
Abstract
The transactive response DNA binding protein of 43 kDa (TDP-43) is an intranuclear protein involved in RNA splicing. Abnormally deposited TDP-43 is found in the brains of patients with frontotemporal lobar degeneration (FTLD). Different morphological characteristics of TDP-43 immunoreactive inclusions define the different variants of FTLD-TDP. Little is known about the relationships between TDP-43 specie (phosphorylated TDP-43, C-terminal fragments and full length TDP-43) and lesion types. Using novel antibodies that recognize phosphorylated TDP-43 (pTDP-43), a neoepitope in the C-terminal fragment of TDP-43 (cTDP-43) and the N-terminal, i.e. full length (nTDP-43) we assess the relative burden of pTDP-43, cTDP-43 and nTDP-43 in 8 different lesion types across FTLD-TDP type A-C. These include neuronal cytoplasmic inclusions, dystrophic neurites, neuronal intranuclear inclusions, fine neurites of the hippocampus, peri-vascular inclusions, Pick body-like inclusions, long thick dystrophic neurites and granular pre-inclusions. We also assess for associations with progranulin (GRN) and C9ORF72 genetic mutations. For all eight lesion types, the highest burden was observed for pTDP-43. In six of the eight lesions studied, cTDP-43 burden was greater than nTDP-43 burden. However, we observed a higher burden of nTDP-43 to cTDP-43 for pre-inclusions. We also noted an equal-to-slightly higher burden of nTDP-43 to cTDP-43 for peri-vascular inclusions. There was not strong evidence for associations to be driven by mutation status although for neuronal cytoplasmic inclusions and dystrophic neurites GRN+ cases had higher burden of pTDP-43, cTDP-43 and nTDP-43 compared to GRN- cases, with nTDP-43 inclusions only observed in GRN+ cases. In addition, for pre-inclusions, cTDP-43 and nTDP-43 burden tended to be higher in C9ORF72- cases compared to C9ORF72+ cases, although this was not the case for pTDP-43. There is clear evidence that phosphorylation and C terminal fragments play an important role in lesion formation in FTLD-TDP. However, for some inclusions, particularly pre-inclusions, full-length TDP-43 appears to be playing a role.
Similar content being viewed by others


Introduction
The TAR DNA binding protein of 43 kDa (TDP-43) is a nuclear protein that is involved with RNA splicing and transcription repression [5, 6]. TDP-43 is present in the brains of individuals with frontotemporal lobar degeneration (FTLD-TDP) with or without concomitant motor neuron disease (MND) [1, 33]. It is known that after translation, TDP-43 undergoes a number of post-translational modifications, such as phosphorylation, ubiquitination and even proteolytic cleavage into abnormal C-terminal specie [1, 33, 46]. Phosphorylated TDP-43 is a characteristic feature of the inclusions in FTLD-TDP and is considered important in the pathogenesis [17]. It has also been shown that inclusions in the brains of cases of FTLD-TDP are characterized by the presence of C-terminal TDP-43 specie, but not full length TDP-43, suggesting that full length TDP-43 may not be a pathologic TDP-43 species in the brains of FTLD-TDP cases [21]. Frontotemporal lobar degeneration with TDP, however, is not a homogeneous entity in terms of morphology of the lesions (lesion type). Over the past decade, many different TDP-43 immunoreactive lesion types have been identified. Lesion types are important, and in fact when combined with the distribution of the lesions defines five different FTLD-TDP variants (A-E) [25, 27, 30, 38]. FTLD-TDP variants A-C are common, accounting for more than 95% of all FTLD-TDP [24], while variants D and E are rare [16, 27]. Little is known regarding differences in phosphorylation specie, C-terminal specie and full length TDP-43 by lesion type. In addition, there is heterogeneity in mutation status in FTLD-TDP with some cases being association with mutations in either the progranulin (GRN) gene [2, 12] or with mutations in the C9ORF72 gene [13, 36] while other cases are not associated with either mutation. A further complicating factor is that there are associations between mutation status and lesion type. For example, GRN mutations are associated with FTLD-TDP type A and hence also neuronal intranuclear inclusions (NII) [16, 23] while C9ORF72 mutations are associated with FTLD-TDP type B and hence also granular pre-inclusions [19, 36, 37]. As with lesion type, little is known about the association between mutation status and TDP-43 specie.
In this study, our primary aim was to determine whether there are any differences in the burden of phosphorylated TDP-43 specie, C-terminal specie and full length TDP-43 across the different lesion types. We also aimed to determine whether there is any evidence for any association with mutation status. We hypothesize that there would be differences across lesion types, as well as evidence for associations with mutation status.
Materials and methods
Case selection
The database of the brain bank at Mayo Clinic, Jacksonville, Florida was queried to identify a random selection of 24 cases received between 1997 to 2013 with a neuropathologic diagnosis of FTLD-TDP type A (n = 8), FTLD-TDP type B (n = 8) and FTLD-TDP type C (N = 8) in which slides were available for review and paraffin blocks were available for additional analyses for this study. All diagnoses were made by one expert neuropathologist (DWD). A diagnosis of FTLD-TDP and types were based on current consensus recommendations [25, 30]. Type A was diagnosed when there were neuronal cytoplasmic inclusions (NCIs) and dystrophic neurites (DNs), as well as NIIs. Type B was diagnosed when there were a predominance of NCIs with minimal DNs and no NIIs. Type C was diagnosed when there were abundant long thick neurites with minimal NCIs in the neocortex and no NIIs.
