
Abstract
Inflammation is becoming increasingly recognized as an important contributor to Alzheimer’s disease (AD) pathogenesis. As a part of the innate immune system, the complement cascade enhances the body’s ability to destroy and remove pathogens and has recently been shown to influence Alzheimer’s associated amyloid and tau pathology. However, little is known in humans about the effects of the complement system and genetic modifiers of AD risk like the ε4 allele of apolioprotein E (APOE ε4) on AD pathobiology. We evaluated cerebrospinal fluid (CSF) protein levels from 267 individuals clinically diagnosed as cognitively normal, mild cognitive impairment, and AD. Using linear models, we assessed the relationship between APOE ε4 genotype, CSF Complement 3 (C3), CSF amyloid-β (amyloid) and CSF hyperphosphorylated tau (ptau). We found a significant interaction between APOE ε4 and CSF C3 on both CSF amyloid and CSF ptau. We also found that CSF C3 is only associated with CSF ptau after accounting for CSF amyloid. Our results support a conceptual model of the AD pathogenic cascade where a synergistic relationship between the complement cascade (C3) and APOE ε4 results in elevated Alzheimer’s neurodegeneration and in turn, amyloid further regulates the effect of the complement cascade on downstream tau pathology.
Similar content being viewed by others


Introduction
Converging experimental and human biomarker evidence suggests that the immune system plays a critical role in Alzheimer’s disease (AD) pathobiology. In rodent models of AD, neuroinflammatory signals have been shown to correlate with amyloid and hyperphosphorylated tau (ptau) deposits as well as cognitive symptoms [23]. Observational studies in humans have shown that neuroinflammation, particularly the innate immune system, contributes to and drives AD pathogenesis [14]. Genetic studies in humans have identified a relationship between inflammatory and immune-associated genes and increased risk for AD [18] and have suggested that immune-mediated genes may contribute to Alzheimer’s pathogenesis [32]. Importantly, several studies have demonstrated that the ε4 allele of apolipoprotein (APOE ε4), the most common genetic risk factor for AD, has an important role in immune and inflammatory processes underlying Alzheimer’s neurodegeneration [6, 29]. Further, a recent study of epigenomic signals in mice and humans has implicated regulatory changes in immune response genes and immune-regulatory genes in AD [13].
Recent work suggests an association between inflammatory pathways, amyloid, and tau pathology. In mouse models, Complement C1q has been shown to be necessary for amyloid-β associated synaptotoxicity [15]. Using microarray data from amyloid and tau transgenic mice, immune gene expression has been shown to be tightly associated with amyloid plaques rather than neurofibrillary tangles suggesting that amyloid dysmetabolism mediates the influence of the immune system on tau pathology [19]. Although several studies have examined the association between inflammation and AD neurodegeneration in humans, it is still unknown whether APOE ε4 influences the relationship between inflammation (specifically the complement cascade) and amyloid and tau pathology.
Here, we investigated the relationship between APOE ε4 and the complement system on amyloid and tau pathology in AD. We evaluated cerebrospinal fluid (CSF) measurements of the immune protein Complement 3 (C3) as well as amyloid β1-42 (amyloid) and ptau181p (ptau), which are sensitive in vivo markers of clinical diagnosis, decline, and pathology in AD [8, 9, 24]. Low CSF amyloid levels are associated with increased amyloid plaques in the brain while high CSF ptau levels are associated with increased ptau tangles in the brain [4, 9]. Low CSF C3 has been associated with worsening cognitive decline in mild cognitive impairment (MCI) [28] and improves diagnostic accuracy when used alongside CSF amyloid and tau [16]. We hypothesized that a statistical interaction between APOE ε4 and CSF C3 levels would be associated with low CSF amyloid (suggesting increased intracranial amyloid plaques) and elevated CSF ptau (suggesting increased intracranial ptau tangles). Further, building on prior work [15, 19], in a mediation analysis, we hypothesized that CSF amyloid would ‘mediate’ the effect of CSF C3 on CSF ptau.
