
Abstract
Background
Sepsis is characterised by diminished vasopressor responsiveness. Vasoconstriction depends upon a balance: Ca2+-dependent myosin light-chain kinase promotes and Ca2+-independent myosin light-chain phosphatase (MLCP) opposes vascular smooth muscle contraction. The enzyme Rho kinase (ROK) inhibits MLCP, favouring vasoconstriction. We tested the hypothesis that ROK-dependent MLCP inhibition was attenuated in late sepsis and associated with reduced contractile responses to certain vasopressor agents.
Methods
This is a prospective, controlled animal study. Sixteen-week-old C57/BL6 mice received laparotomy or laparotomy with caecal ligation and puncture (CLP). Antibiotics, fluids and analgesia were provided before sacrifice on day 5. Vasoconstriction of the femoral arteries to a range of stimuli was assessed using myography: (i) depolarisation with 87 mM K+ assessed voltage-gated Ca2+ channels (L-type, Cav1.2 Ca2+ channels (LTCC)), (ii) thromboxane A2 receptor activation assessed the activation state of the LTCC and ROK/MLCP axis, (iii) direct PKC activation (phorbol-dibutyrate (PDBu), 5 μM) assessed the PKC/CPI-17 axis independent of Ca2+ entry and (iv) α1-adrenoceptor stimulation with phenylephrine (10−8 to 10−4 M) and noradrenaline (10−8 to 10−4 M) assessed the sum of these pathways plus the role of the sarcoplasmic reticulum (SR). ROK-dependent MLCP activity was indexed by Western blot analysis of P[Thr855]MYPT. Parametric and non-parametric data were analysed using unpaired Student's t-tests and Mann-Whitney tests, respectively.
Results
ROK-dependent inhibition of MLCP activity was attenuated in both unstimulated (n = 6 to 7) and stimulated (n = 8 to 12) vessels from mice that had undergone CLP (p < 0.05). Vessels from CLP mice demonstrated reduced vasoconstriction to K+, thromboxane A2 receptor activation and PKC activation (n = 8 to 13; p < 0.05). α1-adrenergic responses were unchanged (n = 7 to 12).
Conclusions
In a murine model of sepsis, ROK-dependent inhibition of MLCP activity in vessels from septic mice was reduced. Responses to K+ depolarisation, thromboxane A2 receptor activation and PKC activation were diminished in vitro whilst α1-adrenergic responses remained intact. Inhibiting MLCP may present a novel therapeutic target to manage sepsis-induced vascular dysfunction.
Similar content being viewed by others


Background
Septic shock arises when infection causes direct tissue injury and a maladaptive host response culminating in refractory vasodilation and loss of vasopressor sensitivity. Clinically, this manifests as septic shock and multi-organ failure, which even in modern intensive care units heralds a 90-day mortality rate of over 30% [1,2].
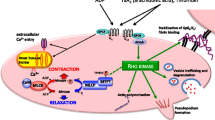
Vascular smooth muscle (VSM) contraction is determined by the balance of two opposing enzymatic processes: (i) myosin light-chain kinase (MLCK) phosphorylation of myosin enables actin-myosin cross bridge cycling and contraction; (ii) myosin light-chain phosphatase (MLCP) dephosphorylation of myosin uncouples actin-myosin, favouring relaxation. Activation of MLCK depends on cytosolic calcium (Ca2+) binding to calmodulin. Cystosolic Ca2+ is derived from either the sarcoplasmic reticulum (SR) or the extracellular space through, for example, voltage-gated L-type Cav1.2 Ca2+ channels (LTCC). Vasoconstrictors variably modulate the cytosolic [Ca2+] and the degree of inhibition of MLCP to regulate vascular tone. MLCP can be inhibited either by (i) RhoA activation of Rho kinase (ROK), which inhibits MLCP by phosphorylating Thr855 of the MYPT regulatory subunit of MLCP [3-5], or (ii) PKC-mediated phosphorylation of Thr38 CPI-17, which directly inhibits MLCP [6-8]. Inhibiting MLCP promotes VSM contraction independent of the prevailing intracellular [Ca2+] - ‘Ca2+ sensitisation’. On the other hand, increasing MLCP activity by reducing Thr855 phosphorylation of MYPT or reduced Thr38 CPI-17 phosphorylation shifts the [Ca2+]:force relationship to the right - effectively reducing Ca2+ sensitivity. Individual vasopressors vasoconstrict through different signalling pathways; for instance, thromboxane is bimodal, activating LTCC and RhoA/ROK [3], whilst α1-adrenergic receptor agonists are multimodal, inducing vasoconstriction through two sources of Ca2+ (SR and LTCC) and two MLCP inhibitory pathways (RhoA/ROK and PKC/CPI-17) [9]. A figure summarising these contractile signalling pathways is provided (Figure 1).

Smooth muscle cell contraction depends on Ca 2+ -dependent and Ca 2+ -independent pathways. Actin-myosin cross bridge cycling and smooth muscle contraction depend on phosphorylation of the myosin light chain by Ca2+: calmodulin (CaM)-dependent myosin light-chain kinase (MLCK). Relaxation is favoured by myosin light-chain phosphatase (MLCP) as the active subunit removes the phosphoryl group from the myosin light chain. Cytosolic [Ca2+] is derived from (i) the extracellular space (through voltage-operated Ca2+ channels (VOCC; predominantly Cav1.2 L-type Ca2+ channels) and receptor-operated non-specific cation channels (ROCC)) or (ii) IP3-dependent release from the sarcoplasmic reticulum (SR). MLCP is inhibited, favouring contraction, either through (i) direct inhibition by PKC-dependent CPI-17 or (ii) inhibitory phosphorylation of the regulatory subunit MYPT by RhoA-dependent Rho kinase. α1-adrenergic G protein-coupled receptors (GPCR) activate all four contractile pathways: VOCC/ROCC, IP3/SR, PKC/CPI-17 and RhoA/ROK; TxA2 GPCR activate VOCC and RhoA/ROK; PDBu activates PKC/CPI-17 whilst high-[K+]-mediated depolarisation activates VOCC. PLC, phospholipase C; PIP2, phosphatidylinositol 4,5-bisphosphate; DAG, diacylglycerol; IP3, inositol triphosphate.