Neuropathologic methods
All cases had undergone neuropathologic assessment by a single neuropathologist (DWD), and had standardized tissue sampling and semi-quantitation of Alzheimer’s disease pathology. Thioflavin-S fluorescent microscopy was used for the evaluation of the distribution of senile plaques and neurofibrillary tangles (NFT) which was then used to determine the Thal β-amyloid phase as previously described [32], and the Braak NFT stage [3]. Immunohistochemistry was also performed on all cases with a α-synuclein antibody (NACP, 1:3000 rabbit polyclonal, Mayo Clinic antibody) and with a phosphorylated tau antibody (PHF-1, 1:1000 mouse monoclonal, gift from Dr. Peter Davies). HpScl was assessed on hematoxylin-eosin stained sections and was diagnosed when neuronal loss and gliosis was identified in the CA1 sector and/or subiculum of the hippocampus [14].
TDP-43 immunohistochemistry & semi-quantitation
In all 24 cases we performed serial sectioning and TDP-43 immunohistochemistry with four different TDP-43 antibodies: phosphorylated TDP-43 (pTDP-43) antibody (pS409/410, 1:5000 mouse monoclonal, Cosmo Bio Co., LTD) that recognizes TDP-43 with phosphorylated epitopes; anti TDP-43 antibody that recognizes a neoepitope in the C terminal fragment of cleaved TDP-43(cTDP-43) (MC2085, 1:2500 rabbit polyclonal, gift from Leonard Petrucelli, Mayo Clinic) [46]; anti TDP-43 antibody that recognizes an epitope in the amino terminus (nTDP-43) (MC1079; 1:2500 rabbit polyclonal, gift from Leonard Petrucelli, Mayo Clinic) [46] and a rabbit polyclonal anti-TDP-43 antibody (ProteinTech Group; Chicago IL; dilution 1:3000) which is a commercially available antibody. Antigens for the three non-commercial antibodies are shown in Fig. 1 and detailed biochemistry regarding these three antibodies have been previously published [7, 9, 10, 46]. Antibodies targeting phosphoserine 409 and 410 have been shown to have very strong immunoreactivity to the inclusions in FTLD-TDP [17, 22]. N-terminal TDP-43 fragments are believed to be rapidly degraded [4, 35] and as expected MC1079 binding represents detection of full length TDP-43 [46]. In all cases, immunohistochemistry was performed using a DAKO Autostainer (Universal Staining System Carpinteria, California). For this study, two investigators (KAJ & DWD) reviewed all 24 cases together and performed semi-quantitation of TDP-43 pathology with the three non-commercial antibodies (pTDP-43, cTDP-43 & nTDP-43) in the entorhinal cortex, CA1 sector, subiculum and dentate nucleus of the hippocampus. Lesions were considered positive, independent of the intensity of lesion staining. Hence, lesions showing slight/mild staining with one antibody were given equal weight to lesions with robust intensity with another antibody and both were assumed positive. This accounts for differences in affinities of the three different antibodies. For each region TDP-43 deposition was assessed independently for eight different types of lesions: (1) small discrete NCIs; (2) short/comma-like DNs; (3) fine neurites in the pyramidal layer of the hippocampus as described by Hatanpaa et al. [18]; (4) perivascular inclusions (typically bilobular and located next to capillary basal lamina) described by Lin et al. [28]; (5) Pick-body like inclusions in the dentate nucleus in FTLD-TDP type C cases as described by Katsuse and Dickson [26] (6) long thick DNs in cortex in FTLD-TDP type C cases, (7) granular pre-inclusions as described by Katsuse and Dickson [26], and (8) NIIs. These lesions were selected given that all have been associated with FTLD-TDP types. The burden of seven of the eight lesions was semi-quantitated on a 5-point scale in order to be able to compare across lesion type: 0 = no inclusions identified, 0.5 = scant number of inclusions; 1 = few inclusions, 2 = moderate inclusions and 3 = marked/frequent number of inclusions. We documented the presence/absence of NIIs given how rarely we identify this inclusion. For each lesion we assessed the burden detected with pTDP-43, cTDP-43 and nTDP-43.

The amino acid sequence of TDP-43 from N terminus (1) to C terminus (414). The highlighted regions are the peptides used to generate the polyclonal antibodies (MC2079, MC2085 and pTDP 409/410)
Genetic screening
In all 24 cases we screened for the presence of a mutation in the GRN and C9ORF72 genes. Briefly, amplification by polymerase chain reaction (PCR) of exons 0-12 and the 3′ untranslated region of the GRN gene was performed using primers and protocols that have been previously described [2]. To assess for the presence of an expanded GGGGCC hexanucleotide repeat in C9ORF72, the repeat primed PCR was also used, as outlined in a previous publication [13].
