
Abstract
Epigenetic modifications are heritable changes in gene expression that do not directly alter DNA sequence. These modifications include DNA methylation, histone post-translational modifications, small and non-coding RNAs. Alterations in epigenetic profiles cause deregulation of fundamental gene expression pathways associated with carcinogenesis. The role of epigenetics in oropharyngeal squamous cell carcinoma (OPSCC) has recently been recognized, with implications for novel biomarkers, molecular diagnostics and chemotherapeutics. In this review, important epigenetic pathways in human papillomavirus (HPV) positive and negative OPSCC are summarized, as well as the potential clinical utility of this knowledge.
This material has never been published and is not currently under evaluation in any other peer-reviewed publication.
Similar content being viewed by others

Background
The field of epigenetics is defined as the study of heritable changes in gene expression without alteration of the DNA sequence. Epigenetic regulation has been implicated in multiple phenomena in both plants and animals. These include embryonic development, cell differentiation, imprinting, X chromosome inactivation, and various other gene expression patterns [1–3]. Aberrations within the epigenome have been implicated in several human diseases and most types of cancer [2, 4–8].
Epigenetic changes influence tumor development, proliferation, metastasis, and resistance to chemoradiotherapies [2, 4–7]. The primary mechanisms of epigenetic carcinogenesis involve DNA methylation, histone modifications, small and non-coding RNAs (ncRNA), which ultimately orchestrate complex gene regulatory pathways [1, 9–12]. Over the past decade, various aspects of these epigenetic processes have been shown to be of diagnostic and prognostic importance in oncology while offering novel therapeutic approaches. In more recent years, the role of epigenetics in oropharyngeal cancer has gained appreciation with new opportunities for translational research. The purpose of this review is to provide a general overview of epigenetic mechanisms in oropharyngeal squamous cell carcinoma (OPSCC) and knowledge of how this could be applied to novel treatment strategies.
Main text
HPV, epigenetics and oropharyngeal cancer
The incidence of HNSCCs has been gradually decreasing since the 1980s in association with the declining use of tobacco products. However, the incidence of OPSCC been steadily increasing over the past decade due to the rise in oncogenic HPV infections [13–16]. Early studies suggested HPV-positive tumors accounted for approximately 40–60% of OPSCCs [15, 17]. These rates have risen to greater than 80% in some regions [14, 18–24].
HPV is a nonenveloped, double-stranded DNA virus that is approximately 8000 base pairs (bp_ [25]. Over 150 serotypes of HPV have been associated with human epidermal and mucosal epithelium. HPV is classified as a sexually transmitted virus, with the majority of infections transmitted human-to-human via genital-to-genital or oral-to genital contact [26–28]. Viruses are categorized as either “low-risk” or “high-risk” with regard to their oncogenic potential. Low-risk types such as HPV 6 and 11 often manifest in the form of epithelial warts or oral papillomas [29]. Approximately 13 HPV serotypes are included in the high-risk category. Serotypes 16, 18, 31, and 33 have causal links with HPV-associated oropharyngeal carcinogenesis; however, HPV-16 has shown a much higher association with OPSCC (>90%) relative to other serotypes [13, 30–36]. Their viral genome consists of eight proteins (E1, E2, E4, E5, E6, E7, L1, L2) involved in viral replication, maintenance, and capsid structure [37, 38]. Viral proteins E6 and E7 have been shown to play an important role in carcinogenesis.
Upon infection of the host cell, HPV replicates its genome as extrachromosomal elements within the nucleus and integrates into the host genome. In cancer cells, chromosomal integration results in an increased expression and stabilization of viral oncoproteins E6 and E7 [39]. HPV integration sites are broadly distributed throughout the human genome, with various serotypes focusing on specific regions. HPV-16 favors integration at chromosomes 1, 2, 3, 5, 8, and 9 [40]. An interesting discovery by Akagi et al. has shown an association between the number of HPV integrants and their effects on neighboring gene expression. They found a higher number of integrants resulted in the direct disruption of neighboring genes via alterations in genomic structures [41]. This area of research is still in its infancy and further investigation into HPV integration-associated mutagenesis is required.
HPV-positive OPSCCs have distinct host gene expression profiles relative to HPV-negative OPSCCs [42–47]. These gene expression differences are thought to involve mechanisms of cancer tumorigenesis, proliferation, invasion, and metastasis. These differences are further reflected by distinct clinical presentations and responses to treatment modalities [13, 14, 17, 21, 48–57]. HPV-positivity is most often determined clinically by p16 overexpression, as an acceptable surrogate marker of this disease. The impact of p16 for diagnostics, prognostics and treatment stratification of OPSCC has highlighted the clinical utility of biomarkers for this disease [57–61]. In other cancers, novel epigenetic biomarkers have shown an increase in popularity for their potential specificity and biomarker-directed therapy [62].
DNA methylation, histone modifications and miRNA modifiers have all been shown to be important biomarkers and their role in OPSCC will be discussed in this review (Table 1). Specific DNA methylation patterns are showing increased promise as biomarkers, with some investigators some claiming superiority to other markers, as they hold higher levels of stability and can be amplified in a cost-effective manner [63, 64] Histone modifications may also have potential utilization as prognostic markers. For example, the methyltransferase enhancer of zeste homolog 2 (EZH2) and its substrate (H3K27 methylation) is overexpressed in numerous cancer types and is frequently indicative of a poor prognosis [7, 58, 65, 66]. However, histone modifications may be limited as biomarkers in isolation without knowing the extent of gene activity changes are associated with [7]. Despite being the most recently discovered of the epigenetic modifiers, miRs have shown some of the greatest potential as prognostic markers. MiRs have been shown to play central roles in tumorigenesis, invasion, metastasis, and responses to therapy [7, 67].
DNA methylation in oropharyngeal cancer
Alterations in DNA methylation occur via three mechanisms; hypomethylation, hypermethylation, and loss of imprinting [68, 69] Hypomethylation of gene promoter regions can result in the activation of various proto-oncogenes and chromatin restructuring [70]. DNA hypermethylation tends to be site-specific, targeting promoter CpG islands catalyzed by a set of enzymes known as DNA methyltransferases (DNMTs). There are three primary DNMTs; DNMT1, responsible for the maintenance of the standard epigenome, DNMT3a and DNMT3b, responsible for de novo methylation patterns [71–73]. DNA hypermethylation in cancers often results in the silencing of various genes, frequently tumor suppressors involved in cell cycle control, DNA repair mechanisms, and apoptosis [1, 74–76].