There is evidence that Ca2+ desensitisation contributes to septic shock: in a rat model, acute endotoxaemia was associated with augmented MLCP activity and reduced sensitivity to an α1-adrenergic agonist [10]. Other models of sepsis have identified a rightward shift in the cytosolic [Ca2+]: force relationship [11] and attenuated vasopressor responses despite the presence of augmented cytosolic [Ca2+] [12], suggesting increased activity of MLCP. Many investigations into the vascular pathology of septic shock have focused on the early, hyperdynamic phase of sepsis. Importantly, however, 70% of deaths from sepsis occur after the first 3 days [2,13,14]. Mechanisms of vascular dysfunction at this later stage of disease remain relatively unexplored, but important differences have been identified; specifically, whilst early sepsis is associated with impaired vascular responses to α1-adrenergic agonists in vitro and a reduction in cytosolic Ca2+ entry, in the later phases of sepsis, the in vitro response to α1-adrenergic agonists and Ca2+ flux are restored [15,16], as is the in vivo blood pressure response to moderate doses of phenylephrine [17]. However, sensitivity to local vasoconstriction by thromboxane A2 (TxA2) and direct electrical coupling of the VSM membrane, significant for their importance in regulating regional blood flow [18], has not been evaluated in sepsis. Unlike α1-adrenergic agonists, TxA2 and membrane depolarisation do not stimulate SR Ca2+ release or PKC/CPI-17-mediated inhibition of MLCP and are therefore potentially more vulnerable to attenuation of the RhoA/ROK pathway [3,19]. Diminished efficacy of endogenous TxA2 and direct depolarisation secondary to dysfunction of the RhoA/ROK pathway may contribute to systemic hypotension in septic shock and dysregulation of regional blood flow [18,20].
To address this gap in knowledge, we used a murine caecal ligation and puncture (CLP) model of sepsis that included an intensive care treatment regimen that simulated the clinical experience of a septic patient [21,22]. CLP is a well-described, reproducible model of sepsis incorporating polymicrobial infection and tissue necrosis [23,24]. As an animal model, it replicates many of the physiological, haemodynamic and metabolic sequelae of sepsis [25-28]. We used a 5-day model of sepsis to identify mechanisms that contribute to vasopressor insensitivity during the late stages of sepsis, noting that most deaths from sepsis occur after the first 3 days [2,13,14]. We hypothesised that CLP sepsis would be associated with increased MLCP activity and Ca2+ desensitisation and that this activity would be associated with a reduction in in vitro vascular responses to vasoconstrictors such as thromboxane A2, phorbol ester and high K+ which are uni- or bimodal, engaging one or two signalling pathways. We hypothesised that responses to α1-adrenergic agonists which invoke multimodal mechanisms of Ca2+ entry (LTCC and SR Ca2+) and Ca2+ sensitisation (PKC/CPI-17 and Rho/ROK) would be relatively preserved in vitro.
Methods
Mice
All animal procedures were approved by the Animal Ethics Committees of the Institute of Medical and Veterinary Science (IMVS; Adelaide, South Australia) and the University of Adelaide (Adelaide, South Australia). Twenty-six, 16-week-old, male C57/BL6 mice purchased from the Animal Resources Centre, WA, were housed in the IMVS Animal Care Facility in individual cages following surgery with food and water ad libitum. Changes in clinical status were determined by cumulative clinical record scores as outlined previously [25].
Caecal ligation and puncture
Sixteen mice underwent a midline laparotomy under general anaesthetic (1% to 1.5% isoflurane in oxygen) followed by location and ligation of the caecum 5 mm from the ileocaecal valve. A 21-gauge needle was used to puncture the caecum midway between the ligature and the tip of the caecum. Faecal material was expressed from the caecum into the abdominal cavity through the puncture site before the perforated caecum was placed back into the abdominal cavity and the incision was sutured. Morbidity associated with CLP surgery was assessed by monitoring for weight loss, abnormal temperatures (normal core temperature range 34 to 37) and other morbidity indicators including ruffled coat, hunched posture, reduced mobility, diarrhoea, abdominal distention, loss of righting ability and laboured respiration; these parameters were scored to calculate a cumulative disease index [25]. Post-mortem was carried out on representative CLP mice demonstrating acute suppurative and necrotising peritonitis. Ten mice underwent laparotomy without CLP as sham controls. All mice underwent an intensive care regime post-surgery: twice-daily subcutaneous administration of antibiotic (Baytril (enrofloxacin), 0.03 mg kg−1, 50 μl), fluid (saline 1 ml) for 5 days and analgesia (butorphanol, 0.05 mg kg−1, 25 μl) for 2 days. On day 5, surviving mice were killed with pentobarbitone (60 mg kg−1 i.p.) before tissue collection.
Isolation of vessels
Femoral and caudal arteries were placed in cold Ca2+-free normal HEPES-Tyrode buffer containing, in mM, 135.5 NaCl, 5.9 KCl, 1.2 MgCl2, 11.6 glucose and 11.6 HEPES, pH 7.4, dissected free of adventitia and cut into 2-mm segments; femoral and caudal arteries were 250 and 300 μm in diameter, respectively. Caudal artery segments from sham and CLP treatment groups were snap-frozen in liquid nitrogen and stored at −80°C prior to SDS-PAGE/Western blot analysis.
Vascular myography
Femoral arterial segments from sham and CLP treatment groups were mounted on a DMT 610M wire myograph (DMT, Aarhus, Denmark) to quantify isometric tension following stimulation. Following length tension analysis, femoral arteries were set at an optimal resting tension of 3 mN and equilibrated in normal HEPES-Tyrode buffer (in mM, 135.5 NaCl, 5.9 KCl, 1.2 MgCl2, 2.5 CaCl2, 11.6 glucose, 11.6 HEPES, pH 7.4). The arteries were equilibrated for 30 min before being stimulated three times with a depolarising stimulus of 87 mM K+-HEPES-Tyrode buffer (K+HT), in which NaCl was substituted with KCl to maintain an iso-osmolar solution. Following relaxation in NHT buffer, the arteries were incubated with the stable thromboxane A2 receptor agonist U46619 (0.1 μM), the direct PKC activator phorbol-dibutyrate (PDBu) (5 μM), the α1-adrenergic agonist phenylephrine (PE) (10−8 to 10−4 M) or noradrenaline (NA) (10−8 to 10−4 M), primarily an α1-adrenergic agonist but with significant β-adrenergic agonist action. Noradrenaline was used because, although less specific in action than phenylephrine, it is more commonly used both in sepsis research and clinical practice.