Results
Demographic and pathological features by FTLD-TDP type
Demographic characteristics of the 24 cases are shown in Table 1. FTLD-TDP type A cases were mainly men, while type B cases were mainly women and type C cases were evenly split among men and women. FTLD-TDP type B cases were on average 7–9 years younger than type A and type C cases. The most common clinical diagnosis in the FTLD-TDP type A cases was behavioral variant frontotemporal dementia (bvFTD), while the most common clinical diagnosis for FTLD-TDP type B was FTD-MND and for type C was primary progressive aphasia (semantic variant). Of the eight FTLD-TDP type A cases, four had a mutation in the GRN gene and one had a mutation in the C9ORF72 gene. Of the eight FTLD-TDP type B cases, four had a mutation in the C9ORF72 gene. The Thal β-amyloid phase and the Braak NFT stage were both low and relatively similar across FTLD-TDP types; none were Braak NFT stage IV or greater and none met intermediate - high probability criteria for Alzheimer’s disease [43]. Hippocampal sclerosis was observed in all FTLD-TDP type A cases, but less than 25% of type B and C cases. All FTLD-TDP type B cases and two FTLD-TDP type A cases had pathological evidence of MND such as bunina bodies, neuronal loss and gliosis and TDP-43 immunoreactive inclusions in hypoglossal nucleus and/or loss of Betz cells from lamina VI of Broadman area 4. None of the FTLD-TDP type C cases had MND.
There were no observable differences in lesion burden between the entorhinal cortex, CA1 sector and subiculum of the hippocampus, and hence these regions were combined for further analyses. In all instances the pTDP-43 antibody detected the greatest number of inclusions compared to cTDP-43 and nTDP-43 (Table 1). There were three cases (13%) where the pTDP-43 antibody detected lesions but neither cTDP-43 or nTDP-43 antibodies detected any lesions. This was observed for fine neurites, perivascular inclusions, NCI’s, DNs and pre-inclusions. We did not observe this phenomenon for long-thick dystrophic neurites. In all instances, except for in the entorhinal/CA1/subiculum in FTLD-TDP type B cases, the cTDP-43 antibody recognized a higher burden of lesions compared to the nTDP-43 antibody (Table 1). For the entorhinal/CA1/subiculum in FTLD-TDP type B cases we instead noted that the nTDP-43 antibody detected more inclusions compared to the cTDP-43 antibody.
Associations of TDP-43 specie with lesion type
In order to better understand the findings regarding the burden of pTDP-43, cTDP-43 and nTDP-43 that we observed, and to allow us to see whether the findings may be driven by any one or more lesion type, we compared pTDP-43, cTDP-43 and nTDP-43 burden for each of the six lesion types. In the entorhinal/CA1/subiculum (Fig. 2), we observed that for most lesion types there was a similar pattern of pTDP-43: cTDP-43: nTDP-43 burden. That is, we saw the greatest burden of inclusions to be detected by the pTDP-43 antibody. We also observed a greater burden of inclusions to be detected with the cTDP-43 antibody compared to the nTDP-43 antibody (Figs. 3 & 4). However, we noticed that pre-inclusions, which are typically observed in FTLD-TDP type B cases, showed a greater burden of inclusions detected with the nTDP-43 antibody compared to the cTDP-43 antibody (Fig. 5). We also noted a slightly higher burden of perivascular inclusions that were detected by the nTDP-43 antibody compared to the cTDP-43 antibody. Neuronal intranuclear inclusions (NIIs) were observed in five of the eight FTLD-TDP type A cases with the pTDP-43 antibody (data not shown). Of these five cases, NIIs were also observed in three cases with the cTDP-43 antibody. We did not detect NIIs with the nTDP-43 antibody.

Histograms showing lesion burden detected using pTDP-43, cTDP-43 and nTDP-43 antibodies across FTLD-TDP types for the entorhinal cortex/CA1/subiculum. Lesion burden is based on semi-quantitation. The boxes represent mean burden with whiskers representing standard deviation

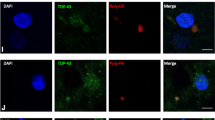
Detection of lesions in FTLD-TDP type A using pTDP-43, cTDP-43, nTDP-43 and the Proteintech antibodies. Top row shows dentate nucleus of the hippocampus, second row shows intranuclear inclusions in entorhinal cortex, third row shows CA1 and fourth row shows subiculum. Arrows depicts perivascular inclusions. Magnification × 20. Note the amount of background staining with the Proteintech antibody which prevents semi-quantitative analysis using this antibody

Detection of lesions in FTLD-TDP type C using pTDP-43, cTDP-43 and nTDP-43 antibodies. Top two rows show dentate nucleus of the hippocampus and bottom row shows entorhinal cortex. Arrow depicts long thick dystrophic neurites. Magnification × 10 for top row, and × 20 for bottom two rows

Detection of lesions in FTLD-TDP type B using pTDP-43, cTDP-43 and nTDP-43 antibodies. Top row shows dentate nucleus of the hippocampus and bottom two rows show entorhinal cortex with magnification × 10 for top row and × 20 for bottom row. Red circles depict pre-inclusions while arrows depict small discrete NCIs
In the dentate nucleus of the hippocampus (Fig. 6), the pTDP-43 antibody detected the highest lesion burden, followed by the cTDP-43 antibody and lastly the nTDP-43 antibody, for both small discrete NCIs (FTLD-TDP types A and B) (Figs. 3 & 5) and Pick-body like NCIs (FTLD-TDP type C) (Fig. 4).