The most well documented epigenetic event occurs directly at the level of DNA, with 5’ methylation of CpG residues, primarily at gene promoter regions. In OPSCC, distinct host methylation profiles can be seen in HPV-positive cancers when compared to HPV-negative cancers [47, 77]. Nearly three times as much differentiation in methylation profiles can be seen between HPV-positive and HPV-negative disease when compared to adjacent somatic cells [78]. HPV-positive cancers have been found to have higher levels of methylation in specific regions of the genome (promoters, genic, and LINE-1). HPV-negative cancers show a much higher degree of genome-wide hypomethylation. It has been suggested that-negative cancers are far less genomically stable relative to their HPV-positive counterparts [47, 72, 79]. Genomic instability in turn leads to widespread deregulation of cellular processes characteristic of aggressive tumors.
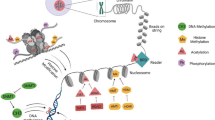
DNMT dysregulation is one potential mechanism for altered DNA methylation in OPSCCs. HPV-positive HNSCCs have shown increased expression in DNMT1 and DNMT3a, a pattern also seen in cervical cancers, suggesting a common mechanism of carcinogenesis by HPV [47, 80, 81]. This process is known to occur through HPV viral oncoproteins E6 and E7 (Fig. 1). HPV viral oncoprotein E6 causes the inhibition of the p53 tumor suppressor protein [48, 72, 82, 83]. As suggested by Anayannis et al. [82], this inhibition allows transcription factor Sp1 to be overexpressed to promote oncogenesis. The more notable interaction is seen by HPV E7 as it has been shown to directly interact with the tumor suppressor pRb, allowing the release of E2F (Fig. 1) from its protein complex to promote the transcription of DNMT1 [84]. E7 has also been shown to directly interact with DNMT1 in vitro, however, its implication requires further investigation [85].

Summary of epigenetic pathways involved in oropharygeal squamous cell carcinoma. Oncogenic human papillomavirus integrated into the human genome, resulting in the expression of HPV-associated proteins E6 and E7. This results in alterations of p53, Rb and Polycomb Repressive Complex (shown here including EZH2, SUZ12, EED and HOXD) related pathways with downstream epigenetic deregulation in OPSCC. Overexpression of P16INK4a occurs as a result of loss of Rb and is used as clinical surrogate marker for HPV-positive OPSCC. *FOXM1 and HOTAIR are presumed to have a role in OPSCC based on studies in OCSCC
Low expression of CDKN2A is seen in HPV-negative cancers, while high expression is found in HPV-associated disease [86]. Schlecht et al. has identified four CDKN2A loci downstream of the p16INK4A and p14ARF transcription start sites that are frequently hypermethylated in HPV-positive OPSCC, suggesting a potential mechanism for p16 overexpression in HPV-positive OPSCC [81]. This study also identified multiple Sp1 binding sites within the CDKN2A locus, further supporting the pRb/E2F pathways role in carcinogenesis (Fig. 1).
Histone post-translational modifications in oropharyngeal cancer
The structure of chromatin is dynamic and involves numerous pathways that regulate cell metabolism. The most basic unit of chromatin is the nucleosome, a 165 bp strand of DNA wrapped in a left-handed supercoil around an octamer core of histones approximately 1.7 turns. The octamer core consists of two of each four globular proteins; H2A, H2B, H3, and H4, with a singular histone (H1) providing linkage alongside DNA [72, 87]. At the amino-terminal ends of the histone proteins, various post-translational modifications can be applied. Through the modification of histone structures, gene expression is regulated via the allowance or blockage of access to various target genes to transcriptional machinery. These include acetylation, phosphorylation, methylation, ubiquitination, sumoylation, and ADP-ribosylation. These modifications, while all able to remodel chromatin structure, frequently show aberrations in acetylation and methylation profiles in human cancers [88–91]. The primary modulators of histone methylation and acetylation processes include histone methyltransferases (HMTs), histone demethylases (HDMs), histone acetyltransferases (HATs), and histone deacetylases (HDACs) [11, 12].
There are two known types of HATs; A-type within the nucleus, involved in the catalysis of transcription-related acetylation, and B-type within the cytosol, associated with newly generated histones. HATs facilitate the opening of chromatin for recruitment of transcriptional machinery by transferring acetyl groups from acetyl-CoA to specific lysine residues [92, 93]. Their overexpression has been associated with various cancers by aberrantly driving gene expression [94, 95]. Conversely, the overexpression of HDACs promotes deacetylation, resulting in abnormal gene silencing in the cancer epigenome [95–97].
Histone methylation works in a comparable process, where methylation, or multiple methylations, of lysine and arginine residues will result in structural rearrangements of chromatin [91, 92]. Like histone acetylation, aberrant expression of histone HMTs (enzymes that add methylation) and HDMs (enzymes that remove methylation) have been associated with carcinogenesis of various cancers [98–104]. Tumor cells frequently show altered methylation profiles on histone H3 at specific lysine sites including K4, K9, K27, K36, K79 and on histone H4 K20 [104–110]. Adding further complexity to this process, mono-, di- or trimethylation can occur on any given histone methylation site.
A recurring histone modifier in cancer literature is the methyltransferase EZH2, a catalytic subunit within the polycomb repressive protein complex 2 (PRC2) [101]. EZH2 catalyzes the trimethylation of lysine 27 on histone 3 (H3K27me3) and appears to have a regulatory role in cell proliferation and cell-cycle progression. Various cancers have displayed overexpression of EZH2 and it has been associated as a marker for malignancy potential and poor clinical prognosis, including HPV-positive OPSCC [111, 112]. HPV status and EZH2 overexpression are closely related, as EZH2 is a downstream target of E7 in vitro via the release of E2F from pocket proteins (Fig. 1) [113]. As proposed by Holland et. al., p53 suppression via E6 may also provide a mechanism for EZH2 overexpression [113]. As expected, HPV-positive (positive for p16INK4A) OPSCCs have genome-wide elevations of H3K27me3 [103]. An additional method of carcinogenesis by EZH2 overexpression may be through DNMT3A, as EZH2 has been shown to recruit DNMT3A; however, the de novo functionality of DNMT3A is not directly activated by this process [111].
