
Abstract
Background
Plant microbiomes and soil are bridged by rhizobacteria, maintaining and improving plant health and growth in different aspects. This study was conducted in the field station of the Guangxi University, Fusui, China. We investigated soil nutrients, root morphology and rhizosphere bacterial composition, and community structures in 18 sugarcane genotypes concerning sugar content under the same environmental condition.
Results
Most of the rhizosphere microbiomes of these genotypes exhibited similar bacterial compositions. However, the evaluated genotypes harbored a significant effect and difference in the abundance of operational taxonomic units and bacterial composition in the rhizosphere compartments. Alpha diversity analysis on the rhizosphere microbiome showed a significant difference in the bacterial diversity (Shannon index, p < 0.001) and OTU richness (Chao1, p < 0.001). The principal coordinate analysis (PCoA) and hierarchical cluster analysis revealed that the genotype replicated samples grouped, indicating their similarity. Besides, these genotypes also differed significantly in terms of root structure and soil properties. A significant genotypic effect (p < 0.05) was found in the root traits except for rooting depth. The soil chemical properties were significantly different among the evaluated genotypes. Furthermore, sucrose content was strongly correlated with the total root length (TRL) and rooting depth. Genotypes (FN-1702, GUC-3, ZZ-13, ZZ-10, ZZ-6) were the best performing and distinct in bacterial diversity, root structure, soil parameters and sucrose content.
Conclusion
The results showed a closely related and highly conserved bacterial community of the rhizosphere microbiome. The rhizosphere microbiome diversity and related bacterial communities were highly associated with the relevant plant taxa, probably at the order level. As a result, it is possible to conclude that the host genotype and the same environmental condition influenced the rhizosphere microbiome via root phenes. Future research regarding plant phenes and microbiome functional groups could be considered an essential factor.
Graphic abstract

Similar content being viewed by others

Background
In soil, terrestrial plants possess complex interconnected microbiota communities such as the root microbiota and rhizosphere adhered to roots [1]. The rhizosphere is the soil adjacent to plant roots ranging 1–3 mm providing habitat for root–microbe interaction that included up to 1011 microbial cells per gram root and more than 30,000 prokaryotic species [2, 3]. Microbial diversity present in rhizosphere contains different types of microorganisms like bacteria, fungi, viruses, and algae exhibiting their interaction with the host plants by influencing the roots and vice versa the plant root secretions effect the microbial activity [3]. The rhizosphere microbiome is presumed to be a bridge between plant and soil microbiomes that benefits the host plant's health and soil fertility [4,5,6].
In sugarcane industry the higher sucrose content incurring is the utmost priority as it provides 70% of the world’s sugar. The photoassimilates are stored in the stalk of sugarcane in the form of sucrose [7]. However, the sucrose accumulation in stalk is affected by factors such as invertase activity, source–sink interaction and more importantly, the genes governing the sucrose synthesis mechanism. Among these factors, the source sink is an integral part of the sucrose accumulation and its activity is decreased as the stalk gains maturity [8]. The second important factor in sucrose accumulation is the quantitative nature of the trait controlled by various genes [9]. Although the genes associated with sugar storage are sucrose metabolism enzymes, sucrose transporters and source–sink, while their genetic mechanism for sucrose accumulation is yet need to explore [7].
Plant growth’s sustainability is dependent on soil health because plants rely on the optimum acquisition of at least 14 essential elements from soil to ample growth [10, 11]. Plant performance can also be affected by the plant genotypes and might change the soil properties. The genetic variation in genotypes results in varying secondary metabolites, which alter the microbial interactions [12]. Microorganisms in the soil represent organic matter and are greatly linked to the rhizosphere’s roots. The plants disposed-off their secondary metabolites in the form of rhizodeposits into the rhizosphere providing an efficient environment for changing nutrient availability, transport and uptake by the plants. Thus, the rhizosphere is an environment formed by the interaction of root exudates and microbes [13]. Biochemical-mediated interactions between plant roots and microbes are now considered a basic phenomenon in the rhizosphere [14] where the plant rhizosphere along with numerous factors affect the microbial communities, such as soil types, root exudates, root phenotypes [15], plant genotypes, plant nutritional status [6], climate factors [16], and plant species [17]. The biotic factors could also influence the rhizosphere microbial diversity such as insect–root phenotypes [18], plant developmental stages [19, 20], and diseases [1, 21]. However, the feedback mechanism investigated through rhizosphere microbiota provides phytohormones synthesis and the bioavailable mineral nutrients to regulate plant growth and disease resistance [1, 2].
The effects of these factors on the composition of rhizosphere and endosphere microbiome communities are drastically different. Therefore, plant species, plant developmental stages and genotypes are predominantly determine the endosphere’s bacterial communities [22]. Comparison of biomass and microorganism activity in the rhizosphere in bulk soil and endosphere exposed an increment in rhizosphere activity owning to the exudation of chemicals by roots [23]. Earlier study on the rhizosphere revealed the existence of species-specific microbial communities and microbiome analyses included the associated host plant's biology, functioning with the microbial ecology, and presuming microorganisms as the sources of additional genes and functions [1].
Research on plant species and cultivars unravel the influential role of plant types on the microbiome communities composition in the rhizosphere, such as the maize [24], rice [25], potato [26], soybean [27], Arabidopsis [28] and intra‐species comparison [20]; whereas, for inter-species interaction the rhizospheres of different genotypes can also have a distinct microbial community. However, in most cases different genotypes of the same crop can only have a minor impact on microbiome assembly [20, 27]. Different genotypes of the same plant species could also affect the bacterial community structure and composition of rhizosphere [26]. The host genotype can cause up to 5.7% variation in the rhizosphere microbiome composition [27]. However, the host genotypes related to their rhizosphere communities and regulatory mechanisms are not entirely understood. Their composition, variation and assembly should be further explored to understand the rhizosphere microbiome activities completely.