Histograms showing lesion burden detected using pTDP-43, cTDP-43 and nTDP-43 antibodies across FTLD-TDP types for the dentate nucleus of the hippocampus. Lesion burden is based on semi-quantitation. The boxes represent mean burden with whiskers representing standard deviation
Demographics and pathological features by mutation
Demographic characteristics of the 16 FTLD-TDP type A and B cases by mutation status are shown in Table 2. The C9ORF72+ cases were the youngest at death. The clinical diagnoses, and β-amyloid and NFT distribution, were relatively similar between GRN+ and GRN- cases and between C9ORF72+ and C9ORF72- cases. Hippocampal sclerosis was present in all GRN+ and GRN- cases and 50% of C9ORF72 + cases, but was not present in any of the C9ORF72- cases. In all instances the pTDP-43 antibody detected the greatest number of inclusions compared to the cTDP-43 and nTDP-43 antibodies. There was little evidence for striking differences in the burden of lesions detected with the pTDP-43, cTDP-43 and nTDP-43 antibodies by mutation status. The higher proportion of lesions detected with nTDP-43 vs cTDP-43 antibody in the entorhinal/CA1/subiculum in FTLD-TDP type B cases was similar for C9ORF72+ and C9ORF72- cases.
Associations of TDP-43 specie with lesion type and mutation status
In order to get a better understanding of whether mutation status may be playing a role in the relationships we observed between the different antibodies and lesion type, we compared pTDP-43, cTDP-43 and nTDP-43 antibody burden for each lesion type, by mutation status. Hence, we compared GRN status within FTLD-TDP type A cases, and C9ORF72 status within FTLD-TDP type B cases, for each lesion type (Fig. 7).

Histograms showing lesion burden detected using pTDP-43, cTDP-43 and nTDP-43 antibodies by mutation status for the entorhinal cortex/CA1/subiculum. Lesion burden is based on semi-quantitation. The boxes represent mean burden with whiskers representing standard deviation
First we describe our findings for FTLD-TDP type A associated lesions. We found that lesion burden was higher with the pTDP-43 antibody for GRN+ cases compared to GRN- cases for all lesions except for fine neurites where burden was about equal. We noted that lesion burden was higher for the cTDP-43 antibody in GRN+ cases compared to GRN- cases for small discrete NCI’s, DNs and fine neurites. We also observed that lesion burden was higher for the nTDP-43 antibody in GRN+ cases compared to GRN- cases for small discrete NCIs, DNs and perivascular inclusions. In fact, the GRN- cases had no nTDP-43 small discrete NCIs or DNs. Comparing across antibodies, there was a higher burden of lesions detected with the cTDP-43 antibody than the nTDp-43antibody in most instances, except for with perivascular inclusions.
For FTLD-TDP type B we investigated mutation status for pre-inclusions. We found that the C9ORF72+ cases had a lower burden of lesions for both nTDP-43 and cTDP-43 antibodies compared to C9ORF72- cases. In fact, we did not see any C9ORF72+ cases with pre-inclusions that were detected with the cTDP-43 antibody. We also found that the C9ORF72+ cases had a slightly higher burden of pre-inclusions that were detected with the pTDP-43 antibody compared to the C9ORF72- cases. Comparing across antibodies, we observed that our finding of higher lesion burden for the nTDP-43 antibody compared to cTDP-43 antibody was observed for both C9ORF72+ and C9ORF72- cases.
Discussion
The results of this study demonstrate that phosphorylated TDP-43, C terminal specie and full length TDP-43 are all present in the inclusions in FTLD-TDP. Given that the characteristics of the different FTLD-TDP variants are dependent on different inclusion types, the results of this study support the notion that there are indeed differences in TDP-43 specie across the different FTLD-TDP variants. The findings improve our understanding of the relationship between TDP-43 specie and FTLD-TDP and may shed light on the cellular mechanisms resulting in lesion formation in the different FTLD-TDP types, with the findings suggesting different underlying mechanisms in FTLD-TDP type B. This knowledge may be helpful for the development of future treatments targeting TDP-43.
Previous studies have demonstrated that the inclusions found in FTLD-TDP consist of phosphorylated TDP-43 [1, 17, 33]. It has also been shown that the inclusions in FTLD-TDP consist mainly of C terminal fragments of TDP-43 [1, 33]. Therefore, one could infer that phosphorylated C terminal fragments are the key to inclusion formation and pathogenicity. However, others have shown that phosphorylation of TDP-43 is not necessary for inclusion formation and that lesions in FTLD-TDP likely consist of phosphorylated and unphosphorylated C terminal specie of TDP-43 [46]. The cTDP-43 antibody utilized in our study detects both phosphorylated and unphosphorylated C terminal fragments [46]. While we cannot determine the degree of phosphorylation in our C terminal fragments, we did find that the burden of inclusions detected with the pTDP-43 antibody was greater than with the cTDP-43 antibody, consistent with pTDP-43 antibody detecting both phosphorylated C-terminal and phosphorylated full-length TDP-43. Full-length TDP-43, therefore appears, to be an important component of inclusions in FTLD-TDP. Indeed, it has been demonstrated that TDP-43 aggregation occurs when full length (endogenous) TDP-43 is prevented from exiting the nucleus or from entering the nucleus [42].