B-cell–specific Moloney murine leukemia virus integration site 1 (BMI1) is a central component of the polycomb repressive complex 1 (PRC1). Overexpression has also been associated in carcinogenesis, functioning by stabilizing H3K27me3 and preventing transcriptional initiation [112, 114, 115]. Huber et al. have shown that BMI1 expression plays a potential role as a prognostic biomarker of OPSSC. Its aberrant expression in conjunction with p16 silencing is negatively correlated with recurrence-free survival in OPSCC [112].
Small and non-coding RNAs in oropharyngeal cancer
ncRNAs have been implicated in carcinogenesis and malignancy progression, with one of the first examples shown in chronic lymphocytic leukemia [116]. ncRNA are categorized based on their size. Nucleic acids less than 200 bp are known as small ncRNAs and greater than 200 bp are known as long non-coding RNAs (lncRNAs) [117]. Included within the small ncRNAs are the small interfering RNAs (siRNAs), micro RNAs (miRs), and PIWI-interacting RNAs (piRNAs) [118]. The majority of cancer research focuses on miRs as they have been shown to promote carcinogenesis through multiple pathways, including the direct interaction with mRNA, either through mRNA translation inhibition or mRNA degradation [67, 119]. Epigenetic silencing of specific miRs may have a causal link in carcinogenesis as they have shown to act as tumor suppressors. lncRNAs have no formal categorization. Most are organized based on the transcripts function; chromatin remodelling and transcription factor modulation. The majority of cancer literature focusing on the former, such as the well Xist transcript [6, 120].
Our current knowledge of the ncRNAs role in carcinogenesis is relatively limited, largely due to the novelty of the molecules discovery. The direct implication ncRNAs in OPSCC further confirm this, as known ncRNAs involved are limited to a few products. For the purposes of this section, the field of study will be expanded slightly to include ncRNAs implicated in other head and neck cancers, in addition to those found in OPSCC.
HPV status in tumors has shown distinct epigenetic profiles and clinical relevance. These distinctions is well outlined in the review by Lajer et al., who compared epigenetic profiles of HPV-positive cervical and head and neck cancers. They found a significant overlap in various miR clusters [121]. Sethi et al. outlined a comprehensive list of multiple miRs with aberrant expression patterns in head and neck cancers in addition to those mentioned by Lajer and colleagues [122]. This suggests distinct miR expressions are associated with HPV-associated cancers. This concept is further enforced by the direct interaction of some miRs, such as miR-15 and miR-16, with viral E6 and E7 [123].
One miR not acknowledged in literature, but which provides great interest, is miR-101. miR-101’s aberrant expression, namely its downregulation, has been involved in multiple cancers and has shown to mediate the overexpression of EZH2 [124–126]. The restoration of miR-101 via DNMT3A inhibition has also been shown to suppress lung tumorigenesis [126]. As both DNMT3A and EZH2 overexpression occurs in HPV-positive OPSCC, it may serve an important role in carcinogenesis [82].
Of the ncRNAs present within head and neck cancer literature, lncRNAs mirror the scarcity of miR counterparts. However, one lncRNA in particular, HOTAIR, has shown great promise as a potential biomarker. HOTAIR is a non-coding RNA transcript of 2.2 kb transcribed from the HOXC locus to transcriptionally silence HOXD [117, 127]. Interactions of HOTAIR have shown the 5’domain to bind to the PRC2 complex described previously as well the 3'domain binding the histone demethylase KDM1A (Fig. 1). These interactions potentially show methods of carcinogenesis, as its overexpression has been demonstrated in multiple cancer types including esophageal, nasopharyngeal, breast, pancreatic, and colorectal cancers [127–129] Overexpression of HOTAIR has been associated with an overall poor clinical prognosis, demonstrating increased lymph node metastasis and resistance to apoptosis. HOTAIR’s direct linkage to OPSCC requires further study. Other lncRNAs of interest are FTH1P3, PDIA3F and GTF2IRD2P1, as they have been associated with the progression and metastasis of oral SCC via the targeting of multiple tumor regulator genes [130].
Epigenetic chemotherapeutics
Aberrant events within the epigenome are suggested to occur more readily than structural gene modification through mutation. Given the reversible nature and specificity of epigenetic modifications, they have become an attractive target for cancer prevention and therapeutic intervention [2, 5, 73, 131]. Epigenetic chemotherapeutics are classified into two primary classes; histone deacetylase (HDAC) inhibitors and DNMT inhibitors. These classes are likely to expand as our knowledge of epigenetics advances and further chemotherapeutics are developed and tested. There are currently five USFDA-approved epigenetic chemotherapeutics on the market. Two are DNMT inhibitors; 5-acactidine (Vidaza) and 5-aza-2’-deoxycitidine (Decitabine). Three are HDAC inhibitors; suberoylanilide hydroxamic acid (Vorinostat), F-228 (Romidepsin), and LAQ-824 (Farydak). Current epigenetic chemotherapeutics in clinical trials or approved by the USFDA are summarized in Table 2.
Both DMNT inhibitors, Vidaza and Decitabine, are the only epidrugs that have been approved for the treatment of patients with acute myeloid leukemia (AML) and myelodyplastic syndrome (MDS) [132]. Vidaza and Decitabine are nucleoside analogs of cytosine modified in position five of their pyrimidine ring [133]. Upon exposure, Vizdaza is incorporated into RNA and Decitabine is incorporated into DNA where they disrupt interactions between DNMTs and DNA. During this process, a covalent bond is formed with DNMT triggering a DNA damage signal and targeting the DNMT for degradation. When utilized in clinical practice, their applicability encountered major limitations. These are characterized by poor bioavailability, poor activity with solid tumors, severe toxic effects, instability in physiological media, and gross non-specific changes to epigenome to both normal and cancer cells. Fortunately, several new specific inhibitors are under development. Of these are [133] MG98, small molecule RG108, nucleoside analog Zebularine, and arsenic trioxide. These inhibitors have shown increased specificity, chemical stability, increased bioavailability, and lower cytotoxic effects [132, 133].
