
Abstract
Prostate cancer (PCa) is one of the main causes of cancer-related death in men. In the present immunotherapy era, several immunotherapeutic agents have been evaluated in PCa with poor results, possibly due to its low mutational burden. The recent development of chimeric antigen receptor (CAR)-T cell therapy redirected against cancer-specific antigens would seem to provide the means for bypassing immune tolerance mechanisms. CAR-T cell therapy has proven effective in eradicating hematologic malignancies and the challenge now is to obtain the same degree of in solid tumors, including PCa. In this study we review the principles that have guided the engineering of CAR-T cells and the specific prostatic antigens identified as possible targets for immunological and non-immunological therapies. We also provide a state-of-the-art overview of CAR-T cell therapy in PCa, defining the key obstacles to its development and underlining the mechanisms used to overcome these barriers. At present, although there are still many unanswered questions regarding CAR-T cell therapy, there is no doubt that it has the potential to become an important treatment option for urological malignancies.
Similar content being viewed by others
Introduction
Prostate cancer (PCa) remains one of the main causes of cancer-related death in men. Although it is often a manageable tumor, around 20% of patients develop metastases and the disease eventually evolves into metastatic castration-resistant PCa (mCRPC) [1]. In the last few years, new drugs have been evaluated for the treatment of mCRPC and, following Food and Drug Administration (FDA) approval of sipuleucel-T (Dendreon Corporation), several studies have been conducted to assess the role of immunotherapeutic agents, including new checkpoint inhibitors, in this setting [2, 3]. No immune checkpoint inhibitor (as monotherapy) has demonstrated efficacy in PCa thus far [4,5,6]. In particular, no overall survival (OS) benefit has been observed in patients treated with ipilimumab [7,8,9], whereas monotherapies directed against PD1 or PD-L1 have only demonstrated limited response in PCa patients, probably due to an immunologically cold PCa microenvironment [10]. Moreover, the role of PD-L1 status in PCa patients is controversial. Recently, Li et al. demonstrated its expression as a negative independent prognostic factor in PCa patients. PD-L1 overexpression has also been correlated with high Gleason scores and androgene receptor positivity [11]. PD-L1 overexpression appears to be higher in metastatic sites than in primary PCa [12], especially in enzalutamide-pretreated patients [13]. Interestingly, Calagua et al. did not find any difference in PD-L1 expression between treated and untreated mPCa patients [14]. At ASCO GU 2019, results from the CheckMate 650 phase II trial (NCT02985957) revealed the efficacy of the combination of the CTLA4-inhibitor ipilimumab and the PD-1-inhibitor nivolumab. In a cohort of mCRPC patients pretreated with taxane and hormone therapy, 10% (3/30) showed a response at a median follow-up of 13.5 months, while in the other cohort pretreated with 2 hormone therapy lines, 25% (8/32) had a response at a median follow-up of 11.9 months [15].
In this scenario, the development of genetically engineered T cells capable of overcoming cancer immunological tolerance would represent an important step forward in cancer research. In the current ‘new era’ of cancer immunotherapy, clinical trials have been carried out to verify the potential for using chimeric antigen receptor (CAR) T cells to identify and eliminate malignant cells. CAR-T is a molecule consisting of a tumor antigen-binding domain fused to an intracellular signaling domain and costimulatory molecules [16]. For this reason, antigen-identification is not major histocompatibility complex (MHC)-restricted, as is the case of T cell receptor (TCR)-mediated antigen recognition.
The first studies were conducted on hematological tumors and showed high response rates and durability of remission in chronic lymphocytic leukemia (CLL) acute lymphoblastic leukemia (ALL), and refractory B cell lymphoma [17,18,19,20,21,22,23]. Such excellent results led to FDA approval of CD19-directed CAR-T cells for the treatment of relapsed/refractory pediatric and young-adult diffuse large B cell lymphoma (DLBCL), also sparking off research into solid tumors. The characteristic of being monoclonal diseases and the consequent identification of the same target antigen for all neoplastic cells is probably the main reason for the success of CAR-T cell therapy in hematological malignancies. In solid tumors, polyclonality, physical barriers and tumor microenvironment probably account for the difficulties in obtaining the same promising results. However, the recent identification of specific PCa membrane antigens can be considered the starting point that has led to the development of cell-directed immunotherapy.