The burden of inclusions detected with the cTDP-43 antibody was higher than the burden of inclusions detected with the nTDP-43 antibody for most inclusion types, including fine neurites of the hippocampus, perivascular inclusions, small discrete NCIs and both short/comma and long thick DNs. These inclusion types, therefore, appear to consist mainly of C terminal TDP-43 and concur with previous studies that have also noted a predominance of C terminal TDP-43 in FTLD-TDP inclusions [1, 21]. A novel finding from our study was that the ratio of C terminal fragments to full-length TDP-43 was reversed in the pre-inclusions identified in FTLD-TDP type B, with a higher burden of inclusions identified with the nTDP-43 antibody. This is intriguing given that FTLD-TDP type B is typically associated with motor neuron disease/amyotrophic lateral sclerosis [25]. This would suggest that the mechanism by which TDP-43 immunoreactive pre-inclusions are formed in cases with motor neuron disease may differ from mechanisms involved with the formation of lesions in FTLD-TDP type A and type C. With-that-said, we did observe pre-inclusions in two FTLD-TDP type A cases. Interestingly, in both cases the pre-inclusions were not detected with the pTDP-43 or cTDP-43 antibodies suggesting the possibility that these inclusions consist of unphosphorylated full length TDP-43. In one of these two cases, there was a mutation in the C9ORF72 gene and the clinical diagnosis was FTD-MND. This case, however, was classified as FTLD-TDP type A given the presence of fine neuritis in the CA1 region of the hippocampus [18, 25]. It should be pointed out that other features of FTLD-TDP type A such s NIIs were not present in this case. Given the uncertainty in classification of this case, it is unclear whether this finding is unique to FTLD-TDP type B, or rather a feature of pre-inclusions, independent of FTLD-TDP type. Previous studies that have assessed C terminal fragments and full-length TDP-43 across FTLD-TDP types did not assess pre-inclusions [1, 21] and hence we are unable to compare results.
We also noted that perivascular inclusions in FTLD-TDP type A cases were slightly more likely to be detected with the nTDP-43 antibody compared to the cTDP-43 antibody. This trend was not as striking as for pre-inclusions, but was very different from the findings across the other inclusions identified in FTLD-TDP type A. In fact there was a case where no perivascular inclusions were detected with the cTDP-43 antibody while they were detected with the nTDP-43 antibody. In this case the pTDP-43 antibody also detected the perivascular inclusions. This finding would support the notion that cellular mechanisms resulting in lesion formation may differ within FTLD-TDP type A. Once again previous studies that have assessed C terminal fragments and full-length TDP-43 across FTLD-TDP types have not assessed perivascular inclusions [1, 15]. Similarly, for FTLD-TDP type B, there may be differences in how pre-inclusions in the cortex are formed versus how small discrete NCI’s are formed in the dentate nucleus of the hippocampus. Hence, although it has been shown that C terminal fragments of TDP-43 are indistinguishable between regions [39], the findings here support another study showing differences between regions when comparing C terminal specie and full length TDP-43 [21].
When we studied NIIs we observed that the highest lesion burden occurred with the pTDP-43 antibody of which only a subset was identified with the C terminal antibody; none were detected with the nTDP-43 antibody. This finding is different from that reported in one previous study where the investigators found evidence for both C terminal specie and full length TDP-43 to be a component of NIIs [21]. Our findings suggest that NIIs consist of C terminal specie TDP-43 that is predominantly phosphorylated but not full length TDP-43. Given that the nuclear localization signal is within the N-terminal half of the protein [45] it is unlikely that TDP-43 is cleaved and phosphorylated in the cytoplasm and then C terminal fragments are shuttled back into the nucleus. Hence, cleavage is most likely occurring inside the nucleus. It has been reported in one study that C-terminal fragments of TDP-43 do not sequester full length TDP-43 [46]. If correct, a likely explanation for the absence of full length TDP-43 in NII’s is that once NIIs are formed in the nucleus, from the cleavage of the full length TDP-43, full length cytoplasmic TDP-43 is blocked from being shuttled inside the nucleus. This would be expected given that TDP-43 pathology interferes with nuclear pore complexes and transport of TDP-43 into the nucleus [11]. On-the-other hand, studies have reported that C terminal fragments can sequester full length TDP-43 [8, 34] which if correct would explain the absence of full length TDP-43 in NII’s.