HDAC inhibitors are regularly divided into four different groups based on their chemical structure. These are hydroximates, cyclic peptides, aliphatic acids, and benzamides. Within the hydroximate class are two USFDA approved agents, Vorinostat and the newly approved Farydak, as well as JNJ-26481585 (Quisinostat) currently in clinical trials [134–136]. Aliphatic acids contain three agents currently in clinical trials; valproic acid, phenylbutyrate, and Belinostat. Cyclic peptides contain the USFDA approved Romidepsin and benzamides contain three agents in the clinical trial stages; MS-275 (entinostat), MGCD-0103 (Mocetinostat), CI-994 [131, 135]. The mechanisms of HDAC inhibitors are not fully understood, but are thought to alter gene expression via regulation at both epigenetic and post-translational modification levels [137, 138]. Evidence also suggests HDAC inhibition may alter tumor progression by inhibiting tumor angiogenesis [138]. HDAC inhibitors are well tolerated relative to other epigenetic chemotherapeutics. However, these drugs still display poor activity against solid tumors when utilized on their own. Suggested application is specific timing in conjunction with current chemotherapeutics [139, 140] 4. Another novel histone modifier inhibitor in the clinical trial stage is EPZ-6438 (Epizyme), an inhibitor of histone methyltransferase DOT1L [141–143]. While still requiring further study for conclusive data, initial studies suggest its efficacy and tolerance.
The use of miR’s as potential targets for chemotherapeutics is still in its infancy. Multiple studies have shown the significant effects of upregulation and downregulation of specific miRs on cancer. Of note is miR-21 s direct role in tumorigenesis following upregulation and reduced tumor survival and progression following its downregulation [144].
Conclusions
As with other cancers, epigenetics has a fundamental role in the pathophysiology of OPSCC. HPV positive and negative OPSCCs have distinct epigenetic profiles, consistent with their pathological and clinical differences. An understanding of epigenetics in OPSCC provides opportunities for the discovery and application of novel biomarkers and treatments.
Abbreviations
- BMI-1:
-
B-cell–specific Moloney murine leukemia virus integration site 1
- Bp:
-
Base pair
- DNA:
-
Deoxyribonucleic acid
- DNMT:
-
DNA methyltransferase
- DZNep:
-
3-deazanoplanocin
- EZH2:
-
Enhancer of zeste homolog 2
- HAT:
-
Histone acetyltransferase
- HDAC:
-
Histone deacetylase
- HDM:
-
Histone demethylase
- HMT:
-
Histone methyltransferase
- HPV:
-
Human papillomavirus
- lncRNA:
-
Long non-coding RNA
- miRNA:
-
micro RNA
- ncRNA:
-
Non-coding RNA
- piRNA:
-
PIWI-interacting RNA
- PRC:
-
Polycomb repressive complex
- RNA:
-
Ribonucleic acid
- siRNA:
-
Small interfering RNA
- USFDA:
-
United States Food and Drug Association
References
Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–53.
Kanwal R, Gupta S. Epigenetic modifications in cancer. Clin Genet. 2012;81:303–11.
Lyon MF. Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature. 1961;190:372–3.
Perroud N, Zewdie S, Stenz L, Adouan W, Bavamian S, Prada P, et al. Methylation of serotonin receptor 3A in ADHD, borderline personality, and bipolar disorders: link with severity of the disorders and childhood maltreatment. Depress Anxiety. 2016;33:45–55.
Kanwal R, Gupta K, Gupta S. Cancer epigenetics: an introduction. Methods Mol Biol. 2015;1238:3–25.
Ducasse M, Brown MA. Epigenetic aberrations and cancer. Mol Cancer. 2006;5:60.
Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36.
Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27.
Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–80.
Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev. 2009;23:781–3.
Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705.
Mattick JS, Makunin IV. Non-coding RNA. Hum Mol Genet. 2006;15(Spec No 1):R17–29.
Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tân PF, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med. 2010;363:24–35.
Gillison ML, Chaturvedi AK, Anderson WF, Fakhry C. Epidemiology of human papillomavirus-positive head and neck squamous cell carcinoma. J Clin Oncol. 2015;33:3235–42.
D'Souza G, Kreimer AR, Viscidi R, Pawlita M, Fakhry C, Koch WM, et al. Case–control study of human papillomavirus and oropharyngeal cancer. N Engl J Med. 2007;356:1944–56.
Chaturvedi AK, Engels EA, Pfeiffer RM, Hernandez BY, Xiao W, Kim E, et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol. 2011;29:4294–301.
Seikaly H, Biron VL, Zhang H, O'Connell DA, Côté DWJ, Ansari K, et al. The role of primary surgery in the treatment of advanced oropharyngeal cancer. Head Neck. 2015;:n/a–n/a.
Biron VL, Kostiuk M, Isaac A, Puttagunta L, O'Connell DA, Harris J, et al. Detection of human papillomavirus type 16 in oropharyngeal squamous cell carcinoma using droplet digital polymerase chain reaction. Cancer. 2016;:n/a–n/a.
Pytynia KB, Dahlstrom KR, Sturgis EM. Epidemiology of HPV-associated oropharyngeal cancer. Oral Oncol. 2014;50:380–6.
Westra WH. The changing face of head and neck cancer in the 21st century: the impact of HPV on the epidemiology and pathology of oral cancer. Head Neck Pathol. 2009;3:78–81.
Clark J, Jeffery CC, Zhang H, Cooper T, O'Connell DA, Harris J, et al. Correlation of PET-CT nodal SUVmax with p16 positivity in oropharyngeal squamous cell carcinoma. J Otolaryngol Head Neck Surg. 2015;44:37.
Cooper T, Biron VL, Fast D, Tam R, Carey T, Shmulevitz M, et al. Oncolytic activity of reovirus in HPV positive and negative head and neck squamous cell carcinoma. J Otolaryngol Head Neck Surg. 2015;44:8.
Wasserman JK, Rourke R, Purgina B, Caulley L, Dimitroulakis J, Corsten M, et al. HPV DNA in saliva from patients with SCC of the head and neck is specific for p16-positive oropharyngeal tumours. J Otolaryngol Head Neck Surg. 2017;46:3.
Isaac A, Kostiuk M, Zhang H, Lindsay C, Makki F, O'Connell DA, et al. Ultrasensitive detection of oncogenic human papillomavirus in oropharyngeal tissue swabs. J Otolaryngol Head Neck Surg. 2017;46:5.
Hausen zur H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2:342–50.
D’Souza G, Cullen K, Bowie J, Thorpe R, Fakhry C. Differences in oral sexual behaviors by gender, age, and race explain observed differences in prevalence of oral human papillomavirus infection. Liu X, editor. PLoS One. 2014;9:e86023.
D'Souza G, Gross ND, Pai SI, Haddad R, Anderson KS, Rajan S, et al. Oral human papillomavirus (HPV) infection in HPV-positive patients with oropharyngeal cancer and their partners. J Clin Oncol. 2014;32:2408–15.