Soil serves as a sole reservoir of bacterial species [4, 21, 29], making it evident that both plant species and soil type highly affect the microbial assembly in the rhizosphere [26, 28]. Rhizobacteria boosts the plant health by protecting from phytopathogens, providing significant nutrients and producing phytohormones to nurture plant growth [30]. Actinobacteria and Proteobacteria are the crucial bacterial groups in the rhizosphere [31], belonging to genus Pseudomonas [32].
Understanding the phenotypic differences in root phenes and their effect on microbiome assemblies in the rhizosphere could provide a basis for constructing a healthy plant rhizosphere microbiome and could be beneficial for plant breeding, improving soil management strategies, and introducing universal biological control agents pathogens and pests to develop more sustainable agricultural practices. Therefore, this study was conducted to investigate the effect of 18 different sugarcane genotypes on the soil-specific parameters, root structure and microbial community in the same environmental conditions.
Materials and methods
Genotypes, experimental design, and sample collection
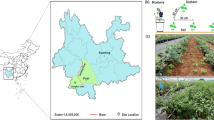
The experiment was carried out in a randomized complete block design (RCBD) with three replicates for each genotype in the field station of the Guangxi University in Fusui, China (22°38′06″N,107°54′15″E), using 18 different sugarcane genotypes including (1) GUC-23, (2) ZZ-1, (3) ZZ-9, (4) GUC-15, (5) GUC-37, (6) ZZ-6, (7) ZZ-5, (8) ZZ-10, (9) TD-11, (10) GUC-3, (11) GUC-8, (12) ZZ-2, (13) ZZ-13, (14) ROC-22, (15) TD-7, (16) ZZ-11, (17) FN-1702, (18) GUC-35, during 2019. These genotypes were cultivated on March 12, 2019 and harvested on January 2, 2020. The average annual temperature and rainfall of the area were 21.3℃ and 1050–1300 mm, respectively. Each row was 30 m long, with a line-to-line distance of 2 m and a plant-to-plant distance of 30 cm. The agronomic practices were kept regular and uniform for all the replications. The soil and roots samples from 18 sugarcane genotypes were collected in the maturation and ripening stage in December 2019.
These genotypes were harvested separately and the roots were shaken gently to remove the large soil particles. The soil particles adhered to the roots were collected carefully with a sterile filter paper strip and used as the rhizosphere soil [28, 33]. The soil samples were divided into two parts: air-dried for nutrient determination in soil, and the second soil sample was kept at − 80 ℃ for < 24 h before DNA extraction.
Measurement of sucrose content
In the late vegetative phase (maturation and ripening), the sucrose content was measured with a digital refractometer ATAGO Pocket PAL-1 (Atago Co. Ltd., Tokyo, Japan) using ten plants per replicate average value was calculated.
Soil chemical analysis and root structure
Soil samples were sieved through a 2-mm mesh and SOC (soil organic carbon), TN (total nitrogen), TP (total phosphorus), and TK (total potassium) were calculated. In detail, the quantity of soil organic carbon (SOC) was measured by the oxidation method with K2Cr2O7.H2SO4 [34]. The total phosphorous and total potassium were measured according to Sommers and Basta [35, 36], respectively. The Kjeldahl method was used to determine the total nitrogen content [37]. Roots were washed with tap water to remove soil cores using a 1-mm sieve mesh to minimize root loss. Root morphology was calculated by scanning root samples with an STD4800 WinRhizo Scanner (Regent Instruments Inc., Canada).
DNA preparation, PCR amplification and high-throughput sequencing
According to the manufacturer’s instructions, the total genomic DNA was extracted from 0.5 g of each soil sample using FastDNA™ Spin Kit for Soil (Co. MP Biomedicals, USA). The extracted metagenomic DNA concentrations were measured using NanoDrop 2000 (Thermo Fisher Scientific, Wilmington, USA). The DNA samples were used as a template for bacterial 16S rRNA gene amplification. The 799F (forward primer, 5-AACMGGATTAGATACCCKG-3) and 1193R (reverse primer 5-ACGTCATCCCCACCTTCC-3) primers set were used for amplification of bacterial V5–V6 region of 16S rRNA [38]. Polymerase chain reaction (PCR) was carried out in a total volume of 50 µL containing 25 µL of 2 × Premix Taq (Takara Biotechnology, Dalian Co., Ltd., China), 1 µL of each primer (10 mM), 3 µL of DNA (20 ng/µL) template and then sterilized deionized water was added to make 50 µl volume. The reaction conditions were: initial denaturation for 5 min at 94 °C; followed by 30 cycles of denaturation at 94 °C for 30 s, annealing at 52 °C for 30 s, extension at 72 °C for 30 s; and a final elongation at 72 °C for 10 min. The resulting PCR products were subjected to sequencing by Magigene Technology (Shanghai, China) using the Illumina Hiseq platform.
Processing of Illumina sequencing data
The paired reads were spliced using FLASH (version 1.2.3) software to merge the sequences before assembling a gene segment [39]. Chimeric sequences were identified and removed with a de novo method using USEARCH (version 8.1.1861) [40]. After the removal of the chimera, high-quality bacterial sequences were collected for subsequent analysis.
Effective bacterial sequences were separately subsampled for each sample for the subsequent statistical analysis. After subsampling, the data were processed using a modified SOP pipeline based on USEARCH and the software package QIIME (Quantitative Insights Into Microbial Ecology v1.8.0) [33]. Briefly, the selected sequences were clustered to operational taxonomic units (OTU) using a two-stage clustering algorithm with USEARCH (version 8.1.1861) at 97% sequence identity [40]. Representative sequences in each OTU were aligned to the SILVA reference alignment [41]. Taxonomy was assigned to each representative sequence using RDP with a minimum confidence of 85%.
Alpha and beta diversity analysis
An OTU-based analysis method was used to evaluate the bacterial diversities in each sample from each plant (alpha diversity). To estimate the diversity index and species richness (alpha diversity) among the genotypes for each sample, OTU richness, Chao1, Simpson and Shannon indices were calculated using QIIME software (v1.8.0), concerning a sequencing depth of 3%. Statistical analysis was performed using ANOVA with p values to determine the significant differences in the diversity indices or species richness among the plant rhizosphere soil samples. The rarefaction curve and rank abundance curves were calculated at a 97% level of similarity of the OTUs.