We should also point out that when we identified inclusions with the nTDP-43 antibody we also noted there was an absence of normal full-length TDP-43 from the nucleus of the associated cells. This implies that full length TDP-43 that is sequestered into the inclusion was unable to shuttle back into the nucleus. Whether this is due to the phosphorylation of the full length TDP-43, given that the pTDP-43 antibody detected the inclusions, or whether it is due to another type of post translational modification, remains unknown.
Our analysis by FTLD mutations showed that our general findings were not driven by mutation status in any of the FTLD-TDP types. This is not surprising as animal models have not found any evidence for GRN mutations to be associated with TDP-43 fragmentation [15, 31] and down regulation of GRN in zebrafish. We did, however, find a suggestion for differences by mutation. In general, the GRN+ cases showed a higher burden of TDP-43 inclusions, independent of the specific antibody, with only a few exceptions. The only demographic difference between the GRN+ and GRN- cases was a younger age in the GRN+ cases. Previous studies have shown that GRN mutations are associated with a more aggressive disease [41], and our findings may imply that this could be due to a higher pathological TDP-43 burden [23, 29]. We did not find this association with the C9ORF72 mutation. Interestingly, we noted that the C9ORF72+ cases had a slightly higher burden of pre-inclusions that were detected with the pTDP-43 antibody compared to the C9ORF72- cases while the C9ORF72- cases had a higher burden of inclusions with the cTDP-43 and nTDP-43 antibodies, suggesting that there were phosphorylated pre-inclusions in the C9ORF72+ cases that must not have been detected with either the cTDP-43 or nTDP-43 antibodies. This is not necessarily surprising given that other C terminal fragments have been reported in FTLD-TDP [20, 34, 40, 44].
Conclusion
A strength of this study was the fact that we looked at eight different inclusions and the fact that all patients underwent genetic screening for a GRN or C9ORF72 mutation. Despite the fact that the number of cases assessed was not large enough to allow for statistical testing, we have identified some interesting trends in the data, particularly in the pre-inclusions in FTLD-TDP type B, which warrant further investigation. Furthermore, the findings have relevance in the interpretation of studies from different laboratories as it is clear that some inclusions may not be seen depending on what kind of anti-TDP-43 antibody is being used for detection.
Availability of data and materials
The datasets generated and analyzed for the current study is available from the corresponding author on reasonable request.
References
Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y et al (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602–611. https://doi.org/10.1016/j.bbrc.2006.10.093
Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S et al (2006) Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442:916–919. https://doi.org/10.1038/nature05016
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 82:239–259
Buratti E (2018) TDP-43 post-translational modifications in health and disease. Expert Opin Ther Targets 22:279–293. https://doi.org/10.1080/14728222.2018.1439923
Buratti E, Baralle FE (2008) Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front Biosci 13:867–878
Buratti E, Dork T, Zuccato E, Pagani F, Romano M, Baralle FE (2001) Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J 20:1774–1784. https://doi.org/10.1093/emboj/20.7.1774
Carlomagno Y, Zhang Y, Davis M, Lin WL, Cook C, Dunmore J, Tay W, Menkosky K, Cao X, Petrucelli L et al (2014) Casein kinase II induced polymerization of soluble TDP-43 into filaments is inhibited by heat shock proteins. PLoS One 9:e90452. https://doi.org/10.1371/journal.pone.0090452
Che MX, Jiang LL, Li HY, Jiang YJ, Hu HY (2015) TDP-35 sequesters TDP-43 into cytoplasmic inclusions through binding with RNA. FEBS Lett 589:1920–1928. https://doi.org/10.1016/j.febslet.2015.06.009
Chew J, Cook C, Gendron TF, Jansen-West K, Del Rosso G, Daughrity LM, Castanedes-Casey M, Kurti A, Stankowski JN, Disney MD et al (2019) Aberrant deposition of stress granule-resident proteins linked to C9orf72-associated TDP-43 proteinopathy. Mol Neurodegener 14:9. https://doi.org/10.1186/s13024-019-0310-z
Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang YJ, Castanedes-Casey M, Lee CW, Jansen-West K, Kurti A, Murray ME et al (2015) Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science 348:1151–1154. https://doi.org/10.1126/science.aaa9344
Chou CC, Zhang Y, Umoh ME, Vaughan SW, Lorenzini I, Liu F, Sayegh M, Donlin-Asp PG, Chen YH, al DDM (2018) TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat Neurosci 21:228–239. https://doi.org/10.1038/s41593-017-0047-3
Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ et al (2006) Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442:920–924. https://doi.org/10.1038/nature05017