Marzouki HZ, Biron VL, Harris J, O'Connell D, Seikaly H. Human papillomavirus-associated oropharyngeal squamous cell carcinoma and anogenital cancers in men: Epidemiologic evaluation of association. Head Neck. 2016;:n/a–n/a.
Markowitz LE, Liu G, Hariri S, Steinau M, Dunne EF, Unger ER. Prevalence of HPV after introduction of the vaccination program in the united states. Pediatrics. 2016;137:1–9.
Lambert R, Sauvaget C, de Camargo Cancela M, Sankaranarayanan R. Epidemiology of cancer from the oral cavity and oropharynx. Eur J Gastroenterol Hepatol. 2011;23:633–41.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29.
Mehanna H, Beech T, Nicholson T, El-Hariry I, McConkey C, Paleri V, et al. Prevalence of human papillomavirus in oropharyngeal and nonoropharyngeal head and neck cancer--systematic review and meta-analysis of trends by time and region. Eisele DW, editor. Head Neck. 2013;35:747–55.
Hartwig S, Syrjänen S, Dominiak-Felden G, Brotons M, Castellsague X. Estimation of the epidemiological burden of human papillomavirus-related cancers and non-malignant diseases in men in Europe: a review. BMC Cancer. 2012;12:30.
Pezzuto F, Buonaguro L, Caponigro F, Ionna F, Starita N, Annunziata C, et al. Update on head and neck cancer: current knowledge on epidemiology, risk factors, molecular features and novel therapies. Oncology. 2015;89:125–36.
Curado MP, Boyle P. Epidemiology of head and neck squamous cell carcinoma not related to tobacco or alcohol. Curr Opin Oncol. 2013;25:229–34.
Gillison ML, Shah KV. Human papillomavirus-associated head and neck squamous cell carcinoma: mounting evidence for an etiologic role for human papillomavirus in a subset of head and neck cancers. Curr Opin Oncol. 2001;13:183–8.
Yugawa T, Kiyono T. Molecular mechanisms of cervical carcinogenesis by high-risk human papillomaviruses: novel functions of E6 and E7 oncoproteins. Rev Med Virol. 2009;19:97–113.
Gillison ML, Restighini C. Anticipation of the impact of human papillomavirus on clinical decision making for the head and neck cancer patient. Hematol Oncol Clin North Am. 2015;29:1045–60.
Jeon S, Lambert PF. Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of E6 and E7 mRNAs: implications for cervical carcinogenesis. Proc Natl Acad Sci U S A. 1995;92:1654–8.
Kumar Gupta A, Kumar M. HPVbase--a knowledgebase of viral integrations, methylation patterns and microRNAs aberrant expression: As potential biomarkers for Human papillomaviruses mediated carcinomas. Sci Rep. 2015;5:12522.
Akagi K, Li J, Broutian TR, Padilla-Nash H, Xiao W, Jiang B, et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res. 2014;24:185–99.
Biron VLV, Mohamed AA, Hendzel MJM, Underhill DDA, Seikaly HH. Epigenetic differences between human papillomavirus-positive and -negative oropharyngeal squamous cell carcinomas. J Otolaryngol Head Neck Surg. 2012;41 Suppl 1:S65–70.
Barber BR, Biron VL, Klimowicz AC, Puttagunta L, Côté DW, Seikaly H. Molecular predictors of locoregional and distant metastases in oropharyngeal squamous cell carcinoma. J Otolaryngol Head Neck Surg. 2013;42:53.
Klussmann JP, Mooren JJ, Lehnen M, Claessen SMH, Stenner M, Huebbers CU, et al. Genetic signatures of HPV-related and unrelated oropharyngeal carcinoma and their prognostic implications. Clin Cancer Res. 2009;15:1779–86.
Martinez I, Wang J, Hobson KF, Ferris RL, Khan SA. Identification of differentially expressed genes in HPV-positive and HPV-negative oropharyngeal squamous cell carcinomas. Eur J Cancer. 2007;43:415–32.
Lohavanichbutr P, Houck J, Fan W, Yueh B, Mendez E, Futran N, et al. Genomewide gene expression profiles of HPV-positive and HPV-negative oropharyngeal cancer: potential implications for treatment choices. Arch Otolaryngol Head Neck Surg. 2009;135:180–8.
Sartor MA, Dolinoy DC, Jones TR, Colacino JA, Prince MEP, Carey TE, et al. Genome-wide methylation and expression differences in HPV(+) and HPV(−) squamous cell carcinoma cell lines are consistent with divergent mechanisms of carcinogenesis. Epigenetics. 2011;6:777–87.
Mantovani F, Banks L. The human papillomavirus E6 protein and its contribution to malignant progression. Oncogene. 2001;20:7874–87.
Cooper T, Biron V, Adam B, Klimowicz AC, Puttagunta L, Seikaly H. Prognostic utility of basaloid differentiation in oropharyngeal cancer. J Otolaryngol Head Neck Surg. 2013;42:57.
Xu CC, Biron VL, Puttagunta L, Seikaly H. HPV Status and second primary tumours in Oropharyngeal Squamous Cell Carcinoma. J Otolaryngol Head Neck Surg. 2013;42:36.
Lohaus F, Linge A, Tinhofer I, Budach V, Gkika E, Stuschke M, et al. HPV16 DNA status is a strong prognosticator of loco-regional control after postoperative radiochemotherapy of locally advanced oropharyngeal carcinoma: results from a multicentre explorative study of the German Cancer Consortium Radiation Oncology Group (DKTK-ROG). Radiother Oncol. 2014;113:317–23.
McDonald JT, Johnson-Obaseki S, Hwang E, Connell C, Corsten M. The relationship between survival and socio-economic status for head and neck cancer in Canada. J Otolaryngol Head Neck Surg. 2014;43:2.
Ziemann F, Arenz A, Preising S, Wittekindt C, Klussmann JP, Engenhart-Cabillic R, et al. Increased sensitivity of HPV-positive head and neck cancer cell lines to x-irradiation ± Cisplatin due to decreased expression of E6 and E7 oncoproteins and enhanced apoptosis. Am J Cancer Res. 2015;5:1017–31.
Zhang H, Seikaly H, Abele JT, Jeffery DT, Harris JR, O'Connell DA. Metabolic tumour volume as a prognostic factor for oral cavity squamous cell carcinoma treated with primary surgery. J Otolaryngol Head Neck Surg. 2014;43:33.