In this review we provide an in-depth overview of CAR-T cell therapy in PCa and suggest strategies to further improve current results.
CAR-T structure
PCa is associated with a low mutational burden. CAR-T cells are synthetic molecules in which the effector function of T lymphocytes combines with the ability of antibodies to identify specific antigens. Thus, CAR T cells do not require antigen presentation by antigen presenting cells (APC) and can recognize intact proteins. Consequently, the creation of genetically engineered T cells redirected to tumor antigens bypasses several mechanisms of immunological tolerance [24]. Recent studies have shown that the “optimal” T cell population for the generation of CAR-T cells are poorly differentiated cells, i.e. the earliest memory T cells (stem cells memory T). The modifications occurring during T cell maturation process (in particular, loss of co- stimulatory receptors and erosion of telomeres) make differentiated T cells less suitable [25,26,27].
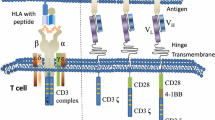
CAR molecules can be divided into 3 components: 1) an extracellular domain, which is involved in antigen identification. This zone is composed of a single-chain fragment variable (scFv) that (specifically) recognizes tumor-associated antigens (TAA). scFV is fixed on T cell by a 2) transmembrane domain, composed of a transmembrane region of CD3, CD8, CD28 or FcεRI. This region is connected to the 3) intracellular zone which is composed of the intracytoplasmic region of CD8, CD28 or CD137 and CD3ζ. This last zone comprises the immune receptor tyrosine-based activation motif (ITAM) which, in turn, plays a fundamental role in signal transduction aimed at activating T cells [28].
To date, in vitro transfection technology is the standard method to transfect CAR molecules into T lymphocytes. Transfection can be achieved through viral (retro- or slow virus) or non-viral (transposon and mRNA electrotransfection) methods.
Generally, CARs are classified into 4 types based on molecular complexity (Fig. 1): the first type comprises CARs with only a simple receptor divided into the above-mentioned 3 components (scFv, transmembrane domain and intracellular zone). These CAR-T constructs permit T cell activation but, given the lack of a costimulatory molecule, this first generation failed to obtain significant results in terms of persistence of T-lymphocyte activation in blood circulation [29,30,31]. To overcome this problem, a second CAR generation was developed by inserting the intracellular domain of a costimulatory protein, such as CD28, CD27, CD134 or CDB7. Another costimulatory molecule (CD28, 4-1BB, or CD3ζ) was added to develop a third CAR generation aimed at increasing the extent of T-cell activation [32]. The fourth generation of these molecules (also known as TRUCK, i.e. T cells redirected for universal cytokine-mediated killing, or CAR-T cells armed with immune stimulatory cytokine) has both a costimulatory element and proinflammatory factor, such as interleukin (IL)-12, which increases T-cell efficacy [33]. In fact, the presence of IL-12 counterbalances the immunosuppressive action of the tumor microenvironment by inducing a shift in the T-cell response towards a T helper-1 type [34, 35]. However, the fourth generation of CAR is not limited to IL-12 alone, different types of molecules having been developed for use in the construction of TRUCKs. These include cytokines such as IL-15 (similar to IL-12, this interleukin enhances the development of T-memory stem cells) [36] and IL-18 [37], and also constitutively active cytokine receptors such as IL-7 receptor (C7R) whose aim is to overcome the risk of cytokine toxicity [38]. Other molecules tested in TRUCKs are knock-out genes (PD-1 or DGK) and knock-in genes (TRAC or CXCR4), their aim to improve CAR expression and anti-tumor activity [39, 40]. Controlled and inducible systems (Syn/Notch) and multiantigen combinations (HER2 + IL13Rα2) have also been used to prevent antigen escape [41].