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J et al (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72:245–256. https://doi.org/10.1016/j.neuron.2011.09.011
Dickson DW, Davies P, Bevona C, Van Hoeven KH, Factor SM, Grober E, Aronson MK, Crystal HA (1994) Hippocampal sclerosis: a common pathological feature of dementia in very old (> or = 80 years of age) humans. Acta Neuropathol 88:212–221
Dormann D, Capell A, Carlson AM, Shankaran SS, Rodde R, Neumann M, Kremmer E, Matsuwaki T, Yamanouchi K, Nishihara M et al (2009) Proteolytic processing of TAR DNA binding protein-43 by caspases produces C-terminal fragments with disease defining properties independent of progranulin. J Neurochem 110:1082–1094. https://doi.org/10.1111/j.1471-4159.2009.06211.x
Forman MS, Mackenzie IR, Cairns NJ, Swanson E, Boyer PJ, Drachman DA, Jhaveri BS, Karlawish JH, Pestronk A, Smith TW et al (2006) Novel ubiquitin neuropathology in frontotemporal dementia with valosin-containing protein gene mutations. J Neuropathol Exp Neurol 65:571–581
Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y, Beach TG, Buratti E, Baralle F, Morita M et al (2008) Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol 64:60–70. https://doi.org/10.1002/ana.21425
Hatanpaa KJ, Bigio EH, Cairns NJ, Womack KB, Weintraub S, Morris JC, Foong C, Xiao G, Hladik C, Mantanona TY et al (2008) TAR DNA-binding protein 43 immunohistochemistry reveals extensive neuritic pathology in FTLD-U: a Midwest-southwest consortium for FTLD study. J Neuropathol Exp Neurol 67:271–279. https://doi.org/10.1097/NEN.0b013e31816a12a6
Hsiung GY, DeJesus-Hernandez M, Feldman HH, Sengdy P, Bouchard-Kerr P, Dwosh E, Butler R, Leung B, Fok A, Rutherford NJ et al (2012) Clinical and pathological features of familial frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p. Brain 135:709–722. https://doi.org/10.1093/brain/awr354
Igaz LM, Kwong LK, Chen-Plotkin A, Winton MJ, Unger TL, Xu Y, Neumann M, Trojanowski JQ, Lee VM (2009) Expression of TDP-43 C-terminal fragments in vitro recapitulates pathological features of TDP-43 Proteinopathies. J Biol Chem 284:8516–8524. https://doi.org/10.1074/jbc.M809462200
Igaz LM, Kwong LK, Xu Y, Truax AC, Uryu K, Neumann M, Clark CM, Elman LB, Miller BL, Grossman M et al (2008) Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am J Pathol 173:182–194
Inukai Y, Nonaka T, Arai T, Yoshida M, Hashizume Y, Beach TG, Buratti E, Baralle FE, Akiyama H, Hisanaga S et al (2008) Abnormal phosphorylation of Ser409/410 of TDP-43 in FTLD-U and ALS. FEBS Lett 582:2899–2904. https://doi.org/10.1016/j.febslet.2008.07.027
Josephs KA, Ahmed Z, Katsuse O, Parisi JF, Boeve BF, Knopman DS, Petersen RC, Davies P, Duara R, Graff-Radford NR et al (2007) Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions with progranulin gene (PGRN) mutations. J Neuropathol Exp Neurol 66:142–151. https://doi.org/10.1097/nen.0b013e31803020cf
Josephs KA, Hodges JR, Snowden JS, Mackenzie IR, Neumann M, Mann DM, Dickson DW (2011) Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol 122:137–153. https://doi.org/10.1007/s00401-011-0839-6
Josephs KA, Stroh A, Dugger B, Dickson DW (2009) Evaluation of subcortical pathology and clinical correlations in FTLD-U subtypes. Acta Neuropathol 118:349–358. https://doi.org/10.1007/s00401-009-0547-7
Katsuse O, Dickson DW (2005) Ubiquitin immunohistochemistry of frontotemporal lobar degeneration differentiates cases with and without motor neuron disease. Alzheimer Dis Assoc Disord 19(Suppl 1):S37–S43
Lee EB, Porta S, Michael Baer G, Xu Y, Suh E, Kwong LK, Elman L, Grossman M, Lee VM, Irwin DJ et al (2017) Expansion of the classification of FTLD-TDP: distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol 134:65–78. https://doi.org/10.1007/s00401-017-1679-9
Lin WL, Castanedes-Casey M, Dickson DW (2009) Transactivation response DNA-binding protein 43 microvasculopathy in frontotemporal degeneration and familial Lewy body disease. J Neuropathol Exp Neurol 68:1167–1176. https://doi.org/10.1097/NEN.0b013e3181baacec
Mackenzie IR, Baker M, Pickering-Brown S, Hsiung GY, Lindholm C, Dwosh E, Gass J, Cannon A, Rademakers R, Hutton M et al (2006) The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain 129:3081–3090. https://doi.org/10.1093/brain/awl271
Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, Perry RH, Trojanowski JQ, Mann DM, Lee VM (2011) A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 122:111–113. https://doi.org/10.1007/s00401-011-0845-8
Moisse K, Mepham J, Volkening K, Welch I, Hill T, Strong MJ (2009) Cytosolic TDP-43 expression following axotomy is associated with caspase 3 activation in NFL−/− mice: support for a role for TDP-43 in the physiological response to neuronal injury. Brain Res 1296:176–186. https://doi.org/10.1016/j.brainres.2009.07.023