Materials and methods
Participant description
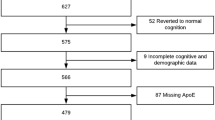
This study utilized samples from cognitively normal older adults (HC; n = 71), individuals diagnosed with amnestic MCI (n = 110), and probable AD (n = 56) from the Alzheimer’s Disease Neuroimaging Initiative (ADNI), which has been used in previously published studies [10, 11]. Clinical severity of symptoms in the MCI and AD groupings was measured using the Clinical Dementia Rating Sum of Boxes (CDR-SB) [22] and Mini Mental State Exam (MMSE) [12]. Diagnostic groupings were assigned using the clinical judgment of each site’s clinicians along with cutoffs based on neuropsychological tests [33]. Briefly, controls were required to have normal memory function on the Logical Memory II subscale of the Weschler Memory Scale – Revised [31], an MMSE score greater than 24, CDR total score equal to 0, and judgment by a clinician that the individual did not have any significant impairment in cognitive function or activities of daily living. Individuals with MCI were required to have abnormal memory function on the Logical Memory II subscale of the Weschler Memory Scale – Revised, an MMSE greater than 24, CDR total score equal to 0.5, and judgment by a clinician that the individual’s general cognition and functional performance was preserved enough that a diagnosis of AD could not be made. Finally, individuals with AD were required to have abnormal memory function on the Logical Memory II subscale of the Weschler Memory Scale – Revised, an MMSE between 20 and 26, CDR total score equal to 0.5 or 1.0, and judgment by a clinician that the individual met NINCDS/ADRDA criteria for probable AD [20]. Informed consent was obtained from all individual participants included in the study. For more information on the ADNI cohort, see Additional file 1.
Biomarker measurements
CSF amyloid β1-42 and CSF ptau181p were measured using the AlzBio3 Luminex xMAP immunoassay (Innogenetics, Ghent, Belgium) according to previously described methods [24]. This method utilizes monoclonal antibodies specific for amyloid β1-42 and ptau181p that are chemically bonded to color-coded beads along with analyte-specific detector antibodies. Baseline CSF C3 levels were measured using a separate multiplex immunoassay panel (Luminex technology), developed by Rules Based Medicine (Myriad RBM; Austin, Texas). CSF measurements in the immunoassay panel were processed and normalized according to previously described methods [7, 26]. Briefly, C3 levels were measured using the Myriad RBM Human DiscoveryMAP panel, which measures CSF protein levels for a range of metabolic, lipid, inflammatory, and other AD-relevant indicators. Myriad RBM used a Luminex 100 instrument for the measurements and analyzed the resulting data using proprietary software. The ADNI staff checked the distributions of analytes in this panel for normality by using Box-Cox analyses, and when appropriate log10 transformed the data to achieve an approximately normal distribution. APOE genotype was previously generated by ADNI using DNA extracted by Cogenics (now Beckman Coulter Inc.; Pasadena, California) from a 3 mL aliquot of EDTA blood. APOE ε4 dosage was scored as count of ε4 allele (0, 1, or 2) in all analyses.
Statistical analysis
We used chi-squared and one-way ANOVA for demographic and biomarker summary statistics and univariate assessments. Multivariate linear models were used to assess all biomarker relationships, covarying for baseline age, baseline CDR-SB, and sex.
We first tested for a statistical interaction between CSF C3 levels and APOE ε4 dosage on CSF amyloid and CSF ptau levels. Prior to our secondary analyses, we verified that there was an association between CSF amyloid and CSF ptau levels.
Next, we evaluated two theoretical models by which CSF C3 levels might modify CSF ptau levels. In Model A, we tested whether CSF amyloid levels ‘mediate’ CSF ptau levels by adding it to a baseline model in which CSF C3 levels predict CSF ptau levels. In Model B, we test whether a statistical interaction between CSF C3 levels and CSF amyloid levels significantly predict CSF ptau levels.
All analyses were performed using R. Plots were made using the ‘visreg’ package in R [5].
Results
Cohort description
Our cohort was balanced with respect to age, sex, and CSF C3 levels as there were no significant differences by diagnostic group (Table 1). There were significant differences in baseline CDR-SB scores, MMSE scores, APOE ε4 dosage, CSF amyloid, and CSF ptau.
An interaction between CSF C3 and APOE ε4 predicts CSF amyloid and CSF ptau
We found a significant statistical interaction between CSF C3 and APOE ε4 dosage on CSF amyloid levels (β-coefficient = -56.2, standard error (SE) = 23.8, p-value = 0.02) suggesting increased intracranial amyloid pathology among individuals with elevated intracranial C3 levels and increased number of APOE ε4 alleles (Fig. 1). With this interaction term in the model, we found a main effect of CSF C3 (β-coefficient = 61.3, SE = 21.9, p-value = 5.55 × 10−3) and APOE ε4 dosage (β-coefficient = -182.0, SE = 62.0, p-value = 3.69 × 10−3).