Roskies M, Kay-Rivest E, Mascarella MA, Sultanem K, Mlynarek A, Hier M. Survival outcomes in patients with oropharyngeal cancer treated with carboplatin/paclitaxel and concurrent radiotherapy. J Otolaryngol Head Neck Surg. 2016;45:50.
Fu TS, Foreman A, Goldstein DP, de Almeida JR. The role of transoral robotic surgery, transoral laser microsurgery, and lingual tonsillectomy in the identification of head and neck squamous cell carcinoma of unknown primary origin: a systematic review. J Otolaryngol Head Neck Surg. 2016;45:28.
Osazuwa-Peters N, Tutlam NT. Knowledge and risk perception of oral cavity and oropharyngeal cancer among non-medical university students. J Otolaryngol Head Neck Surg. 2016;45:5.
Idris S, Lindsay C, Kostiuk M, Andrews C, Côté DWJ, O'Connell DA, et al. Investigation of EZH2 pathways for novel epigenetic treatment strategies in oropharyngeal cancer. J Otolaryngol Head Neck Surg. 2016;45:54.
Murray S, Ha MN, Thompson K, Hart RD, Rajaraman M, Snow SL. A different entity: a population based study of characteristics and recurrence patterns in oropharyngeal squamous cell carcinomas. J Otolaryngol Head Neck Surg. 2015;44:30.
Chung CH, Zhang Q, Kong CS, Harris J, Fertig EJ, Harari PM, et al. p16 protein expression and human papillomavirus status as prognostic biomarkers of nonoropharyngeal head and neck squamous cell carcinoma. J Clin Oncol. 2014;32:3930–8.
Lewis JS. p16 Immunohistochemistry as a standalone test for risk stratification in oropharyngeal squamous cell carcinoma. Head Neck Pathol. 2012;6 Suppl 1:S75–82.
Yan W, Herman JG, Guo M. Epigenome-based personalized medicine in human cancer. Epigenomics. 2016;8:119–33.
Wei SH, Balch C, Paik HH, Kim Y-S, Baldwin RL, Liyanarachchi S, et al. Prognostic DNA methylation biomarkers in ovarian cancer. Clin Cancer Res. 2006;12:2788–94.
Koturbash I, Beland FA, Pogribny IP. Role of epigenetic events in chemical carcinogenesis—a justification for incorporating epigenetic evaluations in cancer risk assessment. Toxicol Mech Methods. 2011;21:289–97.
Zhao L, Yu Y, Wu J, Bai J, Zhao Y, Li C, et al. Role of EZH2 in oral squamous cell carcinoma carcinogenesis. Gene. 2014;537:197–202.
Kidani K, Osaki M, Tamura T, Yamaga K, Shomori K, Ryoke K, et al. High expression of EZH2 is associated with tumor proliferation and prognosis in human oral squamous cell carcinomas. Oral Oncol. 2009;45:39–46.
Orellana EA, Kasinski AL. MicroRNAs in cancer: a historical perspective on the path from discovery to therapy. Cancers (Basel). 2015;7:1388–405.
Robertson KD, Jones PA. DNA methylation: past, present and future directions. Carcinogenesis. 2000;21:461–7.
Bestor TH. The DNA, methyltransferases of mammals. Hum Mol Genet. 2000;9:2395–402.
Ehrlich M, Lacey M. DNA hypomethylation and hemimethylation in cancer. Adv Exp Med Biol. 2013;754:31–56.
Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–57.
van Kempen PM, Noorlag R, Braunius WW, Stegeman I, Willems SM, Grolman W. Differences in methylation profiles between HPV-positive and HPV-negative oropharynx squamous cell carcinoma. Epigenetics. 2014;9(2):194–203.http://doi.org/10.4161/epi.26881.
Bakhtiar SM, Ali A, Barh D. Epigenetics in head and neck cancer. Methods Mol Biol. 2015;1238:751–69.
Gupta GP, Massagué J. Cancer metastasis: building a framework. Cell. 2006;127:679–95.
Teh M-T, Gemenetzidis E, Patel D, Tariq R, Nadir A, Bahta AW, et al. FOXM1 induces a global methylation signature that mimics the cancer epigenome in head and neck squamous cell carcinoma. Yeudall A, editor. PLoS One. 2012;7:e34329.
Kornberg RD. Chromatin structure: a repeating unit of histones and DNA. Science. 1974;184:868–71.
Parfenov M, Pedamallu CS, Gehlenborg N, Freeman SS, Danilova L, Bristow CA, et al. Characterization of HPV and host genome interactions in primary head and neck cancers. Proc Natl Acad Sci U S A. 2014;111:15544–9.
Lleras RA, Smith RV, Adrien LR, Schlecht NF, Burk RD, Harris TM, et al. Unique DNA methylation loci distinguish anatomic site and HPV status in head and neck squamous cell carcinoma. Clin Cancer Res. 2013;19:5444–55.
Richards KL, Zhang B, Baggerly KA, Colella S, Lang JC, Schuller DE, et al. Genome-wide hypomethylation in head and neck cancer is more pronounced in HPV-negative tumors and is associated with genomic instability. Fugmann SD, editor. PLoS One. 2009;4:e4941.
Sawada M, Kanai Y, Arai E, Ushijima S, Ojima H, Hirohashi S. Increased expression of DNA methyltransferase 1 (DNMT1) protein in uterine cervix squamous cell carcinoma and its precursor lesion. Cancer Lett. 2007;251:211–9.
Schlecht NF, Ben-Dayan M, Anayannis N, Lleras RA, Thomas C, Wang Y, et al. Epigenetic changes in the CDKN2A locus are associated with differential expression of P16INK4A and P14ARF in HPV-positive oropharyngeal squamous cell carcinoma. Cancer Med. 2015;4:342–53.
Anayannis NVJ, Schlecht NF, Belbin TJ. Epigenetic mechanisms of human papillomavirus-associated head and neck cancer. Arch Pathol Lab Med. 2015;139:1373–8.
Werness BA, Levine AJ, Howley PM. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science. 1990;248:76–9.
McCance DJ. Transcriptional regulation by human papillomaviruses. Curr Opin Genet Dev. 2005;15:515–9.
Burgers WA, Blanchon L, Pradhan S, de Launoit Y, Kouzarides T, Fuks F. Viral oncoproteins target the DNA methyltransferases. Oncogene. 2007;26:1650–5.