Beta diversity analysis was used among all the samples for the similarity index determination of the community structure. At the OTU-level of genotypes, beta diversity was calculated using weighted UniFrac distances and was visualized through PCoA (Principal co-ordinate analysis). The weighted UniFrac distance matrices were clustered and evaluated by QIIME software (v1.8.0) and showed phylogenetic relationships among various communities and their abundance in the respective samples.
Statistical analysis
Average values from each replicate were used to test the significance and sugarcane genotype effect for all the parameters separately using Graphpad Prism 7 software through analysis of variance (ANOVA). To study and visualize correlation among the variables, principal component analysis (PCA) for the effect of sugarcane genotypes on soil nutrients, root parameters and sucrose content were carried out using PAST4.03 software. Correlation analysis between the soil, root parameters and sucrose content were conducted through PAST 4.03 software.
Results
Measurement of root structure and sucrose content
The genotypes evaluation was mainly focused on the underground parameters (soil chemical properties and roots morphological traits) and the rhizospheric bacterial diversity. The root parameters, such as TRL (total root length), TRV (total root volume), TARD (total average root diameter) and root depth are recorded as shown in Table 1. These genotypes also showed a significant variation (p < 0.05) in the underground traits except rooting depth (Table 1). Genotype FN-1702 resulted in the highest total root length (249.21 m), total root volume of 85.17 cm3, (p < 0.05) and rooting depth (3.13 m), while genotype TD-11 showed the lowest total root length (128.25 m) and genotype ZZ-1 showed the lowest root volume (28.62 cm3) and rooting depth (2.45 m) when compared to the control genotype ROC22. Total average root diameter also varied significantly among these genotypes. Furthermore, the highest (1.42 mm) and lowest (0.95 mm) root diameter were observed in genotype GUC-15 and GUC-8, respectively.
The sucrose content was also measured to confirm the differences among these genotypes. Sucrose content was significantly (p < 0.05) different in these genotypes, such as high sucrose content (22.29Bx) was found in ZZ-6, while the lowest sucrose content (15.19Bx) was found in ZZ-5 (Table 1). Moreover, a PCA plot was drawn to visualize the relation between roots and sugar content (Fig. 1B). The plot indicated that the sugar content positively correlated with TRL and depth, while TRV and TARD do not affect sugar content. The plot also indicated that GUC-3, ZZ-6, ZZ-13, ZZ-10 and FN-1702 lie closer to sugar content, TRL and depth. PCA 1 explained 57.8% variation, while PCA 2 explained 19.1% variation among the genotypes, root parameters and sugar content (Fig. 1B).

A Principal component analysis of soil parameters with 18 different sugarcane genotypes. TK total potassium, SOC Soil organic carbon, TN total nitrogen, TP total phosphorous. Each genotype and biplot is represented by the average PCA scores obtained by running PCA on three replicates. B Principal component analysis of root parameters and sugar content with 18 different sugarcane genotypes
Effect of genotype on soil parameters
The soil-related parameters, such as SOC, TP, TN and TK were measured and the results were shown in Table 2. The concentrations of these parameters varied between genotypes. No single genotype had a dominant effect on soil parameters. For example, SOC concentration ranged from 19.8 g/kg to 48.7 g/kg with minimum and maximum concentrations having genotype ZZ-2 and ZZ-10, respectively (Table 1). Similarly, the highest TN concentration was detected in genotype GUC-37 (2.51 g/kg), and the lowest concentration was noted in ZZ-10 (1.09 g/kg). TP ranged from 0.03 g/kg to 1.97 g/kg in soil samples, with the lowest content in FN-1702 and the highest in GUC-37 (Table 2). Values for TK ranged from 16.35 g/kg to 20.31 g/kg, where the lowest TK values were observed in the genotype ZZ-9, while the highest was observed in genotype TD-11.
The PCA plot indicated a significant effect of genotypes (p < 0.05) on the soil parameters (Fig. 1). Most sugarcane genotypes were clustered in the middle of the plot, whereas genotypes ZZ-2 and GUC-37 were clustered separately on the PC2 axis. Total potassium (TK) and SOC were presented on the PC1 axis, while total nitrogen (TN) and total phosphorus (TP) were presented on the PC2 axis. Moreover, 55.77% and 24.13% variations among the genotypes and soil parameters were explained by PC1 and PC2, respectively.
Correlation among soil parameters, root structure and sucrose content
Sucrose content was highly correlated with the root traits and soil chemical parameters (Table 2). Sucrose content was found to have a strong positive correlation with TRL (0.63), rooting depth (0.52), and TK (0.30). However, the correlation between sucrose content, TRV and TARD was non-significant (0.36, 0.29). All other soil chemical parameters were also non-significant. Total root volume (TRV) was strongly positively correlated with TARD (0.60) and rooting depth (0.70) and negatively correlated with TP (− 0.50). On the other hand, a strong positive correlation (0.93) was found between SOC and TN, while a non-significant correlation was found with all the other parameters. TN was positively correlated with TP (0.42).
Composition and community structure of rhizosphere microbiomes associated with eighteen sugarcane genotypes
After screening, pre-clustering, and chimera removal, a total of 3,333,835 reads of high-quality bacterial 16S rRNA from the V5-V6 region were obtained, with an average of 61,737.7 ± 4091.1 read per sample (min = 54,362, max = 71,714). The number of reads per sample was rarefied to the smallest number of reads. In this case, 50,994 effective bacterial sequences were randomly extracted for subsequent statistical analysis (Additional file 1: Table S1). Operational taxonomic units (OTUs) of each sample were filtered at a minimum count of 2 and 10% prevalence in the samples. After filtering, a total of 3573 OTUs were obtained, in which all the genotypes shared 1287 OTUs (Fig. 2A).