Following a 10-min stimulation with U46619 (0.1 μM) or PDBu (5 μM) or generation of a dose-response relationship with NA or PE (10−8 to 10−4 M), tissues were snap-frozen in dry ice-cold 10% trichloroacetic acid/acetone, washed with dry ice-cold acetone and stored at −80°C prior to SDS-PAGE/Western blot analysis.
Myography data and biochemical analysis of stimulated arterial tissue was derived from the femoral artery. As quantities of the femoral artery were limited, biochemical analysis of unstimulated sham and CLP tissue was derived from the caudal artery (nb caudal segments were always compared with caudal and femoral with femoral). Endothelial integrity of arterial segments was confirmed by phenylephrine challenge followed by an intact 90% ACh relaxation.
Western blot analysis of MYPT
For analysis of total MYPT and P[Thr855]MYPT, proteins were extracted from each 2-mm arterial segment using 100 μl of sample buffer containing 50 mM Tris-HCl, pH 6.8, 1% (w/v) SDS, 1× Complete™ protease inhibitor cocktail (Roche, Mannheim, Germany), 100 μM di-isopropylfluorophosphate (Sigma-Aldrich, Caste Hill, Australia), 10 mM DTT, 10% (w/v) sucrose and 0.1% (w/v) bromophenol blue. Samples were heated to 95°C for 5 min and then agitated for 30 min prior to SDS-PAGE using a Bio-Rad (Sydney, Australia) Mini-PROTEAN II unit at 200 V for 60 min. For analysis of MYPT, proteins were transferred, using a Bio-Rad Mini transfer unit, onto 0.22 μm nitrocellulose (Bio-Rad) at 100 V for 30 min in transfer buffer containing 25 mM Tris-HCl, 192 mM glycine, 0.1% (w/v) SDS and 20% (v/v) methanol.
Following protein transfer to nitrocellulose, non-specific binding sites were blocked with 50% LI-COR Odyssey blocking buffer (LI-COR Biosciences, Lincoln, NE, USA) for 60 min, followed by incubation with TBS-T (20 mM Tris, 150 mM NaCl, 0.05% (v/v) Tween-20) containing either a mouse-derived affinity-purified anti-MYPT antibody (1:1,000) made in-house or a commercially available (Upstate Biotechnology, Lake Placid, NY, USA) rabbit-derived anti-P[Thr855]MYPT antibody (1:1,000) for 60 min. Nitrocellulose membranes were washed three times in TBS-T and incubated with a 1:10,000 dilution (in TBS-T) of biotin-conjugated secondary antibodies (Pierce Thermo Scientific, Rockford, IL, USA): anti-mouse IgG for MYPT and anti-rabbit IgG for P[Thr855]MYPT (1:10,000), for 60 min before another three washes with TBS-T. The nitrocellulose was then incubated for 60 min in TBS-T with a 1:10,000 dilution of streptavidin conjugated to the 800-nm DyLight fluorochome (Pierce Thermo Scientific); fluorescence was detected and quantified using the LI-COR Odyssey system. Western blot signals of the Thr855 phosphorylation state of MYPT of untreated vs. U46619 (0.1 μM)- or PDBu (5 μM)-treated rat caudal artery as previously published [3] were used as controls.
Although consistency of protein content was confirmed with Coomassie blue-stained actin, all Western blot signals were evaluated and found to be within the linear range of detection. Vessel size and protein load were not significantly different; nevertheless, the primary outcome of phosphorylation analysis is expressed as a ratio of P[Thr855]MYPT to total MYPT taken from the same sample.
Statistical analysis
The Mann-Whitney test was used for non-parametric data comparisons, and Student's t-test was used to compare parametric data (both two-tailed); p < 0.05 was considered statistically significant. Asterisks indicate statistically significant differences from control; data are presented as mean ± SEM.
Results
Morbidity and mortality
There were no deaths in sham-operated mice. The mortality rate in CLP-operated mice was 16%, similar to the real-world experience of severe sepsis managed in a critical care environment [29,30]. The cumulative disease index was significantly higher for mice undergoing CLP surgery than for sham-operated controls (p < 0.05, data not shown) [25].
Does the ROK-dependent basal activity state of myosin phosphatase, indexed by Thr855 MYPT phosphorylation, differ between septic and control mice?
To compare the basal activity state of MLCP in sham and CLP mice, the proportion of total MYPT in the [Thr855]-phosphorylated, i.e. ROK-inhibited, state was identified using Western blot analysis of unstimulated mouse caudal artery segments. The proportion of [Thr855]-phosphorylated MYPT in unstimulated vessels was greater in sham than in CLP mice (n = 6 to 7; p < 0.05), indicating that MLCP was more active in CLP mice favouring vasorelaxation (Figure 2).

Sepsis was associated with increased activation state of MLCP. The activity state of MLCP was greater in isolated, unstimulated caudal arteries from 5-day CLP mice than in those from sham mice. The increased activity state of MLCP was indexed by reduction in the ratio of Thr855 phosphorylated to unphosphorylated MYPT determined by Western blot analysis of snap-frozen vessel segments. *p < 0.05, n = 6 to 7.
Does the ability of thromboxane A2 receptor stimulation to inhibit MLCP differ in CLP and sham mice?
Thromboxane activates the RhoA/ROK pathway leading to inhibitory phosphorylation of Thr855 MYPT, the regulatory subunit of MLCP, favouring contraction by Ca2+ sensitisation [3]. To establish whether the ability of the TxA2 receptor to inhibit MLCP was attenuated in CLP compared to sham mice, the [Thr855] phosphorylation state of MYPT was compared in isolated femoral artery segments following challenge with the TxA2 receptor agonist U46619 (0.1 μM). CLP mice showed reduced phosphorylation of [Thr855]MYPT (less MLCP inhibition) in response to TxA2 receptor stimulation than sham-treated mice (Figure 3; n = 9 to 12; p < 0.05).

