
Virtamo J, Valkeila E, Alfthan G et al (1985) Serum selenium and the risk of coronary heart disease and stroke. Am J Epidemiol 122:276–282
Salonen J, Alfthan G, Huttunen J et al (1982) Association between cardiovascular death and myocardial infarction and serum selenium in a matched-pair longitudinal study. Lancet 320:175–179
Flores-Mateo G, Navas-Acien A, Pastor-Barriuso R, Guallar E (2006) Selenium and coronary heart disease: a meta-analysis. Am J Clin Nutr 84:762–773
Liu Y-H, Lu M, Hu L-F et al (2012) Hydrogen sulfide in the mammalian cardiovascular system. Antioxid Redox Signal 17:141–185
Ahmad A, Dempsey S, Daneva Z et al (2018) Role of nitric oxide in the cardiovascular and renal systems. Int J Mol Sci 19:2605
May SW, Pollock SH (1998) Selenium-based antihypertensives: rationale and potential. Drugs 56:959–964
May SW (2002) Selenium-based pharmacological agents: an update. Expert Opin Investig Drugs 11:1261–1269
Blackstone E (2005) H2S Induces a suspended animation-like state in mice. Science 308:518–518
Iwata A, Morrison ML, Blackwood JE, Roth MB (2015) Selenide targets to reperfusing tissue and protects it from injury. Crit Care Med 43:1361–1367
Szabo C, Ransy C, Módis K et al (2014) Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br J Pharmacol 171:2099–2122
Hartmann C, Nussbaum B, Calzia E et al (2017) Gaseous mediators and mitochondrial function: the future of pharmacologically induced suspended animation? Front Physiol 8:691
Samra K, Singer M, Dyson A (2019) Hydrogen selenide as the 4th gasotransmitter: a metabolic modulator with potential therapeutic utility in acute/critical illness states. Intensive Care Med Exp 7:000883
Dyson A, Dal-Pizzol F, Sabbatini G et al (2017) Ammonium tetrathiomolybdate following ischemia/reperfusion injury: chemistry, pharmacology, and impact of a new class of sulfide donor in preclinical injury models. PLOS Med 14:e1002310
Durham T, Zander D, Stomeo N et al (2019) Chemistry, pharmacology, and cellular uptake mechanisms of thiometallate sulfide donors. Br J Pharmacol. https://doi.org/10.1111/bph.14670 [Epub ahead of print]
Parnham MJ, Sies H (2013) The early research and development of ebselen. Biochem Pharmacol 86:1248–1253
Takenori Y, Keiji S, Kintomo T et al (1998) Ebselen in acute ischemic stroke. Stroke 29:12–17
Ogawa A, Yoshimoto T, Kikuchi H et al (1999) Ebselen in acute middle cerebral artery occlusion: a placebo-controlled, double-blind clinical trial. Cerebrovasc Dis 9:112–118
Noguchi N (2016) Ebselen, a useful tool for understanding cellular redox biology and a promising drug candidate for use in human diseases. Arch Biochem Biophys 595:109–112
Kuklinski B, Buchner M, Schweder R, Nagel R (1991) Acute pancreatitis--a free radical disease. Decrease in fatality with sodium selenite (Na2SeO3) therapy. Z Gesamte Inn Med 46:145–149
Lindner D, Lindner J, Baumann G et al (2004) Untersuchung zur antioxidativen Therapie mit Natriumselenit bei akuter Pankreatitis: Eine prospektive, randomisierte Blindstudie. Med Klin 99:708–712
Schmidt T, Pargger H, Seeberger E et al (2018) Effect of high-dose sodium selenite in cardiac surgery patients: a randomized controlled bi-center trial. Clin Nutr 37:1172–1180
Zimmermann T, Albrecht S, Kühne H et al (1997) Selenium administration in patients with sepsis syndrome. A prospective randomized study. Med Klin 92:3–4
Angstwurm MW, Schottdorf J, Schopohl J, Gaertner R (1999) Selenium replacement in patients with severe systemic inflammatory response syndrome improves clinical outcome. Crit Care Med 27:1807–1813
Mishra V, Baines M, Elizabeth Perry S et al (2007) Effect of selenium supplementation on biochemical markers and outcome in critically ill patients. Clin Nutr 26:41–50
Angstwurm MWA, Engelmann L, Zimmermann T et al (2007) Selenium in intensive care (SIC): results of a prospective randomized, placebo-controlled, multiple-center study in patients with severe systemic inflammatory response syndrome, sepsis, and septic shock. Crit Care Med 35:118–126