OTU classification and relative abundance. A Venn diagram representing the total number of OTUs in each genotype and shared number of OTU. B Each bar’s relative abundance of major bacterial phyla represents three replicates’ average value in each sample
Proteobacteria (45% \(\pm\) 4.56%) dominated the sugarcane rhizosphere community, followed by Actinobacteria (23% \(\pm\) 3.46%), Acidobacteria (14% \(\pm\) 4.31%), Chloroflexi (8% \(\pm\) 2.15%), and Firmicutes (2% \(\pm\) 1.21%) (Fig. 2B).
Three classes of Proteobacteria, including Alpha-proteobacteria, Delta-proteobacteria and Gamma-proteobacteria, were observed in the rhizosphere microbiome. Enterobacteriales (4.5% \(\pm\) 0.9%), Xanthomonadales (2.8% \(\pm\) 1.3%) and Betaproteobacteriales (1.8% \(\pm\) 0.9%) of Gamma-proteobacteria (30% \(\pm\) 6.4%) and Rhizobiales (3.9% \(\pm\) 0.8%), Micropepsales (1.5% \(\pm\) 0.7%) and Acetobacterales (1.2% \(\pm\) 0.4%) of Alpha-proteobacteria (14% \(\pm\) 2.2%) and Myxococcales (6.2% \(\pm\) 1.3%), and RCP2-54 (2.1% \(\pm\) 1.3%) of Delta-proteobacteria were highly abundant in Proteobacteria (Fig. 3C, E).

Relative abundance and distribution of taxa. A The relative abundance of major orders from phyla of Acidobacteria. B The relative abundance of major orders from phyla of Actinobacteria. C–E The relative abundance of major bacterial orders from three phyla Proteobacteria classes, i.e., Alpha-proteobacteria, Delta-proteobacteria and Gamma-proteobacteria
For instance, Proteobacteria was the most prominent bacterial group of the rhizosphere microbiome, dominant in the GUC-8 (5.5% \(\pm\) 3.70%) (Fig. 3B). In detail, Rhizobiales (5.7% \(\pm\) 2.08%) of Alpha-proteobacteria were highly present in ZZ-13, Micropepsales (3.7% \(\pm\) 6.65) in GUC-8, Elsterales (2.2% \(\pm\) 0.5%) in GUC-35 (Fig. 3C), while in Delta-proteobacteria, a higher proportion of Myxococcales (7.8% \(\pm\) 3%), RCP2-54 (4.7% \(\pm\) 5.13%) were observed in GUC-37 and GUC-35, respectively (Fig. 3D).
In Gamma-proteobacteria, Enterobacteriales (6.8% \(\pm\) 0.15%) were highly present in ZZ-9, (Fig. 3E). Similar results were also observed in other bacterial groups in the bacterial phyla of Acidobacteria and Actinobacteria (Fig. 3A, B). Frankiales (4.1% \(\pm\) 1.5%) were abundantly present in GUC-35, while Micrococcales (1.9% \(\pm\) 0.5%) were present in ZZ-6. Acidobacteria, Acidobacteriales (6.6% \(\pm\) 0%) and Solibacterales (4.7% \(\pm\) 0.45) were the highly represented orders in ROC-22 and GUC-37, respectively, suggesting that the bacterial composition highly varied at order level in the studied genotypes. The community composition data for each taxonomic level were clustered according to the abundance distribution or the degree of similarity. According to the clustering results, the taxa unit and the genotypes were ranked separately and presented through heat maps. Color gradients were used to distinguish the taxa’s high and low abundance and reflect the community composition’s similarity between the genotypes. The clustering analysis was performed on the top 30 differentially abundant OTUs and was visualized through a heat map (Fig. 4A; Additional file 1: Table S2).

Microbial community composition of top 30 abundant OTUs and their abundance level with cluster analysis. The dendrogram linkages and OTUs’ distances are not phylogenetic but based upon the reads number. The legend and scale represent the abundance of OTU’s (Y-axis) within each sample (X-axis). A The red indicates a more abundant genera and blue represents less abundant genera in the corresponding samples. B Rank abundance curve depicting the abundance of species. The X-axis represents the number of OTUs in each sample and their richness in the community. C Shannon index graph indicating bacterial diversity among the sugarcane genotypes. D Chao1 index graph indicating OTU richness among the sugarcane genotypes
Bacterial diversity and species richness of rhizosphere microbiomes in 18 sugarcane genotypes
The rarefaction curve reflects that all the samples showed a higher sequencing depth and greater diversity (Additional file 1: Fig. S1). The Rank abundance curve was drawn to check the relative abundance of species using R software. The abundance curve visually depicted both species’ richness and evenness in the 18 genotypes. The genotypes TD-11, ROC-22, GUC-35, ZZ-9 and GUC-8 exhibited higher species richness and evenness, while ZZ-13, ZZ-10, GUC-37, and ZZ-6 showed more significant differences in the abundances between OTUs and low homogeneity (Fig. 4B). From the diversity analysis, generally, the plants grown in the same environmental condition significantly affected the diversity and abundance of bacteria species in the rhizosphere. These indices were calculated separately for each sample. Significant differential OTU richness estimated by Chao1 (p < 0.05) and bacterial diversity estimated by the Shannon index (p < 0.05) were observed among the rhizosphere microbiome of 18 sugarcane genotypes (Fig. 4C). Among these, rhizobacteria of the genotype FN-1702 showed higher bacterial diversity (Shannon index: 6.02 ± 0.08) and OTU richness (Chao1: 3334.26 ± 21.37), followed by GUC 3 (Shannon index: 6.31 ± 0.03) and OTU richness (Chao1: 3279.33 ± 111), ZZ 13 (Shannon index: 6.31 ± 0.04) and OTU richness (Chao1: 3205.31 ± 76.08), ZZ 10 (Shannon index: 6.11 ± 0.22) and OTU richness (Chao1: 3279.23 ± 111.13) and ZZ 6 (Shannon index: 6.20 ± 0.13) and OTU richness (Chao1: 3181.93 ± 71.91), while TD-11 and ZZ-9 with smaller Shannon index and OTU richness indicated lower bacterial diversity among these genotypes.