TxA 2 receptor-mediated inhibition of MLCP was attenuated in sepsis. The activity state of MLCP was increased in femoral arteries stimulated with U46619 (0.1 μM) in 5-day CLP mice relative to those in sham mice (indexed by reduction in the ratio of Thr855 phosphorylated to unphosphorylated MYPT on Western blot analysis). *p < 0.05, n = 9 to 12.
Was the reduction in Thr855 MLCP phosphorylation in CLP mice associated with a reduction in vascular contractility?
TxA2 elicits VSM contraction both through Rho kinase-mediated MLCP inhibition and by increasing the open probability of LTCC. If RhoA/ROK activity was disrupted during sepsis, decreasing [Thr855]MYPT phosphorylation, it would be expected that activation of TxA2 receptors would fail to fully restore MLCP inhibition. The consequence would be reduced Ca2+ sensitivity and a diminished contractile response to the TxA2 mimetic U46619. The contraction of isolated femoral artery segments in response to U46619 (0.1 μM) was attenuated in CLP compared to sham mice (Figure 4; n = 10 to 12; p < 0.05), consistent with the hypothesis that reduced MLCP inhibition contributes to septic shock.

The vascular contractile response to the thromboxane receptor agonist U46619 was attenuated in sepsis. Wire myograph-mounted femoral arteries from 5-day CLP mice developed lower tension under stimulation with U46619 (0.1 μM) compared to those from sham mice. *p < 0.05, n = 10 to 12.
Was the contractile response to PDBu, a direct activator of PKC/CPI-17, attenuated in sepsis?
The direct PKC agonist PDBu inhibits MLCP by stimulating PKC-dependent phosphorylation of Thr38 of CPI-17 [31]. In the Thr38-phosphorylated state, CPI-17 is a specific, direct inhibitor of MLCP [6]. Contractile responses to PDBu (5 μM) were reduced in femoral artery segments from CLP mice compared with segments from sham mice (Figure 5; n = 8; p < 0.05). As expected, treatment of artery segments from control mice with PDBu did not directly alter the Thr855 phosphorylation of MYPT (data not shown) [19]. To identify whether the attenuated contractile response to PDBu in CLP mice may have resulted from a sepsis-dependent reduction in baseline Thr855 phosphorylation of MYPT and consequently augmented MLCP activity, we used Western blot analysis to ascertain the Thr855 phosphorylation state of MYPT in control and CLP mice. We identified a reduction in Thr855 phosphorylation of MYPT in the PDBu (5 μM)-treated femoral artery segments from CLP mice compared to segments from sham mice (Figure 6; n = 8; p < 0.05).

The vascular contractile response to the direct PKC activator phorbol-dibutyrate (PDBu) was attenuated in sepsis. Wire myograph-mounted femoral arteries from 5-day CLP mice developed lower tension under stimulation with PDBu (5 μM) compared to those from sham mice. *p < 0.05, n = 8 to 13.

Attenuated PKC-dependent contraction in septic mouse artery was associated with reduced inhibitory phosphorylation of MLCP. The activity state of MLCP was increased in femoral arteries stimulated with PDBu (5 μM) in 5-day CLP mice relative to those in sham mice (indexed by reduction in the ratio of Thr855 phosphorylated to unphosphorylated MYPT on Western blot analysis). *p < 0.05, n = 8.
Could the attenuated contractile response to TxA2 receptor and PKC/CPI-17 stimulation in CLP mice be wholly explained by changes in membrane potential during sepsis?
A significant body of work has demonstrated that augmented ATP-sensitive potassium (KATP) channel function causes VSM hyperpolarisation with consequent vasodilation and vasopressor resistance in sepsis [32,33]. Contractile responses to U46619 and PKC/CPI-17 depend on both the basal activity of MLCP and baseline conductance of the KATP channel as it modulates membrane potential and hence the activity of LTCC. In order to isolate the contributions made by increased MLCP and KATP channel activities to attenuated functional vascular contractile responses, we made use of a high-K+ depolarising solution, which directly activates LTCC independent of KATP channel conductance. The contractile response to high-K+-mediated depolarisation of isolated femoral artery segments from CLP mice was reduced compared to those from sham mice independent of KATP activity (Figure 7; n = 9 to 13; p < 0.05).

The vascular contractile response to depolarisation with high-K + HT solution was attenuated in sepsis. Wire myograph-mounted femoral arteries from 5-day CLP mice developed lower tension under stimulation with a depolarising high-K+ HT solution compared to those from sham mice. *p < 0.05, n = 9 to 13.
Is the contractile response to α1-adrenergic stimulation intact during the later phase of sepsis?
VSM stimulation by α1-adrenergic agonists involves Ca2+ entry through LTCC and from SR as well as Ca2+ sensitisation mediated by both PKC/CPI-17 and RhoA/ROK activation [9]. Contrary to acute endotoxaemia models, studies analysing vessels after longer periods of sepsis have found recovery of contractile responses to α1-adrenergic stimulation [16]. We used isolated femoral vessels from a 5-day model of polymicrobial sepsis, somewhat akin to a clinical scenario, to identify whether survivors had recovered α1-adrenergic response. Figure 8 shows that the contractile response to the α1-adrenergic agonists noradrenaline (Figure 8A) and phenylephrine (Figure 8B) (10−8 to 10−4 M) was not significantly different in CLP compared to sham mice (E max and EC50 p = NS, n = 7 to 12).

Vasoconstrictor responses to stimulation with phenylephrine and noradrenaline remained intact in arteries from septic mice. Dose-response curves to stimulation with NA (10−8 to 10−4 M) (A) and PE (10−8 to 10−4 M) (B) were generated using wire myograph-mounted femoral arteries. For both agonists, dose-response curves in 5-day CLP mice (filled circles) and sham mice (hollow circles) did not differ with regard to EC50 or E max (p > 0.05). (n = 7 to 12).