CSF amyloid versus CSF C3 by APOE ε4 dosage. CSF amyloid levels are plotted against CSF C3 levels by APOE ε4 dosage. C3 levels are quality controlled and transformed as described in [26]. Non-carriers of the APOE ε4 allele (blue) have the highest level of amyloid in CSF, reflecting lower levels of amyloid pathology in the brain, and display a positive slope. Carriers of one copy of the APOE ε4 allele (green) have intermediate levels of amyloid and display an approximately flat slope. Carriers of two copies of the APOE ε4 allele (red) have the lowest levels of CSF amyloid, suggesting the highest levels of amyloid pathology in the brain, and display a negative slope. The plotted points are partial residuals with 95 % confidence bands provided in shading
Similarly, we found a significant statistical interaction between CSF C3 and APOE ε4 dosage on CSF ptau levels (β-coefficient = 22.5, SE = 8.6, p-value = 9.44 × 10−3) suggesting increased neurofibrillary pathology among individuals with elevated C3 levels and increased number of APOE ε4 alleles (Fig. 2). With this interaction term in the model, we found a main effect of APOE ε4 dosage (β-coefficient = 65.4, SE = 22.4, p-value = 3.86 × 10−3) but not CSF C3 (β-coefficient = -4.3, SE = 7.9, p-value = 0.59).

CSF ptau versus CSF C3 by APOE ε4 dosage. CSF ptau levels are plotted against CSF C3 levels by APOE ε4 dosage. C3 levels are quality controlled and transformed as described in [26]. Non-carriers of the APOE ε4 allele (blue) have the lowest levels of ptau and display a slightly negative slope. Carriers of one copy of the APOE ε4 allele (green) have intermediate levels of ptau and display a modestly positive slope. Carriers of two copies of the APOE ε4 allele (red) have the highest levels of ptau and display a strongly positive slope. The plotted points are partial residuals with 95 % confidence bands provided in shading
Amyloid statistically mediates the effect of CSF C3 on CSF ptau
We assessed whether CSF amyloid influences the relationship between CSF C3 and CSF ptau by statistically testing two theoretical models (illustrated in Fig. 3). Model A proposes that amyloid mediates the effect of CSF C3 on CSF ptau. Model B proposes that an interaction between CSF C3 and CSF amyloid alters CSF ptau levels.

Two possible models for the relationship between C3, amyloid, and ptau. Two possible models for the effects of C3, amyloid β, and APOE ε4 on ptau are illustrated as A and B. Solid arrows represent main effects of each variable and dotted arrows represent the interaction effect of two variables, all assessed in multivariate linear models which included age, sex, and CDR-SB score as covariates. Results for the two illustrated models are in Table 2. a Model A hypothesizes that C3’s effect on ptau will be mediated by amyloid. Roman numerals represent the following effects: (I) direct effect of C3 to predict ptau and (II) the hypothesized mediating effect of amyloid when added to the model in (I). b Model B suggests an alternate relationship whereby C3 and amyloid act synergistically to alter ptau. Roman numeral (I) corresponds to a hypothesized statistical interaction between C3 and amyloid to predict ptau levels
Full regression results from Models A and B (shown in Fig. 3) are provided in Table 2. We first tested Model A. We found that CSF C3 did not predict CSF ptau in the baseline model (p = 0.35). When CSF amyloid levels were added to the baseline model as a mediating variable, CSF C3 was significantly associated with CSF ptau (p = 0.04). Next we assessed Model B. In contrast to Model A, we found that there was no statistical interaction between CSF C3 and CSF amyloid on CSF ptau (p = 0.41).
Discussion
Among individuals with elevated intracranial C3 and increased APOE ε4 dosage, we found clinical evidence of elevated intracranial amyloid and neurofibrillary tangle pathology. We also found statistical evidence that amyloid mediates the effect of C3 on ptau pathology. Considered together, our results support a conceptual model of the AD pathogenic cascade where a synergistic relationship between the complement cascade (C3) and APOE ε4 results in elevated Alzheimer’s associated pathology and in turn, amyloid further regulates the effect of the complement cascade on ptau pathology (Fig. 4). These results suggest that a combination of inflammatory and pathologic biomarkers may help elucidate the neurodegenerative process underlying clinical AD.