The similarity of the rhizosphere microbiomes in the 18 genotypes of sugar cane
Beta diversity in the microbial structure and composition was assessed by PCoA, which revealed that the three replicates from the same genotype demonstrated a clear tendency to the group except for ZZ-13, which tended to differ from other genotypes in terms of the rhizosphere microbial community. Moreover, 31.5% of the total variation in the rhizosphere microbial population was explained by PCoA 1, while 16.9% of the total variation was explained by PCoA 2 (Fig. 5B). Similar results were also observed in the Unifrac-based hierarchical cluster analysis (Fig. 5A). The results showed that all the samples were significantly clustered into different groups based on their taxonomic divergence, although not for each genotype. In Fig. 5A, the samples were significantly clustered into two different main groups. The replicated samples from ZZ-13, GUC-3 FN-1702, ZZ-10 and ZZ-6 clustered into one group, while ZZ-3, ZZ-1, ZZ-11, TD-11, ZZ-9, ROC-22, GUC-35, GUC-23, ZZ-5, GUC-15, GUC-37, GUC-8 and TD-7 were clustered in the other groups.

Analysis of beta diversity for estimating similarity or dissimilarity between the genotypes. A Weighted unifrac distances matrix, clustering is performed based on similarity. The samples are more similar to each other in terms of branching distance, i.e., smaller branch distance indicates similarity among the two samples. B PCoA (principal coordinate analysis) of weighted unifrac distance
Discussion
The rhizosphere microbiome has been evolved rapidly since last few decades and become a topic of interest for researchers around the globe. Microbial interactions between soil and roots are crucial for improving plant performance [5]. Phytohormones released by plants roots fascinate the nearby soil microbes. However, variations do exist in the microbial community composition among different plant species or even within genotypes [42]. The influential involvement of microbial communities and engrossed mechanism contributing to the plant improvement is still not well understood due to limited number of available studies on the effect of the plant hosts microbiome at the interspecies and intraspecies level [5].
In this study, rhizosphere bacterial community along with their composition in addition to root morphology and soil nutrients of 18 randomized and replicated modern sugarcane genotypes was evaluated. The results of this study presented that different sugarcane genotypes had substantial variations in root parameters as the long root genotypes had higher bacterial diversity and significantly high sucrose content except for genotype ZZ-13. Different genotypes were characterized based on root architecture and soil properties, for instance, the genotype by environment (G × E) interactions could attribute variations observed in different genotypes [10, 43]. Our study design assessed the effect of sugarcane genotypes with in same environmental conditions and agronomic practices. The rhizosphere microbiome differed in alpha and beta diversity among genotypes; although the effect was small, the results interpreted the significant influence of genotypes on the rhizosphere microbiome.
Considering the fact that the host genotype might influence rhizosphere microbiota, our work also added a general view though demonstrating that the rhizosphere microbiota was enriched in replicated samples of the host genotypes even in the absence of unique agronomic treatments. Further, our study also highlighted the importance of interspecies effect on the rhizosphere microbiota diversity. In this study, sampling was done at the maturation stage, indicating that the root exudates were either decreased or stopped. In this case, the rhizosphere microorganisms must acquire their nutrients from soil [44] and microbial communities are expected to closely associate because of the same agronomic practices and, the same type and amount of fertilizers (N, P, K). However, different microbial communities and compositions suggested that the sugarcane host genotype was the primary factor in shaping bacterial communities. Most probably the phenotypic differences observed among the root morphology in this study. While those sugarcane genotypes having extensive in-depth roots structure could be associated with the variable and distinct bacterial community. The available carbon increases photosynthetic activity, which causes root elongation and branching so, the root growth and movement directly dependent on the availability of carbon concentration [45]. Genotypes with higher sucrose contents availability enhances the root exudates which cover more area to interact with rhizosphere microbes [46, 47]. Plants might release their photosynthates into the soil up to 20%, thereby increasing the plant–microorganism interaction and improving plant growth through mineral availability [48]. The carbon cycling enzymatic activities depends on the disposed concentrations of inorganic nitrogen N and vice versa, suggesting an increment in total nitrogen and total carbon contents in the root area might be related to the soil enzyme activities [49].
Moreover, we also analyzed the rhizosphere microbial composition in different sugarcane genotypes and is demonstrated that most of the genotype’s rhizosphere microbiome contained Proteobacteria, Actinobacteria, and Acidobacteria, which were consistent in the rhizosphere of tomato [28], maize [24], soybean [27] and rice [25]. Our research findings were also in line with Bruto and Vurukonda [48, 49], who also reported several PGPR that mainly comprised phylum Proteobacteria, that indorsed the acquisition of nutritional elements (N, P, and Fe) to accomplish the plant growth activities [50, 51]. This study also found the enriched order of Rhizobiales and the genus of Burkholderia and Bradyrhizobium, the members of well-known plant growth-promoting bacteria [44]. Thus, proteobacteria members could effectively use the carbon source from metabolites generated by the primary assimilators in the sugarcane rhizosphere [48, 52].
The studied genotypes revealed various bacterial orders, including an abundance of Sphingomonadales in ZZ-6, Rhizobiales and Myxococcales in ZZ-13, frankiales in ZZ-2, Enterobacteriales in ZZ-9, Xanthomonadales in GUC-37, Acidobacteriales and Solibacteriales in ZZ-11 and GUC-37, respectively. Therefore, supporting the hypothesis that the more phylogenetically distant plant hosts, the more variability in their bacterial communities [53, 54]. Our work provided sufficient evidence for the variation in bacterial communities due to host sugarcane genotype under natural environmental conditions. However, the question that remains to be addressed is: what specific alleles of sugarcane are involved in altering the microbial community?