Discussion
Hospital mortality from septic shock remains around 30%, highlighting the need for a more complete understanding of the mechanisms of pathological vasodilation [1]. Proposed mechanisms include reduced α1-receptor density and reduced α1-adrenergic signal transduction [34], activation of vascular KATP channels [33], impaired SR Ca2+ release [15,35], relative corticosteroid and vasopressin deficiency [36,37] and activation of inducible nitric oxide synthase [38] and vasodilating inflammatory mediators [39-42]. We tested the hypothesis that reduced inhibition of MLCP promotes Ca2+ desensitisation, contributing to pathological vasodilation in a murine CLP model of sepsis. Normally, MLCP inhibition sensitises the vascular contractile apparatus to Ca2+, favouring vasoconstriction [19,43]. Events that release MLCP inhibition by reducing either (i) RhoA/ROK-mediated inhibitory Thr855 phosphorylation of the regulatory subunit MYPT or (ii) PKC-mediated Thr38 phosphorylation of CPI-17 favour vasorelaxation [19]. Using a 5-day murine model of polymicrobial sepsis, we identified that both the baseline phosphorylation state of Thr855 MYPT and the Thr855 phosphorylation state of MYPT following stimulation with the TxA2 receptor agonist U46619 (0.1 μM) were attenuated in septic animals compared to sham-operated controls (Figures 2 and 3). Reduced Thr855 MYPT phosphorylation was associated with reduced vasoconstrictor response to U46619, PDBu or high-[K+]-mediated depolarisation (Figures 4, 5 and 7). These data are consistent with late sepsis being associated with Ca2+ desensitisation, rendering the vasculature less responsive to certain endogenous vasopressors.
Previous data are consistent with the view that vasopressor responses can be attenuated in sepsis due to Ca2+ desensitisation, rather than as a consequence of reduced flux of Ca2+ into the cytosol [Ca2+]cyt. For example, the [Ca2+]cyt content in the aortae of rats with confirmed CLP sepsis was actually elevated relative to that of non-septic animals [12]. Isolated mesenteric arteries from rats injected with Escherichia coli lipopolysaccharide (LPS) also had elevated VSM [Ca2+]cyt and a force:intracellular Ca2+ relationship shifted to the right, consistent with Ca2+ desensitisation [11]. More recently, in an acute (6 to 24 h) rat model of LPS-induced endotoxaemia, da Silva-Santos and colleagues identified reduced inhibitory phosphorylation of MLCP in the mesenteric vasculature, associated with reduced vasopressor sensitivity [10].
Sepsis in humans typically lasts longer than most experimental animal models of sepsis, which, for practical reasons, are often less than 24 h. In clinical practice, most deaths occur after several days of sepsis [2,13,14]. It is arguable that a CLP model, whereby tissue necrosis and polymicrobial infection with host enteric organisms develop over several days after traumatic perforation of a viscus, reflects a more common clinical paradigm than sepsis induced by injection of bacterial inoculum or LPS [44,45]. We evaluated the functional and molecular features of murine vascular dysfunction 5-days post caecal ligation and puncture.
Our data identified an important difference in vascular response between acute endotoxaemia and a 5-day peritonitis model: the first 2 to 6 h of acute endotoxaemia models typically demonstrate reduced vasoconstriction in response to α1-adrenoceptor stimulation [10,16,33,46]. In contrast, after 5 days of polymicrobial sepsis, we identified normal in vitro α1-adrenoceptor-mediated vasoconstriction, consistent with endotoxaemia models, which found recovery of in vitro and in vivo sensitivity to moderate doses of α1-adrenoceptor agonist between 6 and 24 h [16,17]. However, our data identifies clear insensitivity to other vasoconstrictor mechanisms, including TxA2 receptor activation and direct membrane depolarisation (Figures 4, 5 and 7). α1-adrenoceptor agonist responses may demonstrate resilience by recruiting multiple, parallel signalling pathways to achieve contraction (Figure 1), specifically 1) LTCC opening, 2) SR Ca2+ release, 3) inhibition of MLCP by PKC/CPI-17 and 4) inhibition of MLCP by RhoA/ROK phosphorylation of MYPT. In particular, α1-adrenoceptor agonists increase cytosolic [Ca2+]cyt through SR Ca2+ release, a mechanism not shared by TxA2 receptor, PDBu activation or high-K+-mediated contraction [3,19], and have two mechanisms by which to inhibit MLCP through both PKC/CPI-17 and RhoA/ROK. These data provide a mechanistic explanation for the effectiveness of α1-adrenoceptor agonists as vasoconstrictors in sepsis.
The results of our study together with those of da Silva-Santos and colleagues [10] indicate two salient findings: 1) disinhibition of MLCP favouring Ca2+ desensitisation has been identified at different time points and in different models of sepsis, suggesting a potentially important role in the pathogenesis of septic shock, and 2) preservation of in vitro α1-adrenoceptor agonist sensitivity may not necessarily reflect intact vasoconstrictor responses to other physiologically significant stimuli.
Several potential mechanisms by which sepsis might promote MLCP disinhibition and Ca2+ desensitisation can be postulated. Pathogenic bacteria produce toxins capable of directly interfering with RhoA/ROK-dependent phosphorylation of MYPT, thereby disinhibiting MLCP and opposing cell contraction [19]: E. coli and Clostridium botulinum derived exotoxins EDIN and C3-transferase ADP-ribosylate and inactivate RhoA, Clostridium difficile toxin B glycosylates and inactivates RhoA whilst Yersinia spp. produces toxin Yop T which prevents RhoA from co-localising in the cell membrane with Rho kinase [47-49]. In contrast, SpA from Staphylococcus aureus and CNF1 from E. coli promote RhoA/ROK activity [50,51]. Endogenous mediators might also contribute to Ca2+ desensitisation: nitric oxide, kynurenine and ANP levels are elevated in septic shock [40,52], potentially promoting protein kinase G (PKG)-dependent disinhibition of MLCP and Ca2+ desensitisation [10,53,54].
Our findings of impaired response to TxA2 and membrane depolarisation may be of particular clinical significance for two reasons: Firstly, it is well established that restoration of normal systemic circulatory parameters is not necessarily indicative of restored regional perfusion, and indeed, ongoing regional ischaemia despite restoration of systemic blood pressure is associated with poor clinical outcome [55-58]. Microvascular splanchnic perfusion is not only reduced but highly heterogeneous in septic shock even within areas of uniform metabolic demand [20], indicating a failure of local vasomotor regulation. Since TxA2 [59] and local VSM membrane potential [60,61] play a key role in local vasomotor regulation, our evidence that their effectiveness is impaired in late sepsis and might be restored by MLCP inhibition opens a potentially useful area of investigation. Secondly, the experimentally observed restoration of α1-adrenergic responses in vitro may not be borne out in the septic patient because in vivo endogenous vasoconstrictors such as constitutively secreted TxA2 and direct depolarisation potentiate the response to an α1-adrenergic agonist [62]. The apparently attenuated response to α1-adrenergic agonists in vivo may reflect diminished response to these normally synergistic vasopressor pathways.