Forceville X, Laviolle B, Annane D et al (2007) Effects of high doses of selenium, as sodium selenite, in septic shock: a placebo-controlled, randomized, double-blind, phase II study. Crit Care 11:R73
González CM (2009) Efecto antiinflamatorio del selenio en pacientes sépticos. Rev Asoc Mex Med Crítica Ter Intensiva 23:199–205
Andrews PJD, Avenell A, Noble DW et al (2011) Randomised trial of glutamine, selenium, or both, to supplement parenteral nutrition for critically ill patients. BMJ 342:d1542
Manzanares W, Biestro A, Torre MH et al (2011) High-dose selenium reduces ventilator-associated pneumonia and illness severity in critically ill patients with systemic inflammation. Intensive Care Med 37:1120–1127
Valenta J, Brodska H, Drabek T et al (2011) High-dose selenium substitution in sepsis: a prospective randomized clinical trial. Intensive Care Med 37:808–815
Janka V, Ladislav K, Jozef F, Ladislav V (2013) Restoration of antioxidant enzymes in the therapeutic use of selenium in septic patients. Wien Klin Wochenschr 125:316–325
Woth G, Nagy B, Mérei Á et al (2014) The effect of Na-selenite treatment on the oxidative stress–antioxidants balance of multiple organ failure. J Crit Care 29:883.e7–883.e11
Chelkeba L, Ahmadi A, Abdollahi M et al (2015) The effect of parenteral selenium on outcomes of mechanically ventilated patients following sepsis: a prospective randomized clinical trial. Ann Intensive Care 5:29
Bloos F, Trips E, Nierhaus A et al (2016) Effect of sodium selenite administration and procalcitonin-guided therapy on mortality in patients with severe sepsis or septic shock: a randomized clinical trial. JAMA Intern Med 176:1266–1276
Chelkeba L, Ahmadi A, Abdollahi M et al (2017) The effect of high-dose parenteral sodium selenite in critically ill patients following sepsis: a clinical and mechanistic study. Indian J Crit Care Med 21:287–293
Khalili H, Ahl R, Cao Y et al (2017) Early selenium treatment for traumatic brain injury: does it improve survival and functional outcome? Injury 48:1922–1926
Moghaddam OM, Lahiji MN, Hassani V, Mozari S (2017) Early administration of selenium in patients with acute traumatic brain injury: a randomized double-blinded controlled trial. Indian Soc Crit Care Med 21:75–79
Zhao Y, Yang M, Mao Z et al (2019) The clinical outcomes of selenium supplementation on critically ill patients: a meta-analysis of randomized controlled trials. Medicine (Baltimore) 98:e15473
Sun X, Wang W, Dai J et al (2017) A long-term and slow-releasing hydrogen sulfide donor protects against myocardial ischemia/reperfusion injury. Sci Rep 7:3541
Rose P, Dymock BW, Moore PK (2015) GYY4137, a novel water-soluble, H2S-releasing molecule. In: Methods in Enzymology. Elsevier, pp 143–167
Huang CW, Feng W, Peh MT et al (2016) A novel slow-releasing hydrogen sulfide donor, FW1256, exerts anti-inflammatory effects in mouse macrophages and in vivo. Pharmacol Res 113:533–546
Powell CR, Dillon KM, Matson JB (2018) A review of hydrogen sulfide (H2S) donors: chemistry and potential therapeutic applications. Biochem Pharmacol 149:110–123
Wallace JL, Wang R (2015) Hydrogen sulfide-based therapeutics: exploiting a unique but ubiquitous gasotransmitter. Nat Rev Drug Discov 14:329–345
Acknowledgements
None.
Funding
UCLH/UCL receives a proportion of funding from the UK Department of Health’s NIHR Biomedical Research funding scheme. The funders were not involved in the experimental work performed, conclusions draw, or the preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
AD supervised the work. All authors contributed towards writing and approved the final manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
We have referenced pilot studies using biological tissue obtained from male Wistar rats, performed according to local ethics committee and UK Home Office guidelines under the 1986 Scientific Procedures Act.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kuganesan, M., Samra, K., Evans, E. et al. Selenium and hydrogen selenide: essential micronutrient and the fourth gasotransmitter?. ICMx 7, 71 (2019). https://doi.org/10.1186/s40635-019-0281-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40635-019-0281-y