Murray ME, Lowe VJ, Graff-Radford NR, Liesinger AM, Cannon A, Przybelski SA, Rawal B, Parisi JE, Petersen RC, Kantarci K et al (2015) Clinicopathologic and 11C-Pittsburgh compound B implications of Thal amyloid phase across the Alzheimer's disease spectrum. Brain 138:1370–1381. https://doi.org/10.1093/brain/awv050
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133
Nonaka T, Kametani F, Arai T, Akiyama H, Hasegawa M (2009) Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum Mol Genet 18:3353–3364. https://doi.org/10.1093/hmg/ddp275
Pesiridis GS, Tripathy K, Tanik S, Trojanowski JQ, Lee VM (2011) A “two-hit” hypothesis for inclusion formation by carboxyl-terminal fragments of TDP-43 protein linked to RNA depletion and impaired microtubule-dependent transport. J Biol Chem 286:18845–18855. https://doi.org/10.1074/jbc.M111.231118
Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L et al (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72:257–268. https://doi.org/10.1016/j.neuron.2011.09.010
Stewart H, Rutherford NJ, Briemberg H, Krieger C, Cashman N, Fabros M, Baker M, Fok A, DeJesus-Hernandez M, Eisen A et al (2012) Clinical and pathological features of amyotrophic lateral sclerosis caused by mutation in the C9ORF72 gene on chromosome 9p. Acta Neuropathol 123:409–417. https://doi.org/10.1007/s00401-011-0937-5
Tan RH, Shepherd CE, Kril JJ, McCann H, McGeachie A, McGinley C, Affleck A, Halliday GM (2013) Classification of FTLD-TDP cases into pathological subtypes using antibodies against phosphorylated and non-phosphorylated TDP43. Acta Neuropathol Commun 1:33. https://doi.org/10.1186/2051-5960-1-33
Tsuji H, Arai T, Kametani F, Nonaka T, Yamashita M, Suzukake M, Hosokawa M, Yoshida M, Hatsuta H, Takao M et al (2012) Molecular analysis and biochemical classification of TDP-43 proteinopathy. Brain 135:3380–3391. https://doi.org/10.1093/brain/aws230
Tsuji H, Nonaka T, Yamashita M, Masuda-Suzukake M, Kametani F, Akiyama H, Mann DM, Tamaoka A, Hasegawa M (2012) Epitope mapping of antibodies against TDP-43 and detection of protease-resistant fragments of pathological TDP-43 in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Biochem Biophys Res Commun 417:116–121. https://doi.org/10.1016/j.bbrc.2011.11.066
Whitwell JL, Boeve BF, Weigand SD, Senjem ML, Gunter JL, Baker MC, DeJesus-Hernandez M, Knopman DS, Wszolek ZK, Petersen RC et al (2015) Brain atrophy over time in genetic and sporadic frontotemporal dementia: a study of 198 serial magnetic resonance images. Eur J Neurol 22:745–752. https://doi.org/10.1111/ene.12675
Winton MJ, Igaz LM, Wong MM, Kwong LK, Trojanowski JQ, Lee VM (2008) Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J Biol Chem 283:13302–13309. https://doi.org/10.1074/jbc.M800342200
Working Group (1997) Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. The National Institute on Aging, and Reagan institute working group on diagnostic criteria for the Neuropathological assessment of Alzheimer's disease. Neurobiol Aging 18:S1–S2
Xiao S, Sanelli T, Chiang H, Sun Y, Chakrabartty A, Keith J, Rogaeva E, Zinman L, Robertson J (2015) Low molecular weight species of TDP-43 generated by abnormal splicing form inclusions in amyotrophic lateral sclerosis and result in motor neuron death. Acta Neuropathol 130:49–61. https://doi.org/10.1007/s00401-015-1412-5
Zhang YJ, Caulfield T, Xu YF, Gendron TF, Hubbard J, Stetler C, Sasaguri H, Whitelaw EC, Cai S, Lee WC et al (2013) The dual functions of the extreme N-terminus of TDP-43 in regulating its biological activity and inclusion formation. Hum Mol Genet 22:3112–3122. https://doi.org/10.1093/hmg/ddt166
Zhang YJ, Xu YF, Cook C, Gendron TF, Roettges P, Link CD, Lin WL, Tong J, Castanedes-Casey M, Ash P et al (2009) Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci U S A 106:7607–7612
Acknowledgements
We wish to thank Linda Rousseau, Virginia Phillips and Monica Casey Castanedes for pathological support.
Funding
The study was funded by National Institute of Aging grant R01 AG37491.
Author information
Authors and Affiliations
Contributions
KAJ was responsive for screening all the cases and for drafting the manuscript. YJZ was responsible for antibody development and critical revision of the manuscript. MB was responsible for screening all the cases for GRN and C9ORF72 mutations. RR was responsible for screening all the cases for GRN and C9ORF72 mutations. LP was responsible for antibody development and critical revision of the manuscript. DWD was responsible for performing the immunohistochemistry, screening the frontotemporal lobar degeneration cases and critical revision of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Mayo institutional Review Board (IRB) has deem this study exempt from IRB review and approval given that it utilizes postmortem brain tissue only.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Josephs, K.A., Zhang, YJ., Baker, M. et al. C-terminal and full length TDP-43 specie differ according to FTLD-TDP lesion type but not genetic mutation. acta neuropathol commun 7, 100 (2019). https://doi.org/10.1186/s40478-019-0755-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-019-0755-x