Implications for clinical research and management
Our data, from a murine model, identify the association between MLCP disinhibition and vasopressor insensitivity in a murine model of sepsis. Future studies relating MLCP activity and regional blood flow in vivo in large animals and ultimately human sepsis patients will provide valuable insights into the specific clinical consequences of myosin phosphatase dysregulation.
Advances in our understanding of the pathobiology of sepsis have characterised sepsis as a heterogeneous disease, and optimal therapy will need to be tailored to particular unique patient/pathogen disease phenotypes [63,64]. Our findings suggest that in certain patients, excessive MLCP activity could contribute to the pathogenesis of sepsis. Identifying these patients would allow targeted therapeutic inhibition of MLCP or use of vasopressors less dependent upon RhoA/ROK signalling. Restoring vascular tone by directly inhibiting MLCP circumvents receptor downregulation and avoids promoting injurious increases in cytosolic [Ca2+] [12]. In addition, targeting MLCP-dependent Ca2+ sensitisation could be particularly effective in re-establishing local responsiveness to TxA2 and membrane depolarisation, thereby restoring regulation of regional perfusion [18].
The role of RhoA/ROK-mediated inhibition of MLCP extends beyond vascular smooth muscle: thrombin, a key mediator of the cross-talk between coagulation and inflammation in sepsis has, been shown to inactivate MLCP and therefore contraction in a RhoA-dependent manner in human endothelial cells [65]. Given the prominence of endothelial dysfunction in sepsis, investigating the role of MLCP disinhibition in the pathogenesis of increased endothelial permeability is a research priority.
Study limitations
We studied an animal model of sepsis at a single time point, and translating our results to patients mandates caution. To reflect human sepsis as closely as possible, we used a CLP model, which has advantages over bacterial inoculum and lipopolysaccharide models [45], incorporating fluid and antibiotic therapy to recreate haemodynamic and metabolic phases of treated human polymicrobial sepsis. Mortality in the intervention arm was 16%, comparable to mortality in human studies of severe sepsis, but approximately half the mortality rate found amongst patients with septic shock [30]. In addition, to maintain consistency, we analysed vessels from sacrificed mice rather than mice dying of their sepsis. For these reasons, our data may be derived from somewhat healthier animals than typical patients with septic shock.
Conclusions
Mechanisms of pathological vasodilation and vasopressor resistance in septic shock are diverse. Our observations in a murine caecal ligation and puncture model are consistent with the hypothesis that during sepsis disinhibition of myosin phosphatase promotes insensitivity to certain physiologically important vasoconstrictor mechanisms, such as thromboxane A2 and membrane depolarisation. Approaches aimed at directly inhibiting MLCP might prove to be an effective therapeutic strategy to counter the pathological vasodilatation of septic shock.
References
Sprung CL, Annane D, Keh D, Moreno R, Singer M, Freivogel K, Weiss YG, Benbenishty J, Kalenka A, Forst H, Laterre PF, Reinhart K, Cuthbertson BH, Payen D, Briegel J (2008) Hydrocortisone therapy for patients with septic shock. N Engl J Med 358(2):111–124
The Pro CI (2014) A randomized trial of protocol-based care for early septic shock. N Engl J Med 370:1683–1693
Wilson DP, Susnjar M, Kiss E, Sutherland C, Walsh MP (2005) Thromboxane A2-induced contraction of rat caudal arterial smooth muscle involves activation of Ca2+ entry and Ca2+ sensitization: Rho-associated kinase-mediated phosphorylation of MYPT1 at Thr-855, but not Thr-697. Biochem J 389(Pt 3):763–774
Muranyi A, Derkach D, Erdodi F, Kiss A, Ito M, Hartshorne DJ (2005) Phosphorylation of Thr695 and Thr850 on the myosin phosphatase target subunit: inhibitory effects and occurrence in A7r5 cells. FEBS Lett 579(29):6611–6615
Stevenson AS, Matthew JD, Eto M, Luo S, Somlyo AP, Somlyo AV (2004) Uncoupling of GPCR and RhoA-induced Ca2+-sensitization of chicken amnion smooth muscle lacking CPI-17. FEBS Lett 578(1–2):73–79
Eto M, Ohmori T, Suzuki M, Furuya K, Morita F (1995) A novel protein phosphatase-1 inhibitory protein potentiated by protein kinase C: isolation from porcine aorta media and characterization. J Biochem 118(6):1104–1107
Eto M, Kitazawa T, Brautigan DL (2004) Phosphoprotein inhibitor CPI-17 specificity depends on allosteric regulation of protein phosphatase-1 by regulatory subunits. Proc Natl Acad Sci U S A 101(24):8888–8893
Hayashi Y, Senba S, Yazawa M, Brautigan DL, Eto M (2001) Defining the structural determinants and a potential mechanism for inhibition of myosin phosphatase by the protein kinase C-potentiated inhibitor protein of 17 kDa. J Biol Chem 276(43):39858–39863
Dimopoulos GJ, Semba S, Kitazawa K, Eto M, Kitazawa T (2007) Ca2+-dependent rapid Ca2+ sensitization of contraction in arterial smooth muscle. Circ Res 100(1):121–129
da Silva-Santos JE, Chiao CW, Leite R, Webb RC (2009) The Rho-A/Rho-kinase pathway is up-regulated but remains inhibited by cyclic guanosine monophosphate-dependent mechanisms during endotoxemia in small mesenteric arteries. Crit Care Med 37(5):1716–1723
Martinez MC, Muller B, Stoclet JC, Andriantsitohaina R (1996) Alteration by lipopolysaccharide of the relationship between intracellular calcium levels and contraction in rat mesenteric artery. Br J Pharmacol 118(5):1218–1222