Conclusions
Experimental evidence signified the intricate association between plant roots and soil microbes because plants with distinct root architecture have distinct microbial communities. Understanding these microbes’ activity, such as their role in plant growth and resistance to various abiotic factors, is crucial for future research and the development of relevant bioproducts. Exploring the exact rhizosphere microbiome colonization mechanisms was still complicated. According to our findings, genotype and the same environmental condition influenced the rhizosphere microbiome and subsequently the plant phenes. Future research regarding plant root phenes and microbiome functional microbial groups should also be addressed instead of taxonomy. However, an additional large-scale approach would be needed to study further information and serve sustainable agricultural practices.
Availability of data and materials
Additional data are available on request from the corresponding author.
Abbreviations
- OTU:
-
Operational taxonomic unit
- SOC:
-
Soil organic carbon
- TRL:
-
Total root length
- TRV:
-
Total root volume
- TARD:
-
Total average root diameter
- TN:
-
Total nitrogen
- TP:
-
Total phosphorous
- TK:
-
Total potassium
References
Qu Q, Zhang Z, Peijnenburg WJ, Liu W, Lu T, Hu B, Chen J, Chen J, Lin Z, Qian H. Rhizosphere microbiome assembly and its impact on plant growth. J Agric Food Chem. 2020;68(18):5024–38. https://doi.org/10.1021/acs.jafc.0c00073.
Mendes R, Kruijt M, De Bruijn I, Dekkers E, van der Voort M, Schneider JH, Piceno YM, DeSantis TZ, Andersen GL, Bakker PA, Raaijmakers JM. Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science. 2011;332(6033):1097–100. https://doi.org/10.1126/science.1203980.
Philippot L, Raaijmakers JM, Lemanceau P, Van Der Putten WH. Going back to the roots: the microbial ecology of the rhizosphere. Nat Rev Microbiol. 2013;11(11):789–99. https://doi.org/10.1038/nrmicro3109.
Bulgarelli D, Schlaeppi K, Spaepen S, Van Themaat EV, Schulze-Lefert P. Structure and functions of the bacterial microbiota of plants. Annu Rev Plant Biol. 2013;64:807–38. https://doi.org/10.1146/annurev-arplant-050312-120106.
Lei S, Xu X, Cheng Z, Xiong J, Ma R, Zhang L, Yang X, Zhu Y, Zhang B, Tian B. Analysis of the community composition and bacterial diversity of the rhizosphere microbiome across different plant taxa. MicrobiologyOpen. 2019;8(6): e00762. https://doi.org/10.1002/mbo3.762.
Turner TR, James EK, Poole PS. The plant microbiome. Genome Biol. 2013;14(6):1–10. https://doi.org/10.1186/gb-2013-14-6-209.
Wang J, Zhao T, Yang B, Zhang S. Sucrose metabolism and regulation in sugarcane. J Plant Physiol Pathol. 2017;5:2. https://doi.org/10.4172/2329-955X.1000167.
McCormick AJ, Watt DA, Cramer MD. Supply and demand: sink regulation of sugar accumulation in sugarcane. J Exp Bot. 2009;60(2):357–64. https://doi.org/10.1093/jxb/ern310.
Papini-Terzi FS, Rocha FR, Vêncio RZ, Felix JM, Branco DS, Waclawovsky AJ, Del Bem LE, Lembke CG, Costa MD, Nishiyama MY, Vicentini R. Sugarcane genes associated with sucrose content. BMC Genom. 2009;10(1):1–21. https://doi.org/10.1186/1471-2164-10-120.
Corneo PE, Suenaga H, Kertesz MA, Dijkstra FA. Effect of twenty four wheat genotypes on soil biochemical and microbial properties. Plant Soil. 2016;404(1):141–55. https://doi.org/10.1007/s11104-016-2833-1.
White PJ, Brown PH. Plant nutrition for sustainable development and global health. Ann Bot. 2010;105(7):1073–80. https://doi.org/10.1093/aob/mcq085.
Mueller LO, Borstein SR, Tague ED, Dearth SP, Castro HF, Campagna SR, Bailey JK, Schweitzer JA. Populations of Populus angustifolia have evolved distinct metabolic profiles that influence their surrounding soil. Plant Soil. 2020;8:1–3. https://doi.org/10.1007/s11104-019-04405-2.
Lagomarsino A, Moscatelli MC, Di Tizio A, Mancinelli R, Grego S, Marinari S. Soil biochemical indicators as a tool to assess the short-term impact of agricultural management on changes in organic C in a Mediterranean environment. Ecol Indic. 2009;9(3):518–27. https://doi.org/10.1016/j.ecolind.2008.07.003.
Schmidt R, Ulanova D, Wick LY, Bode HB, Garbeva P. Microbe-driven chemical ecology:past, present and future. ISME J. 2019;13(11):2656–63. https://doi.org/10.1038/s41396-019-0469-x.
Massalha H, Korenblum E, Malitsky S, Shapiro OH, Aharoni A. Live imaging of root–bacteria interactions in a microfluidics setup. Proc Natl Acad Sci. 2017;114(17):4549–54. https://doi.org/10.1073/pnas.1618584114.
Jin H, Yang X, Liu R, Yan Z, Li X, Li X, Su A, Zhao Y, Qin B. Bacterial community structure associated with the rhizosphere soils and roots of Stellera chamaejasme L. along a Tibetan elevation gradient. Ann Microbiol. 2018;68(5):273–86. https://doi.org/10.1007/s13213-018-1336-0.
Lima AB, Cannavan FS, Navarrete AA, Teixeira WG, Kuramae EE, Tsai SM. Amazonian dark earth and plant species from the Amazon region contribute to shaping rhizosphere bacterial communities. Microb Ecol. 2015;69(4):855–66. https://doi.org/10.1007/s00248-014-0472-8.
Cotton TA, Pétriacq P, Cameron DD, Al Meselmani M, Schwarzenbacher R, Rolfe SA, Ton J. Metabolic regulation of the maize rhizobiome by benzoxazinoids. ISME J. 2019;13(7):1647–58. https://doi.org/10.1038/s41396-019-0375-2.