Different characteristics of chimeric antigen receptor (CAR) generations. scFv, single-chain fragment variable
Prostate TAAs and known immunotherapy strategies
The identification of prostate TAAs is the first step towards developing an effective CAR-T cell therapy. An ideal antigen should be constitutive and specifically expressed by cancer cells to enable CAR-T cells to develop a cancer-specific immunologic response, thus sparing healthy tissue [42, 43]. In PCa the group of protein preferentially expressed by malignant cells are prostate-specific antigen (PSA), prostatic acid phosphatase (PAP), prostate stem cell antigen (PSCA), T-cell receptor gamma alternate reading frame protein (TARP), transient receptor potential (trp)-p8 and prostate-specific membrane antigen (PSMA). In recent years, several studies have used prostate TAAs as a target for the induction of an immunological response in PCa patients [44, 45] (Table 1).
PSA
Preclinical studies in transgenic mice have shown that PSA, a kallikrein-like serin-protease almost exclusively expressed by prostate epithelial cells, induces a specific T-cell response. Arredouani et al. generated a transgenic mouse expressing human PSA in the prostate and crossed it to the human leucocyte antigen (HLA-A2.1 transgenic mouse to assess whether androgen deprivation affects T-cell response, observing a significant increase in PSA-specific cytotoxic lymphocytes, especially after androgen ablation [46].
PAP
PAP is secreted by benign and malignant prostate cells and is more highly expressed in Gleason score 6 and 7 tumors then in higher Gleason score tumors. It is not really a specific prostate antigen because it is expressed in the placenta, kidneys and testes, and also in gastric, breast and colon cancer. Kantoff et al. presented the results of a phase III trial that led to the FDA approval of sipuleucel-T for the treatment of asymptomatic or minimally symptomatic mCRPC. In the trial, PCa patients in the experimental arm were treated with APCs pre-exposed in vitro to PA2024, a fusion protein consisting of human granulocyte-macrophage colony-stimulating factor and PAP [47]. The sipuleucel-T patient group experienced a 22% relative reduction in risk of death compared to the placebo group, the reduction representing a 4.1-month improvement in median survival.
Patients enrolled in the experimental arm experienced chills, fever, and headache as adverse events.
PSCA
PSCA is a cell surface glycoprotein expressed by prostate cells and carcinomas with a higher Gleason score. Several studies have evaluated the activity of in vitro-generated tumor-reactive CTL response by HLA-A2-restricted anti-prostate stem cell antigen (PSCA) peptides [48,49,50]. Other studies have been conducted on the TRAMP mouse model with PSCA-expressing PCa. Following vaccination with a viral vector encoding PSCA, TRAMP mice developed an antigen-specific CTL response that subsequently inhibited PCa progression [51, 52]. PSCA has also been evaluated as a target for antibody-based immunotherapy. Both conjugated and unconjugated anti-PSCA antibodies have shown activity against PCa cells, resulting in cytotoxicity and regression of xenografts in mice [53,54,55]. Taking into account the potential immunological effect of PSCA, Morgenroth et al. modified T cells by transducing chimeric antigen receptors that specifically recognize PSCA. The engineered T cells efficiently lysed PSCA-expressing cells [56].
PSMA
PSMA is a transmembrane glycoprotein (also known as FOLH1) with relative specificity as a PCa cell-surface ligand [57]. Moreover, its expression progressively increases as higher grade tumors [58] and correlates with castration-resistant disease. Its role in positron emission tomography (PET) was confirmed by Caroli et al. in a prospective series of patients with biochemical recurrence of PCa, the authors reporting the superior performance and safety of 68Ga-PSMA PET/CT over choline PET/CT [59].