Song SK, Karl IE, Ackerman JJ, Hotchkiss RS (1993) Increased intracellular Ca2+: a critical link in the pathophysiology of sepsis? Proc Natl Acad Sci U S A 90(9):3933–3937
Hotchkiss RS, Monneret G, Payen D (2013) Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis 13(3):260–268
Ranieri VM, Thompson BT, Barie PS, Dhainaut JF, Douglas IS, Finfer S, Gardlund B, Marshall JC, Rhodes A, Artigas A, Payen D, Tenhunen J, Al-Khalidi HR, Thompson V, Janes J, Macias WL, Vangerow B, Williams MD (2012) Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med 366(22):2055–2064
Farmer MR, Roberts RE, Gardiner SM, Ralevic V (2003) Effects of in vivo lipopolysaccharide infusion on vasoconstrictor function of rat isolated mesentery, kidney, and aorta. J Pharmacol Exp Ther 306(2):538–545
Bennett T, Mahajan RP, March JE, Kemp PA, Gardiner SM (2004) Regional and temporal changes in cardiovascular responses to norepinephrine and vasopressin during continuous infusion of lipopolysaccharide in conscious rats. Br J Anaesth 93(3):400–407
Guarido KL, Goncalves RP, Junior AG, da Silva-Santos JE (2014) Increased activation of the Rho-A/Rho-kinase pathway in the renal vascular system is responsible for the enhanced reactivity to exogenous vasopressin in endotoxemic rats. Crit Care Med 42(6):e461–e471
Segal SS (2005) Regulation of blood flow in the microcirculation. Microcirculation 12(1):33–45
Somlyo AP, Somlyo AV (2003) Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev 83(4):1325–1358
Vincent JL, De Backer D (2005) Microvascular dysfunction as a cause of organ dysfunction in severe sepsis. Crit Care 9(Suppl 4):S9–12
Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA (2009) Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc 4(1):31–36
Doi K, Leelahavanichkul A, Yuen PS, Star RA (2009) Animal models of sepsis and sepsis-induced kidney injury. J Clin Invest 119(10):2868–2878
Buras JA, Holzmann B, Sitkovsky M (2005) Animal models of sepsis: setting the stage. Nat Rev Drug Discov 4(10):854–865
Hubbard WJ, Choudhry M, Schwacha MG, Kerby JD, Rue LW 3rd, Bland KI, Chaudry IH (2005) Cecal ligation and puncture. Shock 24(Suppl 1):52–57
Deane AM, Rayner CK, Keeshan A, Cvijanovic N, Marino Z, Nguyen NQ, Chia B, Summers MJ, Sim JA, van Beek T, Chapman MJ, Horowitz M, Young RL (2013) The effects of critical illness on intestinal glucose sensing, transporters, and absorption. Crit Care Med.
Tao W, Deyo DJ, Traber DL, Johnston WE, Sherwood ER (2004) Hemodynamic and cardiac contractile function during sepsis caused by cecal ligation and puncture in mice. Shock 21(1):31–37
van de Sandt AM, Windler R, Godecke A, Ohlig J, Zander S, Reinartz M, Graf J, van Faassen EE, Rassaf T, Schrader J, Kelm M, Merx MW (2013) Endothelial NOS (NOS3) impairs myocardial function in developing sepsis. Basic Res Cardiol 108(2):330
Hollenberg SM, Dumasius A, Easington C, Colilla SA, Neumann A, Parrillo JE (2001) Characterization of a hyperdynamic murine model of resuscitated sepsis using echocardiography. Am J Respir Crit Care Med 164(5):891–895
Blanco J, Muriel-Bombin A, Sagredo V, Taboada F, Gandia F, Tamayo L, Collado J, Garcia-Labattut A, Carriedo D, Valledor M, De Frutos M, Lopez MJ, Caballero A, Guerra J, Alvarez B, Mayo A, Villar J (2008) Incidence, organ dysfunction and mortality in severe sepsis: a Spanish multicentre study. Crit Care 12(6):R158
Kaukonen KM, Bailey M, Suzuki S, Pilcher D, Bellomo R (2014) Mortality related to severe sepsis and septic shock among critically ill patients in Australia and New Zealand, 2000-2012. JAMA 311(13):1308–1316
Walsh MP, Susnjar M, Deng J, Sutherland C, Kiss E, Wilson DP (2007) Phosphorylation of the protein phosphatase type 1 inhibitor protein CPI-17 by protein kinase C. Methods Mol Biol 365:209–223
Buckley JF, Singer M, Clapp LH (2006) Role of KATP channels in sepsis. Cardiovasc Res 72(2):220–230
Landry DW, Oliver JA (1992) The ATP-sensitive K+ channel mediates hypotension in endotoxemia and hypoxic lactic acidosis in dog. J Clin Invest 89(6):2071–2074
Carcillo JA, Litten RZ, Suba EA, Roth BL (1988) Alterations in rat aortic alpha 1-adrenoceptors and alpha 1-adrenergic stimulated phosphoinositide hydrolysis in intraperitoneal sepsis. Circ Shock 26(3):331–339
El-Awady MS, Smirnov SV, Watson ML (2009) Voltage-independent calcium channels mediate lipopolysaccharide-induced hyporeactivity to endothelin-1 in the rat aorta. Am J Physiol Heart Circ Physiol 296(5):H1408–H1415
Landry DW, Levin HR, Gallant EM, Ashton RC Jr, Seo S, D'Alessandro D, Oz MC, Oliver JA (1997) Vasopressin deficiency contributes to the vasodilation of septic shock. Circulation 95(5):1122–1125
Annane D, Maxime V, Ibrahim F, Alvarez JC, Abe E, Boudou P (2006) Diagnosis of adrenal insufficiency in severe sepsis and septic shock. Am J Respir Crit Care Med 174(12):1319–1326
Hollenberg SM, Cunnion RE, Zimmerberg J (1993) Nitric oxide synthase inhibition reverses arteriolar hyporesponsiveness to catecholamines in septic rats. Am J Physiol 264(2 Pt 2):H660–H663
Tracey KJ, Beutler B, Lowry SF, Merryweather J, Wolpe S, Milsark IW, Hariri RJ, Fahey TJ 3rd, Zentella A, Albert JD (1986) Shock and tissue injury induced by recombinant human cachectin. Science 234(4775):470–474
Landry DW, Oliver JA (2001) The pathogenesis of vasodilatory shock. N Engl J Med 345(8):588–595
Chernow B, Roth BL (1986) Pharmacologic manipulation of the peripheral vasculature in shock: clinical and experimental approaches. Circ Shock 18(2):141–155