Proposed biological model for the relationships between C3, APOE ε4, and amyloid on ptau. A proposed model for the relationships between C3, APOE ε4, and amyloid on ptau based on results from our statistical analyses. Solid arrows represent main effects of each variable and dotted arrows represent the interaction effect of two variables, all assessed in multivariate linear models which included age, sex, and CDR-SB score as covariates. Roman numerals represent the following effects: (I) interaction effect of C3 and APOE ε4 on amyloid (II) interaction effect of C3 and APOE ε4 on ptau, and (III) the effect of C3 on ptau, which is mediated by amyloid
Building on prior genetic evidence implicating a relationship between APOE and immune dysfunction, we found signs of elevated Alzheimer’s pathology in the presence of both APOE ε4 and elevated complement activation indicating that genetic propensity and inflammation may act synergistically to accelerate amyloid and tau pathology. In the presence of the statistical interaction, we found no effect of C3 on ptau suggesting that APOE may play an important role in influencing the relationship between inflammation and tau pathology. Interestingly, even with the interaction term in place, we found a significant effect of C3 on amyloid indicating that the complement cascade could influence amyloid pathology via both APOE-dependent and APOE-independent mechanisms. Additional experimental work will be required to delineate the precise mechanistic relationship between complement activation, APOE, amyloid and tau pathology.
Building on empirical research from prior studies [15, 19], our mediation analyses suggest that amyloid may influence the relationship between C3 and ptau. These results support the hypothesis that amyloid dysmetabolism regulates downstream pathological processes via inflammatory mechanisms. Recent work has shown that inflammatory processes are required for amyloid related synaptotoxicity [15] and other work has shown that amyloid can incite an inflammatory response [27] and specifically activate the complement cascade [1]. Taken together, our findings draw attention to the role of C3 in combination with amyloid and APOE as a potential regulator of downstream pathology.
Our findings suggest that a pro-inflammatory environmental milieu alters AD pathology in conjunction with genotype. This work further illustrates that inflammatory biomarkers along with genetic information may predict increased risk for AD [32] and other neurodegenerative disorders with genetic and immune components [21]. Clinically, this is important because it suggests that modifying specific components of the innate immune system or their downstream signaling pathways may serve as a target, which could ameliorate AD pathology. In addition to amyloid and tau metabolism and clearance, these results, in combination with prior studies [2, 17, 25, 30], underscore the importance of targeting inflammatory (specifically the complement cascade) processes for AD prevention.
Our study benefits from its use of a well-established, thoroughly characterized cohort of AD, MCI and cognitively normal healthy controls. In addition, our study benefits from using CSF, which is a biologically proximate measure of disease status and progression [3, 4]. A limitation of our study is its observational nature, which prevents us from determining a causative relationship. We cannot determine whether C3 causes, results from, or merely correlates with amyloid deposition. The specific biological mechanisms by which C3 and APOE ε4 influence ptau and amyloid remain to be determined. Our results will require follow-up in a larger, independent population-based cohort to determine the generalizability of this finding to diverse communities. Finally, our study highlights the need for future longitudinal studies that can directly test the causal effects suggested by our correlative analyses.
Conclusion
In summary, we found a statistical interaction between elevated C3 and APOE ε4 dosage on biomarkers indicative of elevated amyloid and tau pathology. Further, our mediation analyses indicate that C3 is associated with ptau only after adjusting for amyloid suggesting that amyloid may mediate the relationship between inflammation and tau pathology. Our findings underscore the importance of APOE ε4 in regulating the putative effects of C3 on both amyloid and ptau. More generally, this study highlights the importance of the innate immune system in modulating levels of the pathological proteins most associated with AD and its relevance as a biomarker and potential therapeutic target.
References
Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WST, Hampel H, Hull M, Landreth G, Lue LF, Mrak R, MacKenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi:10.1016/S0197-4580(00)00124-X.
Anand K, Sabbagh M. Early investigational drugs targeting tau protein for the treatment of Alzheimer’s disease. Expert Opin Investig Drugs. 2015;24:1355–60. doi:10.1517/13543784.2015.1075002.
Anoop A, Singh PK, Jacob RS, Maji SK. CSF Biomarkers for Alzheimer’s Disease Diagnosis. Int J Alzheimers Dis. 2010;2010:1–12. doi:10.4061/2010/606802
Blennow K. CSF biomarkers for Alzheimer’s disease: use in early diagnosis and evaluation of drug treatment. Expert Rev Mol Diagn. 2005;5:661–72. doi:10.1586/14737159.5.5.661.