The potential of PSMA has been investigated in targeted therapy and in immunotherapy, some studies showing that HLA-A2-restricted PSMA-derived peptides induce antitumoral CTL responses in vitro [60,61,62,63]. Other studies in vitro and in xenograft models have evaluated PSMA as a target molecule for immunotherapy with conjugated and unconjugated antibodies directed against PSMA-expressing PCa cells [64,65,66,67]. Over the last decade, PSMA has been studied in vitro and in vivo to optimize antigenic stimulation of T-cell response through engineered T-cells expressing chimeric anti-PSMA immunoglobulin-T-cell-receptor constructs. In our Institute, PSMA conjugated with 177Lutetium (177Lu-PSMA) is being evaluated for safety and efficacy in an ongoing single-arm phase II trial of radiometabolic therapy for advanced castration-resistant PCa (NCT03454750). The phase III open label VISION Trial is currently recruiting 750 patients with progressive PSMA-positive PCa pretreated with abiraterone or enzalutamide and one or 2 lines of taxane-based chemotherapies. Patients are randomized to receive either 177Lu-PSMA-617 plus best supportive/best standard care or best supportive/best standard care alone, the aim being to compare overall survival (OS) between the two arms (NCT03511664). Other studies, not yet recruiting, have been designed to examine the safety, tolerability and efficacy of the combination of 177Lu-PSMA with pembrolizumab (NCT0365844) or olaparib (NCT03874884).
Prostein, TARP, trp-p8
Prostein and trp-p8 are transmembrane proteins expressed in normal and malignant prostate tissue, while TARP is present in the mitochondria of PCa cells. Several preclinical trials have evaluated their efficacy in stimulating CTL response [68,69,70,71]. Recently, a pilot study of PSMA and TARP peptide vaccine with poly IC-LC (Hiltonol) as adjuvant was performed in HLA-A2 (+) hormone-naive PCa patients with elevated PSA after initial definitive treatment (NCT00694551). The aim of the study was to establish the safety and toxicity of varying doses of the vaccine and to assess its impact on PSA. The results are still incomplete but no serious adverse events have been recorded to date.
CAR-T cells in metastatic PCa
Few studies evaluating CAR-T cell therapy in metastatic prostate cancer (mPCa) have been conducted to date (Table 2), PSMA and PSCA representing the most important candidates as CAR-T cell-targeted antigens.
PSMA-CAR-T cells
In vitro and in vivo models have shown that PSMA-CAR-T cells proliferate and recognize PSMA+ cells [72, 73]. An in vivo study by Zuccolotto et al. on the activity of PSMA-CAR-T cells in mPCa revealed that these cells can survive in mice with diabetes/severe combined immunodeficiency. The treatment proved capable of eradicating mPCa in the preclinical setting [74].
Second-generation CAR-T cells show a better killing effect than those of the previous generation and represent a novel immune-targeted approach for mPCa [75]. Slovin et al. investigated an anti-PSMA CAR-T cell therapy in a phase I clinical trial of mPCa patients (NCT01140373). The authors assessed the safety of various doses and developed a protocol for ex-vivo transduction, expansion and clinical administration of the treatment [76]. Another phase I trial (NCT03089203) is currently testing the safety and feasibility of dual PSMA-specific/TGFβ-resistant, lentivirally transduced, CAR-modified autologous T cells (CART-PSMA-TGFβRDN cells) [77].
Ma et al. fabricated a second-generation anti-PSMA CAR-T cell therapy by inserting the co-stimulator CD28, testing it in mice [75]. Tumor volume decreased significantly (virtually disappearing after 3 weeks) in mice inoculated with anti-PSMA CAR-T cells with respect to those inoculated with non-transduced T cells. Zhang et al. recently developed a CAR-T cell therapy specific for PSMA and resistant to transforming growth factor β (TGF-β) by infecting CD8+ T cells from mCRPC patients with a retroviral construct. The construct carried an anti-PSMA chimeric T-cell receptor (TCR) gene and a dominant negative TGF-β type II gene, the former conferring T-cell specificity and the latter, resistance to TGF-β-mediated suppression of cytotoxic T lymphocytes. The engineered CAR-T cells had ganciclovir as a safety mechanism thanks to their expression of HSV1 thymidine kinase. The CAR-T cells increased 23.4-fold in 21 days and ganciclovir decreased survival to 1.5% in 5 days. In a mouse xenograft model, treatment with PSMA-specific and TGF-β-insensitive CAR-T cells led to lysis of PSMA-expressing PC3 tumors but not of normal PC3 tumors. Tumor apoptosis, CD8+ cell infiltration and increased interferon-gamma (IFNγ) and interleukin-2 (IL-2) levels were only seen in PSMA-positive PC3 tumors [78].