Schlemper TR, Leite MF, Lucheta AR, Shimels M, Bouwmeester HJ, van Veen JA, Kuramae EE. Rhizobacterial community structure differences among sorghum cultivars in different growth stages and soils. FEMS Microbiol Ecol. 2017;93:(8). https://doi.org/10.1093/femsec/fix096.
Zhang J, Zhang N, Liu YX, Zhang X, Hu B, Qin Y, Xu H, Wang H, Guo X, Qian J, Wang W. Root microbiota shift in rice correlates with resident time in the field and developmental stage. Sci China Life Sci. 2018;61(6):613–21. https://doi.org/10.1007/s11427-018-9284-4.
Berendsen RL, Vismans G, Yu K, Song Y, de Jonge R, Burgman WP, Burmølle M, Herschend J, Bakker PA, Pieterse CM. Disease-induced assemblage of a plant-beneficial bacterial consortium. ISME J. 2018;12(6):146–507. https://doi.org/10.1038/s41396-018-0093-1.
Fitzpatrick CR, Copeland J, Wang PW, Guttman DS, Kotanen PM, Johnson MT. Assembly and ecological function of the root microbiome across angiosperm plant species. Proc Natl Acad Sci. 2018;115(6):1157–65. https://doi.org/10.1073/pnas.1717617115.
Stringlis IA, Yu K, Feussner K, De Jonge R, Van Bentum S, Van Verk MC, Berendsen RL, Bakker PA, Feussner I, Pieterse CM. MYB72-dependent coumarin exudation shapes root microbiome assembly to promote plant health. Proc Natl Acad Sci. 2018;115(22):5213–22. https://doi.org/10.1073/pnas.1722335115.
Peiffer JA, Spor A, Koren O, Jin Z, Tringe SG, Dangl JL, Buckler ES, Ley RE. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc Natl Acad Sci. 2013;110(16):6458–553. https://doi.org/10.1073/pnas.1302837110.
Edwards J, Johnson C, Santos-Medellín C, Lurie E, Podishetty NK, Bhatnagar S, Eisen JA, Sundaresan V. Structure, variation and assembly of the root-associated microbiomes of rice. Proc Natl Acad Sci. 2015;112(8):911–20. https://doi.org/10.1073/pnas.1414592112.
Inceoglu O, Al-Soud WA, Salles JF, Semenov AV, van Elsas JD. Comparative analysis of bacterial communities in a potato field as determined by pyrosequencing. PLoS ONE. 2011;6(8):8:e23321. https://doi.org/10.1371/journal.pone.0023321.
Mendes LW, Kuramae EE, Navarrete AA, Van Veen JA, Tsai SM. Taxonomical and functional microbial community selection in soybean rhizosphere. ISME J. 2014;8(8):1577–87. https://doi.org/10.1038/ismej.2014.17.
Bulgarelli D, Rott M, Schlaeppi K, van Themaat EV, Ahmadinejad N, Assenza F, Rauf P, Huettel B, Reinhardt R, Schmelzer E, Peplies J. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature. 2012;488(7409):91–5. https://doi.org/10.1038/nature11336.
Dombrowski N, Schlaeppi K, Agler MT, Hacquard S, Kemen E, Garrido-Oter R, Wunder J, Coupland G, Schulze-Lefert P. Root microbiota dynamics of perennial Arabis alpina are dependent on soil residence time but independent of flowering time. ISME J. 2017;11(1):43–55. https://doi.org/10.1038/ismej.2016.109.
Pii Y, Mimmo T, Tomasi N, Terzano R, Cesco S, Crecchio C. Microbial interactions in the rhizosphere: beneficial influences of plant growth-promoting rhizobacteria on the nutrient acquisition process. A review. Biol Fertil Soils. 2015;51(4):403–15. https://doi.org/10.1007/s00374-015-0996-1.
Teixeira LC, Peixoto RS, Cury JC, Sul WJ, Pellizari VH, Tiedje J, Rosado AS. Bacterial diversity in rhizosphere soil from Antarctic vascular plants of Admiralty Bay, maritime Antarctica. ISME J. 2010;4(8):989–1001. https://doi.org/10.1002/9781118297674.ch105.
Yang Y, Wang N, Guo X, Zhang Y, Ye B. Comparative analysis of bacterial community structure in the rhizosphere of maize by high-throughput pyrosequencing. PLoS ONE. 2017;12(5): e0178425. https://doi.org/10.1371/journal.pone.0178425.
Tian BY, Cao Y, Zhang KQ. Metagenomic insights into communities, functions of endophytes and their associates with infection by root-knot nematode, Meloidogyne incognita, in tomato roots. Sci Rep. 2015;5(1):1–5. https://doi.org/10.1038/srep17087.
Jiang F, Wu X, Xiang W, Fang X, Zeng Y, Ouyang S, Lei P, Deng X, Peng C. Spatial variations in soil organic carbon, nitrogen and phosphorus concentrations related to stand characteristics in subtropical areas. Plant Soil. 2017;413(1–2):289–301. https://doi.org/10.1007/s11104-016-3101-0.
Sommers LE, Nelson DW. Determination of total phosphorus in soils: a rapid perchloric acid digestion procedure. Soil Sci Soc Am J. 1972;36(6):902–4. https://doi.org/10.2136/sssaj1972.03615995003600060020x.
Basta NT, Tabatabai MA. Determination of total potassium, sodium, calcium and magnesium in plant materials by ion chromatography. Soil Sci Soc Am J. 1985;49(1):76–81. https://doi.org/10.2136/sssaj1985.03615995004900010015x.
Bremner JM, Tabatabai MA. Use of an ammonia electrode for determination of ammonium in Kjeldahl analysis of soils. Commun Soil Sci Plant Anal. 1972;3(2):159–65. https://doi.org/10.1080/00103627209366361.