Breheny P, Burchett W. Visualization of regression models using visreg. R Package. 2013;1–15.
Colton CA, Brown CM, Cook D, Needham LK, Xu Q, Czapiga M, Saunders AM, Schmechel DE, Rasheed K, Vitek MP. APOE and the regulation of microglial nitric oxide production: A link between genetic risk and oxidative stress. Neurobiol Aging. 2002;23:777–85. doi:10.1016/S0197-4580(02)00016-7.
Craig-Schapiro R, Kuhn M, Xiong C, Pickering EH, Liu J, Misko TP, Perrin RJ, Bales KR, Soares H, Fagan AM, Holtzman DM. Multiplexed immunoassay panel identifies novel CSF biomarkers for alzheimer’s disease diagnosis and prognosis. PLoS One. 2011. doi:10.1371/journal.pone.0018850.
Desikan RS, McEvoy LK, Thompson WK, Holland D, Brewer JB, Aisen PS, Sperling RA, Dale AM, Alzheimer's Disease Neuroimaging Initiative. Amyloid-β–Associated Clinical Decline Occurs Only in the Presence of Elevated P-tau. Arch Neurol. 2012;69:709–13. doi:10.1001/archneurol.2011.3354.
Desikan RS, McEvoy LK, Thompson WK, Holland D, Roddey JC, Blennow K, Aisen PS, Brewer JB, Hyman BT, Dale AM. Amyloid-β associated volume loss occurs only in the presence of phospho-tau. Ann Neurol. 2011;70:657–61. doi:10.1002/ana.22509.
Desikan RS, Thompson WK, Holland D, Hess CP, Brewer JB, Zetterberg H, Blennow K, Andreassen OA, McEvoy LK, Hyman BT, Dale AM. Heart fatty acid binding protein and Aβ-associated Alzheimer’s neurodegeneration. Mol Neurodegener. 2013;8:39. doi:10.1186/1750-1326-8-39.
Desikan RS, Thompson WK, Holland D, Hess CP, Brewer JB, Zetterberg H, Blennow K, Andreassen OA, McEvoy LK, Hyman BT, Dale AM. The role of clusterin in amyloid-β-associated neurodegeneration. JAMA Neurol. 2014;71:180–7. doi:10.1001/jamaneurol.2013.4560.
Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–98.
Gjoneska E, Pfenning AR, Mathys H, Quon G, Kundaje A, Tsai L-H, Kellis M. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nature. 2015;518:365–9. doi:10.1038/nature14252.
Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. 2015;16:358–72. doi:10.1038/nrn3880.
Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352(80-):712–6.
Hu WT, Watts KD, Tailor P, Nguyen TP, Howell JC, Lee RC, Seyfried NT, Gearing M, Hales CM, Levey AI, Lah JJ, Lee EK. CSF complement 3 and factor H are staging biomarkers in Alzheimer’s disease. Acta Neuropathol Commun. 2016;4:14. doi:10.1186/s40478-016-0277-8.
Huang Y, Mahley R. Apolipoprotein E4 Structure Correctors to Treat Alzheimer’s Disease and Other Neurodegenerative Disorders. San Francisco: Gladstone Institute; 2013
Karch CM, Cruchaga C, Goate AM. Alzheimer’s disease genetics: From the bench to the clinic. Neuron. 2014;83:11–26. doi:10.1016/j.neuron.2014.05.041.
Matarin M, Salih DA, Yasvoina M, Cummings DM, Guelfi S, Liu W, Nahaboo Solim MA, Moens TG, Paublete RM, Ali SS, Perona M, Desai R, Smith KJ, Latcham J, Fulleylove M, Richardson JC, Hardy J, Edwards FA. A Genome-wide Gene-Expression Analysis and Database in Transgenic Mice during Development of Amyloid or Tau Pathology. Cell Rep. 2015;10:633–44. http://dx.doi.org/10.1016/j.celrep.2014.12.041.
McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–44. doi:10.1212/WNL.34.7.939.