Hassani et al. recently constructed a CAR-T cell therapy against PSMA using camelid nanobody (VHH) [79]. For the first time scFvs of murine origin were not used in the CAR-T cell structure because of its limitations with regard to the immunogenicity of mouse antigens in humans and the relatively large size of scFvs. The specificity of VHH-CAR-T cells against PSMA+cells was confirmed by the increase observed in interleukin-2 (IL-2) cytokine and in CD69 expression (around 38%) [79].
PSCA-CAR-T cells
With regard to PSCA, a first-generation CAR with scFv of 7F5 antibody led to the activation of an anti-tumor response in mice [80]. In a recent study, Priceman et al. evaluated the role of co-stimulation in PSCA-CAR-T cell activity. Comparing the co-stimulation activity of both CD28 and 4-1BB, the authors found that the latter molecule was more effective in activating T-cells than the former, thus paving the way for further analyses in this field [81].
A PSCA-CAR-T cell-mediated delay in tumor growth was obtained in mice using 1G8 and Ha1–4.117 antibodies [82], suggesting that CAR-T cell cytotoxicity may not be sufficient for in vivo treatment. A potential solution might be to develop a combined low-affinity PSCA-CAR-T and high-affinity PSMA-CAR-T cell therapy. Tested by Kloss et al., this combination proved capable of eliminating double-positive T cells, suggesting its potential as a new therapeutic strategy for PCa [83].
Diabodies and bispecific T-cell engagers (BITEs)
Another approach could be to use bispecific antibodies (diabodies) or BITEs [85]. These constructs not only bind to the minimal binding domains (single-chain fragment variables, scFvs) of monoclonal antibodies for CD3ϵ T-cell receptor-associated molecule on the T-cell surface, but also to a specific antigen expressed on the surface of cancer cells. Concurrent engagement of both the specific antigen and CD3 leads to tumor cell lysis through the activation of cytotoxic T-cells, regardless of the TCR-mediated specificity of these cells [86]. The relative specificity and sensitivity of BiTE and CAR constructs has been compared in preclinical models [87]. Given that BiTEs may be beneficial in cancers in which a specific epitope is overexpressed compared with normal tissue, as described by Stone et al., this approach has also been studied in PCa.
Several studies developed and evaluated in vitro the efficacy of these novel antibodies in targeting PSCA and PSMA [83, 88, 89]. However, some failed to block cancer cell proliferation in animal models, only delaying tumor growth, suggesting that diabodies used as a single treatment do not achieve a durable cellular memory response [34]. Despite this, administration of the humanized bispecific antibody MOR209/ES414 in murine xenograft models of human PCa led to the inhibition of tumor growth and increased survival, decreasing PSA expression only in adoptively transferred human T cells [90] A phase I study is ongoing to identify the maximum tolerated dose and to test the clinical activity of ES414 in mPCa patients [NCT02262910].
More recently, AMG 160, a fully human, half-life extended (HLE) BiTE targeting PSMA in PCa cells and CD3 in T cells, demonstrated antitumor activity in xenograft models [91]. Based on these data, a phase I study is underway to evaluate its activity in mPCa patients (NCT03792841). At the 2019 Annual ASCO Meeting, Hummel et al. reported that the PSMA x CD3 BiTE pasotuxizumab demonstrated an acceptable safety profile and dose-dependent clinical activity in mPCa patients [92]. Moreover, theirs was the first study demonstrating BITE clinical activity in solid tumors, 2 long-term responses described in the dose escalation cohort (NCT01723475).
Epithelial cell adhesion molecules (EpCAM)
EpCAM, also known as CD326, is a stem cell antigen expressed by several solid tumors, including PCa [93, 94]. An EpCAM-CD3 bispecific antibody was recently approved in Europe for patients with malignant ascites. Using this molecule as a TAA, Deng et al. developed EpCAM-specific CARs which not only proved capable of killing PC3M prostate cells (overexpressing EpCAM) but also of prolonging survival in PC3 prostate cells (underexpressing EpCAM). Further investigation is warranted into the role of this molecule in mPCa [84].