Hafkamp HC, Speel EJM, Haesevoets A, Bot FJ, Dinjens WNM, Ramaekers FCS, et al. A subset of head and neck squamous cell carcinomas exhibits integration of HPV 16/18 DNA and overexpression of p16INK4A and p53 in the absence of mutations in p53 exons 5–8. Int J Cancer. 2003;107:394–400.
Hammoud SS, Cairns BR, Jones DA. Epigenetic regulation of colon cancer and intestinal stem cells. Curr Opin Cell Biol. 2013;25:177–83.
Fenrick R, Hiebert SW. Role of histone deacetylases in acute leukemia. J Cell Biochem Suppl. 1998;30–31:194–202.
Dueñas-González A, Lizano M, Candelaria M, Cetina L, Arce C, Cervera E. Epigenetics of cervical cancer. An overview and therapeutic perspectives. Mol Cancer. 2005;4:38.
Brownell JE, Allis CD. An activity gel assay detects a single, catalytically active histone acetyltransferase subunit in Tetrahymena macronuclei. Proc Natl Acad Sci U S A. 1995;92:6364–8.
Baylin SB, Höppener JW, de Bustros A, Steenbergh PH, Lips CJ, Nelkin BD. DNA methylation patterns of the calcitonin gene in human lung cancers and lymphomas. Cancer Res. 1986;46:2917–22.
Qiu W, Schönleben F, Li X, Ho DJ, Close LG, Manolidis S, et al. PIK3CA mutations in head and neck squamous cell carcinoma. Clin Cancer Res. 2006;12:1441–6.
Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–47.
O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–43.
Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449:682–8.
Pogo BG, Allfrey VG, Mirsky AE. RNA synthesis and histone acetylation during the course of gene activation in lymphocytes. Proc Natl Acad Sci U S A. 1966;55:805–12.
Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, Coetzee GA, et al. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell. 2006;9:435–43.
Gannon OM, Merida de Long L, Endo-Munoz L, Hazar-Rethinam M, Saunders NA. Dysregulation of the repressive H3K27 trimethylation mark in head and neck squamous cell carcinoma contributes to dysregulated squamous differentiation. Clin Cancer Res. 2013;19:428–41.
Højfeldt JW, Agger K, Helin K. Histone lysine demethylases as targets for anticancer therapy. Nat Rev Drug Discov. 2013;12:917–30.
Young LC, Hendzel MJ. The oncogenic potential of Jumonji D2 (JMJD2/KDM4) histone demethylase overexpression. Biochem Cell Biol. 2013;91:369–77.
McCabe MT, Creasy CL. EZH2 as a potential target in cancer therapy. Epigenomics. 2014;6:341–51.
Verma SK, Tian X, LaFrance LV, Duquenne C, Suarez DP, Newlander KA, et al. Identification of potent, selective, cell-active inhibitors of the histone lysine methyltransferase EZH2. ACS Med Chem Lett. 2012;3:1091–6.
Biron VL, Côté DWJ, Seikaly H. Oropharyngeal squamous cell carcinoma and human papillomavirus-associated cancers in women: epidemiologic evaluation of association. J Otolaryngol Head Neck Surg. 2011;40 Suppl 1:S65–9.
Biron VL, McManus KJ, Hu N, Hendzel MJ, Underhill DA. Distinct dynamics and distribution of histone methyl-lysine derivatives in mouse development. Dev Biol. 2004;276:337–51.
Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37:391–400.
Yang H, Mizzen CA. The multiple facets of histone H4-lysine 20 methylation. Biochem Cell Biol. 2009;87:151–61.
Du Y, Carling T, Fang W, Piao Z, Sheu JC, Huang S. Hypermethylation in human cancers of the RIZ1 tumor suppressor gene, a member of a histone/protein methyltransferase superfamily. Cancer Res. 2001;61:8094–9.
Koturbash I, Simpson NE, Beland FA, Pogribny IP. Alterations in histone H4 lysine 20 methylation: implications for cancer detection and prevention. Antioxid Redox Signal. 2012;17:365–74.
Thompson LL, Guppy BJ, Sawchuk L, Davie JR, McManus KJ. Regulation of chromatin structure via histone post-translational modification and the link to carcinogenesis. Cancer Metastasis Rev. 2013;32:363–76.
McManus KJ, Biron VL, Heit R, Underhill DA, Hendzel MJ. Dynamic changes in histone H3 lysine 9 methylations: identification of a mitosis-specific function for dynamic methylation in chromosome congression and segregation. J Biol Chem. 2006;281:8888–97.
Rush M, Appanah R, Lee S, Lam LL, Goyal P, Lorincz MC. Targeting of EZH2 to a defined genomic site is sufficient for recruitment of Dnmt3a but not de novo DNA methylation. Epigenetics. 2009;4:404–14.
Huber GF, Albinger-Hegyi A, Soltermann A, Roessle M, Graf N, Haerle SK, et al. Expression patterns of Bmi-1 and p16 significantly correlate with overall, disease-specific, and recurrence-free survival in oropharyngeal squamous cell carcinoma. Cancer. 2011;117:4659–70.
Holland D, Hoppe-Seyler K, Schuller B, Lohrey C, Maroldt J, Dürst M, et al. Activation of the enhancer of zeste homologue 2 gene by the human papillomavirus E7 oncoprotein. Cancer Res. 2008;68:9964–72.
Hyland PL, McDade SS, McCloskey R, Dickson GJ, Arthur K, McCance DJ, et al. Evidence for alteration of EZH2, BMI1, and KDM6A and epigenetic reprogramming in human papillomavirus type 16 E6/E7-expressing keratinocytes. J Virol. 2011;85:10999–1006.
Shao Z, Raible F, Mollaaghababa R, Guyon JR, Wu CT, Bender W, et al. Stabilization of chromatin structure by PRC1, a Polycomb complex. Cell. 1999;98:37–46.
Pekarsky Y, Balatti V, Palamarchuk A, Rizzotto L, Veneziano D, Nigita G, et al. Dysregulation of a family of short noncoding RNAs, tsRNAs, in human cancer. Proc Natl Acad Sci U S A. 2016;113:5071–6.
Maass PG, Luft FC, Bähring S. Long non-coding RNA in health and disease. J Mol Med. 2014;92:337–46.
González-Ramírez I, Soto-Reyes E, Sánchez-Pérez Y, Herrera LA, García-Cuellar C. Histones and long non-coding RNAs: the new insights of epigenetic deregulation involved in oral cancer. Oral Oncol. 2014;50:691–5.
Melo SA, Sugimoto H, O'Connell JT, Kato N, Villanueva A, Vidal A, et al. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell. 2014;26:707–21.