Seybold H, Demetrowitsch TJ, Hassani MA, Szymczak S, Reim E, Haueisen J, Lübbers L, Rühlemann M, Franke A, Schwarz K, Stukenbrock EH. A fungal pathogen induces systemic susceptibility and systemic shifts in wheat metabolome and microbiome composition. Nat Commun. 2020;11(1):1–2. https://doi.org/10.1038/s41467-020-15633-x.
Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27(21):2957–63. https://doi.org/10.1093/bioinformatics/btr507.
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460–1. https://doi.org/10.1093/bioinformatics/btq461.
Yilmaz P, Parfrey LW, Yarza P, Gerken J, Pruesse E, Quast C, Schweer T, Peplies J, Ludwig W, Glöckner FO. The SILVA and “all-species living tree project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014;42(D1):D643–8. https://doi.org/10.1093/nar/gkt1209.
Ullah A, Akbar A, Luo Q, Khan AH, Manghwar H, Shaban M, Yang X. Microbiome diversity in cotton rhizosphere under normal and drought conditions. Microb Ecol. 2019;77(2):429–39. https://doi.org/10.1007/s00248-018-1260-7.
Rich SM, Watt M. Soil conditions and cereal root system architecture: review and considerations for linking Darwin and Weaver. J Exp Bot. 2013;64(5):1193–208. https://doi.org/10.1093/jxb/ert043.
Dong M, Yang Z, Cheng G, Peng L, Xu Q, Xu J. Diversity of the bacterial microbiome in the roots of four saccharum species: S. spontaneum, S. robustum, S. barberi and S. officinarum. Front Microbiol. 2018;9:267. https://doi.org/10.3389/fmicb.2018.00267.
Hennion N, Durand M, Vriet C, Doidy J, Maurousset L, Lemoine R, Pourtau N. Sugars en route to the roots. Transport, metabolism and storage within plant roots and towards microorganisms of the rhizosphere. Physiol Plant. 2019;165(1):44–57. https://doi.org/10.1111/ppl.12751.
Miotto-Vilanova L, Jacquard C, Courteaux B, Wortham L, Michel J, Clement C, Barka EA, Sanchez L. Burkholderia phytofirmans PsJN confers grapevine resistance against Botrytis cinerea via a direct antimicrobial effect combined with better resource mobilization. Front Plant Sci. 2016;7:1236. https://doi.org/10.1371/journal.pone.0029382.
Schwachtje J, Karojet S, Thormahlen I, Bernholz C, Kunz S, Brouwer S, Schwochow M, Kohl K, van Dongen JT. A naturally associated rhizobacterium of Arabidopsis thaliana induces a starvation-like transcriptional response while promoting growth. PLoS ONE. 2011;6(12): e29382. https://doi.org/10.1371/journal.pone.0029382.
Zhao X, Jiang Y, Liu Q, Yang H, Wang Z, Zhang M. Effects of drought-tolerant Ea-DREB2B transgenic sugarcane on bacterial communities in soil. Front Microbiol. 2020;11:704. https://doi.org/10.3389/fmicb.2020.00704.
Bowles TM, Acosta-Martinez V, Calderon F, Jackson LE. Soil enzyme activities, microbial communities and carbon and nitrogen availability in organic agroecosystems across an intensively managed agricultural landscape. Soil Biol Biochem. 2014;68:252–62. https://doi.org/10.1016/j.soilbio.2013.10.004.
Bruto M, Prigent-Combaret C, Muller D, Moenne-Loccoz Y. Analysis of genes contributing to plant-beneficial functions in plant growth-promoting rhizobacteria and related Proteobacteria. Sci Rep. 2014;4(1):1. https://doi.org/10.1038/srep06261.
Vurukonda SS, Vardharajula S, Shrivastava M, SkZ A. Enhancement of drought stress tolerance in crops by plant growth-promoting rhizobacteria. Microbiol Res. 2016;184:13–24. https://doi.org/10.1016/j.micres.2015.12.003.
Da Costa DP, Dias AC, Cotta SR, Vilela D, De Andrade PA, Pellizari VH, Andreote FD. Changes of bacterial communities in the rhizosphere of sugarcane under the elevated concentration of atmospheric CO2. Glob Change Biol Bioenergy. 2018;10(2):137–45. https://doi.org/10.1111/gcbb.12476.
Lambais MR, Lucheta AR, Crowley DE. Bacterial community assemblages associated with the phyllosphere, dermosphere, and rhizosphere of tree species of the Atlantic forest are host taxon dependent. Microb Ecol. 2014;68(3):567–74. https://doi.org/10.1007/s00248-014-0433-2.
Perez-Jaramillo JE, Mendes R, Raaijmakers JM. Impact of plant domestication on rhizosphere microbiome assembly and functions. Plant Mol Biol. 2016;90(6):635–44. https://doi.org/10.1007/s11103-015-0337-7.
Acknowledgements
We thank our cooperates from Guangxi University for the help in conducting and managing this research.
Funding
This research was financially supported by the China Agricultural Research System of MOF and MARA (CARS-170109).
Author information
Authors and Affiliations
Contributions
MQ and ZW conceived the main idea of research. AK wrote the manuscript. MQ, ZC, and ZW revised the manuscript and provided suggestions. In addition, MA, JB, and AK analyzed the data. AK, MA, JB, ZW and ZC assessed and data collection. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval and consent to participate were not required for this research, as it does not involve humans or animals.
Consent for publication
Not applicable.
Competing interests
The authors have no conflict of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig S1.
Rarefaction curve of the sugarcane genotypes depicting species richness and number of sequences. Table S1. Summary of Species richness estimator and alpha diversity indices. Table S2. Taxonomic identification of the most 30 abundant OTUs in eighteen sugarcane genotypes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Khan, A., Wang, Z., Chen, Z. et al. Investigation of soil nutrients and associated rhizobacterial communities in different sugarcane genotypes in relation to sugar content. Chem. Biol. Technol. Agric. 8, 59 (2021). https://doi.org/10.1186/s40538-021-00244-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40538-021-00244-5