Problems relating to use of CAR-T cell therapy in PCa
The use of CAR-T cells for the treatment of non-hematological tumors exposes the patient to risks that could limit their use in clinical practice. Perhaps the most important risk is the presence of several structures in solid tumors (i.e. extracellular matrix, tumor stroma) that limit the contact between CAR-T cells and the tumor itself [95]. For example, bone is the most frequent site of PCa metastasis. Within this context, the tumor microenvironment enhances aberrant angiogenesis mediated by vascular endothelial growth factor receptor (VEGF) [96]. Shi et al. demonstrated that a combination of immunotherapy and angiogenesis-normalizing treatments increases the efficacy of immunotherapeutic agents [97].
Another issue is the inhibitory tumor microenvironment. Several studies have demonstrated that solid tumors express a higher concentration of programmed death-ligand 1 PD-L1, tryptophan 2,3-dioxygenase, indoleamine 2,3-dioxygenase, IL-10 and regulatory T-cells (Tregs) [98,99,100,101,102,103,104]. As Tregs overexpress TGF-β, blocking TGF-β activity could help to improve T cell activity [105]. Kloss et al. evaluated TGF-β overexpression in mice models of aggressive mPCa, reporting enhanced T-cell proliferation, cytokine secretion, in vivo survival and efficacy in destroying cancer cells [77]. As previously reported, bone is the most frequent site of PCa metastasization and different cytokines have been studied for their potential to enable T cells to access bone metastases. In 2000, Kantele et al. used mild radiation treatment or cyclophosphamide chemotherapy to stimulate mPCa cells to express chemokine (C-X-C motif) ligand (CXCL) 12, also known as stromal cell-derived factor (SDF)-1 [106], which is involved in T cell migration to and adhesion on activated endothelium [107].
More recently, some authors evaluated the possibility of inserting a chemokine receptor gene into CAR-T cells. For example, engineering CXCL12 ligand, i.e. C-X-C motif receptor (CXCR)-4, into CAR-T cells could increase the percentage of CAR molecules reaching reach tumor cells [108, 109].
Based on the same hypothesis, other studies have evaluated CAR-T cells engineered to secrete different chemokines, such as CCL2 (involved in tumor homing and vascularization) [110]. Another way of enhancing T cell activity could be to add an immune checkpoint inhibitor to treatment. Combination therapy with CAR-T cells and an anti-PD1 antibody demonstrated higher T-cell activation in a transgenic Her2 mouse model [111]. In PCa, androgen-deprivation therapy combined with T-cells has been evaluated, an in vitro study demonstrating higher cytotoxic activity and proliferation rates of T cells using this treatment strategy. Sanchez et al. showed the feasibility in vitro of a combination of androgen-deprivation therapy and CAR-T cells [112]. Such findings may be attributable to androgen-mediated apoptosis and, consequently, to an increase in TAAs which, in turn, stimulates T-helper activation. For the same reasons, using radiotherapy to induce apoptosis could help to overcome immune inhibition by the tumor microenvironment [34].
However, the solution to CAR-T-related problems is not limited to removing physical or chemical “barriers”. In fact, toxicities caused by the new immunological approach are sometimes difficult to manage. The majority of data on CAR-T-related toxicities originate from hematological trials. Neurological and cardiovascular toxicities, cytokine release syndrome, tumor lysis syndrome and macrophage activation syndrome have all been observed in studies using CD19 CAR-T cells [113,114,115,116]. In PCa, the use of prostate-specific antigens could limit systemic immune-related adverse events (IRAEs). Moreover, several molecular options are currently being developed to further reduce the risks of such adverse events. For example, the abovementioned study by Kloss et al. [77] evaluated a combination of low-affinity PSCA-CAR-T cells with high-affinity PSMA-CAR-T cells. Another way of overcoming potential toxicities is to insert an inducible suicide gene into CAR-T cells with the aim of destroying CAR-T cells in the event of serious toxicity. Within this context, Di Stasi et al. demonstrated the role of caspase-9 in inducing T-cell apoptosis [117]. Moreover, some studies suggest that inserting CARs into NK cells or into γδ T-cells could substantially limit the risk of IRAEs [118, 119]. Other recent findings on hematological [120] and solid tumors, including PCa, indicate that the problem might be resolved by developing nanoparticles for CAR-T delivery [28, 121]. However, all of the above issues also limit the use of CAR-T cell therapy in mPCa [122].