Brown CJ, Lafreniere RG, Powers VE, Sebastio G, Ballabio A, Pettigrew AL, et al. Localization of the X inactivation centre on the human X chromosome in Xq13. Nature. 1991;349:82–4.
Lajer CB, Garnæs E, Friis-Hansen L, Norrild B, Therkildsen MH, Glud M, et al. The role of miRNAs in human papilloma virus (HPV)-associated cancers: bridging between HPV-related head and neck cancer and cervical cancer. Br J Cancer. 2012;106:1526–34.
Sethi N, Wright A, Wood H, Rabbitts P. MicroRNAs and head and neck cancer: reviewing the first decade of research. Eur J Cancer. 2014;50:2619–35.
Zheng Z-M, Wang X. Regulation of cellular miRNA expression by human papillomaviruses. Biochim Biophys Acta. 1809;2011:668–77.
Friedman JM, Jones PA, Liang G. The tumor suppressor microRNA-101 becomes an epigenetic player by targeting the polycomb group protein EZH2 in cancer. Cell Cycle. 2009;8:2313–4.
Friedman JM, Liang G, Liu C-C, Wolff EM, Tsai YC, Ye W, et al. The putative tumor suppressor microRNA-101 modulates the cancer epigenome by repressing the polycomb group protein EZH2. Cancer Res. 2009;69:2623–9.
Yan F, Shen N, Pang J, Xie D, Deng B, Molina JR, et al. Restoration of miR-101 suppresses lung tumorigenesis through inhibition of DNMT3a-dependent DNA methylation. Cell Death Dis. 2014;5:e1413.
Ge X-S, Ma H-J, Zheng X-H, Ruan H-L, Liao X-Y, Xue W-Q, et al. HOTAIR, a prognostic factor in esophageal squamous cell carcinoma, inhibits WIF-1 expression and activates Wnt pathway. Cancer Sci. 2013;104:1675–82.
Li C-H, Chen Y. Targeting long non-coding RNAs in cancers: progress and prospects. Int J Biochem Cell Biol. 2013;45:1895–910.
Li C-H, Xiao Z, Tong JH-M, To K-F, Fang X, Cheng AS, et al. EZH2 coupled with HOTAIR to silence MicroRNA-34a by the induction of heterochromatin formation in human pancreatic ductal adenocarcinoma. Int J Cancer. 2017;140:120–9.
Zhang S, Tian L, Ma P, Sun Q, Zhang K, GuanchaoWang, et al. Potential role of differentially expressed lncRNAs in the pathogenesis of oral squamous cell carcinoma. Arch Oral Biol. 2015;60:1581–7.
Pan LN, Lu J, Huang B. HDAC inhibitors: a potential new category of anti-tumor agents. Cell Mol Immunol. 2007;4:337–43.
Gros C, Fleury L, Nahoum V, Faux C, Valente S, Labella D, et al. New insights on the mechanism of quinoline-based DNA Methyltransferase inhibitors. J Biol Chem. 2015;290:6293–302.
Gros C, Fahy J, Halby L, Dufau I, Erdmann A, Gregoire J-M, et al. DNA methylation inhibitors in cancer: recent and future approaches. Biochimie. 2012;94:2280–96.
Venugopal B, Baird R, Kristeleit RS, Plummer R, Cowan R, Stewart A, et al. A phase I study of quisinostat (JNJ-26481585), an oral hydroxamate histone deacetylase inhibitor with evidence of target modulation and antitumor activity, in patients with advanced solid tumors. Clin Cancer Res. 2013;19:4262–72.
Duvic M. Histone deacetylase inhibitors for cutaneous T-cell lymphoma. Dermatol Clin. 2015;33:757–64.
Duvic M, Dimopoulos M. The safety profile of vorinostat (suberoylanilide hydroxamic acid) in hematologic malignancies: A review of clinical studies. Cancer Treat Rev. 2016;43:58–66.
Gui C-Y, Ngo L, Xu WS, Richon VM, Marks PA. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc Natl Acad Sci U S A. 2004;101:1241–6.
Sandor V, Senderowicz A, Mertins S, Sackett D, Sausville E, Blagosklonny MV, et al. P21-dependent g(1)arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br J Cancer. 2000;83:817–25.
Modesitt SC, Sill M, Hoffman JS, Bender DP, Gynecologic Oncology Group. A phase II study of vorinostat in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol. 2008;109:182–6.
Mascolo M, Siano M, Ilardi G, Russo D, Merolla F, De Rosa G, et al. Epigenetic disregulation in oral cancer. Int J Mol Sci. 2012;13:2331–53.
Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield VM, et al. hDOT1L links histone methylation to leukemogenesis. Cell. 2005;121:167–78.
Stein RA. Epigenetic therapies - a new direction in clinical medicine. Int J Clin Pract. 2014;68:802–11.
Stein EM, Tallman MS. Mixed lineage rearranged leukaemia: pathogenesis and targeting DOT1L. Curr Opin Hematol. 2015;22:92–6.
Medina PP, Nolde M, Slack FJ. OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature. 2010;467:86–90.
Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science. 1996;272:408–11.
Guo M, Yan W. Epigenetics of gastric cancer. Methods Mol Biol. 2015;1238:783–99.
la Rosa AH-D, Acker M, Swain S, Manoharan M. The role of epigenetics in kidney malignancies. Cent European J Urol. 2015;68:157–64.
Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang K-C, Xiao Y, et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol Cancer Ther. 2014;13:842–54.
Autophagic effect of SAM-competitive EZH2 inhibitors on cancer cells. Can Cell Microenviron [Internet]. 2015;:1–4. Available from: http://www.smartscitech.com/index.php/CCM/article/view/551.
Chi P, Allis CD, Wang GG. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer. 2010;10:457–69.
Felsenfeld G. A brief history of epigenetics. Cold Spring Harb Perspect Biol. 2014;6:a018200.
Acknowledgements
Not applicable.
Funding
Not applicable.
Availability of data and materials
Not applicable.
Authors’ contributions
CL performed primary aspects of the literature review. HS and VLB were involved in manuscript preparation. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lindsay, C., Seikaly, H. & Biron, V.L. Epigenetics of oropharyngeal squamous cell carcinoma: opportunities for novel chemotherapeutic targets. J of Otolaryngol - Head & Neck Surg 46, 9 (2017). https://doi.org/10.1186/s40463-017-0185-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40463-017-0185-3