Miller ZA, Rankin KP, Graff-Radford NR, Takada LT, Sturm VE, Cleveland CM, Criswell LA, Jaeger PA, Stan T, Heggeli KA, Hsu SC, Karydas A, Khan BK, Grinberg LT, Gorno-Tempini ML, Boxer AL, Rosen HJ, Kramer JH, Coppola G, Geschwind DH, Rademakers R, Seeley WW, Wyss-Coray T, Miller BL. TDP-43 frontotemporal lobar degeneration and autoimmune disease. J Neurol Neurosurg Psychiatry. 2013;84:956–62. doi:10.1136/jnnp-2012-304644.
Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412–4. doi:10.1212/WNL.43.11.2412-a.
Nazem A, Sankowski R, Bacher M, Al-Abed Y. Rodent models of neuroinflammation for Alzheimer’s disease. J Neuroinflammation. 2015;12:74. doi:10.1186/s12974-015-0291-y.
Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, Blennow K, Soares H, Simon A, Lewczuk P, Dean R, Siemers E, Potter W, Lee VMY, Trojanowski JQ. Cerebrospinal fluid biomarker signature in alzheimer’s disease neuroimaging initiative subjects. Ann Neurol. 2009;65:403–13. doi:10.1002/ana.21610.
Siemers ER, Sundell KL, Carlson C, Case M, Sethuraman G, Liu-Seifert H, Dowsett SA, Pontecorvo MJ, Dean RA, Demattos R. Phase 3 solanezumab trials: Secondary outcomes in mild Alzheimer’s disease patients. Alzheimer’s Dement. 2015;1–11. doi:10.1016/j.jalz.2015.06.1893
Siuciak J. Biomarkers Consortium Data Primer. 2011.
Stancu I-C, Vasconcelos B, Terwel D, Dewachter I. Models of β-amyloid induced Tau-pathology: the long and “folded” road to understand the mechanism. Mol Neurodegener. 2014;9:51. doi:10.1186/1750-1326-9-51.
Toledo JB, Korff A, Shaw LM, Trojanowski JQ, Zhang J. Low levels of cerebrospinal fluid complement 3 and factor H predict faster cognitive decline in mild cognitive impairment. Alzheimers Res Ther. 2014;6:36. doi:10.1186/alzrt266.
Vitek MP, Brown CM, Colton CA. APOE genotype-specific differences in the innate immune response. Neurobiol Aging. 2009;30:1350–60. doi:10.1016/j.neurobiolaging.2007.11.014.
Walker D, Lue L-F. Anti-inflammatory and immune therapy for Alzheimer’s disease: current status and future directions. Curr Neuropharmacol. 2007;5:232–43. doi:10.2174/157015907782793667.
Wechsler D. Manual for Wechsler Memory Scale - Revised. Psychol Corp. 1987. doi: PCA-Converted #56
Yokoyama JS, Wang Y, Schork AJ, Thompson WK, Karch CM, Cruchaga C, McEvoy LK, Witoelar A, Chen C-H, Holland D, Brewer JB, Franke A, Dillon WP, Wilson DM, Mukherjee P, Hess CP, Miller Z, Bonham LW, Shen J, Rabinovici GD, Rosen HJ, Miller BL, Hyman BT, Schellenberg GD, Karlsen TH, Andreassen O a., Dale AM, Desikan RS. Association Between Genetic Traits for Immune-Mediated Diseases and Alzheimer Disease. JAMA Neurol. 2016;94158:1–7. doi:10.1001/jamaneurol.2016.0150.
ADNI General Procedures Manual. 2006. http://adni.loni.usc.edu/wp-content/uploads/2010/09/ADNI_GeneralProceduresManual.pdf
Acknowledgements
We thank the ADNI research participants for their longstanding dedication. Primary support for data analyses was provided by the Larry L. Hillblom Foundation 2012-A-015-FEL (JSY), AFTD Susan Marcus Memorial Fund Clinical Research Grant (JSY), NIA K01 AG049152 (JSY), and the Tau Consortium.
Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Author information
Authors and Affiliations
Consortia
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
LWB analyzed the data and wrote the manuscript; RSD obtained the data, designed the study, and edited the manuscript; JSY designed the study and edited the manuscript. All authors read and approved the final manuscript.
Additional file
Additional file 1:
ADNI methods. (DOCX 12 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Bonham, L.W., Desikan, R.S., Yokoyama, J.S. et al. The relationship between complement factor C3, APOE ε4, amyloid and tau in Alzheimer’s disease. acta neuropathol commun 4, 65 (2016). https://doi.org/10.1186/s40478-016-0339-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-016-0339-y