Conclusions
Although TAA-targeting CARs have shown interesting results in pre-clinical studies on mPCa, their clinical use is associated with significant risks for the patient and requires further in-depth investigation. It is therefore essential to draw up toxicity management plans and to identify biomarkers that can predict toxicities such as cytokine-release syndrome. It is still open to debate whether clinical CAR-T cell programs should be managed by bone marrow transplantation teams or by disease-specific teams. This is especially important for solid tumors, where the ideal situation would be to have a team whose expertise comprises bone marrow transplantation in specific diseases.
Numerous issues remain to be resolved, e.g. best TAA to induce safe and effective T-cell activation; best CAR-T cells to use (NK, αβ T cell, γδ T cell); best way to reduce IRAEs in mPCa treated with CAR-T cell treatment. Moreover, is CAR-T cell treatment better than monotherapy, and if not, what is the best combination treatment to improve T-cell activation (CAR-T + antiandrogens; CAR-T + radiotherapy)? Are these combinations safe? Which kind of patient could benefit from CAR-T treatment and which might not? Why? The development of this promising treatment strategy in PCa will depend on these questions being answered, hopefully in the near future.
Availability of data and materials
Not applicable.
Abbreviations
- ALL:
-
Acute lymphoblastic leukemia
- APC:
-
Antigen presenting cells
- BITEs:
-
Bispecific T-cell engagers
- CAR:
-
Chimeric antigen receptor
- CLL:
-
Chronic lymphocytic leukemia
- DLBCL:
-
Diffuse large B cell lymphoma
- IRAEs:
-
Immune-related adverse events
- ITAM:
-
Immune receptor tyrosine-based activation motif
- mCRPC:
-
Metastatic castration-resistant PCa
- MHC:
-
Major histocompatibility complex
- mPCa:
-
Metastatic prostate cancer
- NED:
-
Neuroendocrine differentiation
- PAP:
-
Prostatic acid phosphatase
- PCa:
-
Prostate cancer
- PSA:
-
Prostate-specific antigen
- PSCA:
-
Prostate stem cell antigen
- PSMA:
-
Prostate-specific membrane antigen
- scFv:
-
Single-chain fragment variable
- SDF:
-
Stromal cell-derived factor
- TAA:
-
Tumor-associated antigens
- TARP:
-
T-cell receptor gamma alternate reading frame protein
- TCR:
-
T cell receptor
- VEGF:
-
Vascular endothelial growth factor receptor
References
Siegel RL, Miller KD, Fedewa SA, Ahnen DJ, Meester RGS, Barzi A, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin. 2017;67:177–93. https://doi.org/10.3322/caac.21395.
Drake CG. Prostate cancer as a model for tumour immunotherapy. Nat Rev Immunol. 2010;10:580–93. https://doi.org/10.1038/nri2817.
Schepisi G, Farolfi A, Conteduca V, Martignano F, De Lisi D, Ravaglia G, et al. Immunotherapy for prostate cancer: Where we are headed. Int J Mol Sci. 2017;18:e2627. https://doi.org/10.3390/ijms18122627.
Topalian S, Hodi F. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54. https://doi.org/10.1056/NEJMoa1200690.
Kwon ED, Drake CG, Scher HI, Fizazi K, Bossi A, van den Eertwegh AJM, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184–043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014;15:700–12. https://doi.org/10.1016/S1470-2045(14)70189-5.
Hansen AR, Massard C, Ott PA, Haas NB, Lopez JS, Ejadi S, et al. Pembrolizumab for advanced prostate adenocarcinoma: Findings of the KEYNOTE-028 study. Ann Oncol. 2018;29:1807–13. https://doi.org/10.1093/annonc/mdy232.
Gao J, Ward JF, Pettaway CA, Shi LZ, Subudhi SK, Vence LM, et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat Med. 2017;23:551–5. https://doi.org/10.1038/nm.4308.