Hollenberg SM (2011) Vasoactive drugs in circulatory shock. Am J Respir Crit Care Med 183(7):847–855
Wilson DP (2011) Vascular smooth muscle structure and function. In: Fitridge RTM (ed) Mechanisms of vascular disease. University of Adelaide Press, Adelaide
Wichterman KA, Baue AE, Chaudry IH (1980) Sepsis and septic shock—a review of laboratory models and a proposal. J Surg Res 29(2):189–201
Dejager L, Pinheiro I, Dejonckheere E, Libert C (2011) Cecal ligation and puncture: the gold standard model for polymicrobial sepsis? Trends Microbiol 19(4):198–208
Bucher M, Kees F, Taeger K, Kurtz A (2003) Cytokines down-regulate alpha1-adrenergic receptor expression during endotoxemia. Crit Care Med 31(2):566–571
Fujihara H, Walker LA, Gong MC, Lemichez E, Boquet P, Somlyo AV, Somlyo AP (1997) Inhibition of RhoA translocation and calcium sensitization by in vivo ADP-ribosylation with the chimeric toxin DC3B. Mol Biol Cell 8(12):2437–2447
Sorg I, Goehring UM, Aktories K, Schmidt G (2001) Recombinant Yersinia YopT leads to uncoupling of RhoA-effector interaction. Infect Immun 69(12):7535–7543
Lucius C, Arner A, Steusloff A, Troschka M, Hofmann F, Aktories K, Pfitzer G (1998) Clostridium difficile toxin B inhibits carbachol-induced force and myosin light chain phosphorylation in guinea-pig smooth muscle: role of Rho proteins. J Physiol 506(Pt 1):83–93
Soong G, Martin FJ, Chun J, Cohen TS, Ahn DS, Prince A (2011) Staphylococcus aureus protein A mediates invasion across airway epithelial cells through activation of RhoA GTPase signaling and proteolytic activity. J Biol Chem 286(41):35891–35898
Boquet P (2001) The cytotoxic necrotizing factor 1 (CNF1) from Escherichia coli. Toxicon 39(11):1673–1680
Wang Y, Liu H, McKenzie G, Witting PK, Stasch JP, Hahn M, Changsirivathanathamrong D, Wu BJ, Ball HJ, Thomas SR, Kapoor V, Celermajer DS, Mellor AL, Keaney JF Jr, Hunt NH, Stocker R (2010) Kynurenine is an endothelium-derived relaxing factor produced during inflammation. Nat Med 16(3):279–285
Kitazawa T, Semba S, Huh YH, Kitazawa K, Eto M (2009) Nitric oxide-induced biphasic mechanism of vascular relaxation via dephosphorylation of CPI-17 and MYPT1. J Physiol 587(Pt 14):3587–3603
Surks HK, Mochizuki N, Kasai Y, Georgescu SP, Tang KM, Ito M, Lincoln TM, Mendelsohn ME (1999) Regulation of myosin phosphatase by a specific interaction with cGMP- dependent protein kinase Ialpha. Science 286(5444):1583–1587
Sakr Y, Dubois MJ, De Backer D, Creteur J, Vincent JL (2004) Persistent microcirculatory alterations are associated with organ failure and death in patients with septic shock. Crit Care Med 32(9):1825–1831
Hernandez G, Regueira T, Bruhn A, Castro R, Rovegno M, Fuentealba A, Veas E, Berrutti D, Florez J, Kattan E, Martin C, Ince C (2012) Relationship of systemic, hepatosplanchnic, and microcirculatory perfusion parameters with 6-hour lactate clearance in hyperdynamic septic shock patients: an acute, clinical-physiological, pilot study. Ann Intensive Care 2(1):44
Howell MD, Donnino M, Clardy P, Talmor D, Shapiro NI (2007) Occult hypoperfusion and mortality in patients with suspected infection. Intensive Care Med 33(11):1892–1899
De Backer D, Creteur J, Dubois MJ, Sakr Y, Koch M, Verdant C, Vincent JL (2006) The effects of dobutamine on microcirculatory alterations in patients with septic shock are independent of its systemic effects. Crit Care Med 34(2):403–408
Huang RJ, Liao CX, Chen DZ (2003) Effect of tetramethylpyrazine on endothelin, von Willebrand factor and thromboxane A2 during cardiopulmonary bypass in patients of congenital heart disease with pulmonary hypertension. Zhongguo Zhong Xi Yi Jie He Za Zhi 23(4):268–271
Segal SS, Duling BR (1986) Flow control among microvessels coordinated by intercellular conduction. Science 234(4778):868–870
Welsh DG, Segal SS (1998) Endothelial and smooth muscle cell conduction in arterioles controlling blood flow. Am J Physiol 274(1 Pt 2):H178–H186
Errasti AE, Luciani LI, Cesio CE, Tramontano J, Boveris D, Daray FM, Nowak W, Pelorosso FG, Rothlin RP (2007) Potentiation of adrenaline vasoconstrictor response by sub-threshold concentrations of U-46619 in human umbilical vein: involvement of smooth muscle prostanoid TP(alpha) receptor isoform. Eur J Pharmacol 562(3):227–235
Angus DC, van der Poll T (2013) Severe sepsis and septic shock. N Engl J Med 369(9):840–851
Artenstein AW, Higgins TL, Opal SM (2013) Sepsis and scientific revolutions. Crit Care Med 41(12):2770–2772
Essler M, Amano M, Kruse HJ, Kaibuchi K, Weber PC, Aepfelbacher M (1998) Thrombin inactivates myosin light chain phosphatase via Rho and its target Rho kinase in human endothelial cells. J Biol Chem 273(34):21867–21874
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BR participated in study design, myography and biochemical analysis, data analysis and manuscript preparation. JB participated in study design. RY developed and generated animal model and assisted in manuscript preparation. DW assisted with experimental design, myography and biochemical analysis, data analysis and manuscript preparation. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Reddi, B.A., Beltrame, J.F., Young, R.L. et al. Calcium desensitisation in late polymicrobial sepsis is associated with loss of vasopressor sensitivity in a murine model. ICMx 3, 2 (2015). https://doi.org/10.1186/s40635-014-0036-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40635-014-0036-8