Beer TM, Kwon ED, Drake CG, Fizazi K, Logothetis C, Gravis G, et al. Randomized, double-blind, phase III trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naive castration-resistant prostate cancer. J Clin Oncol. 2017;35:40–7.
Reese Z, Straubhar A, Pal SK, Agarwal N. Ipilimumab in the treatment of prostate cancer. Futur Oncol. 2015;11:27–37. https://doi.org/10.2217/fon.14.196.
Bilusic M, Madan RA, Gulley JL. Immunotherapy of prostate cancer: Facts and hopes. Clin Cancer Res. 2017;23:6764–70. https://doi.org/10.1158/1078-0432.CCR-17-0019.
Li Y, Huang Q, Zhou Y, He M, Chen J, Gao Y, et al. The clinicopathologic and prognostic significance of programmed cell death ligand 1 (PD-L1) expression in patients with prostate cancer: A systematic review and meta-analysis. Front Pharmacol. 2019;9:1494. https://doi.org/10.3389/fphar.2018.01494.
Haffner MC, Guner G, Taheri D, Netto GJ, Palsgrove DN, Zheng Q, et al. Comprehensive evaluation of programmed death-ligand 1 expression in primary and metastatic prostate cancer. Am J Pathol. 2018;188:1478–85. https://doi.org/10.1016/j.ajpath.2018.02.014.
Bishop JL, Sio A, Angeles A, Roberts ME, Azad AA, Chi KN, et al. PD-L1 is highly expressed in Enzalutamide resistant prostate cancer. Oncotarget. 2015;6:234–42.
Calagua C, Russo J, Sun Y, Schaefer R, Lis R, Zhang Z, et al. Expression of PD-L1 in hormone-naïve and treated prostate cancer patients receiving neoadjuvant abiraterone acetate plus prednisone and leuprolide. Clin Cancer Res. 2017;23:6812–22. https://doi.org/10.1158/1078-0432.CCR-17-0807.
Sharma P, Pachynski RK, Narayan V, Flechon A, Gravis G, Galsky MD, et al. Initial results from a phase II study of nivolumab (NIVO) plus ipilimumab (IPI) for the treatment of metastatic castration-resistant prostate cancer (mCRPC; CheckMate 650). J Clin Oncol. 2019;37(Suppl):142(Abstr.). https://doi.org/10.1200/JCO.2019.37.7_suppl.142.
Morello A, Sadelain M, Adusumilli PS. Mesothelin-targeted CARs: Driving T cells to solid Tumors. Cancer Discov. 2016;6(2):133–46. https://doi.org/10.1158/2159-8290.CD-15-0583.
Porter DL, Levine BL, Kalos M, Bagg A, June CH, Abramson T. Chimeric Antigen Receptor-Modified T Cells in Chronic Lymphoid Leukemia From the Abramson Cancer Center (D). N Engl J Med. 2011;365:725–33.
Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7:303ra139. https://doi.org/10.1126/scitranslmed.aac5415.
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N Engl J Med. 2014;371:1507–17. https://doi.org/10.1056/NEJMoa1407222.
Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med. 2018;378:439–48. https://doi.org/10.1056/NEJMoa1709866.
Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med. 2018;378:449–59. https://doi.org/10.1056/NEJMoa1709919.
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. 2017;377:2531–44. https://doi.org/10.1056/NEJMoa1707447.
Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N Engl J Med. 2017;377:2545–54. https://doi.org/10.1056/NEJMoa1708566.
Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci. 1993;90:720–4. https://doi.org/10.1073/pnas.90.2.720.
Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17:1290–7. https://doi.org/10.1038/nm.2446.
Röth A, Yssel H, Pène J, Chavez EA, Schertzer M, Lansdorp PM, et al. Telomerase levels control the lifespan of human T lymphocytes. Blood. 2003;102:849–57. https://doi.org/10.1182/blood-2002-07-2015.
Topp MS, Riddell SR, Akatsuka Y, Jensen MC, Blattman JN, Greenberg PD. Restoration of CD28 Expression in CD28 − CD8 + Memory Effector T Cells Reconstitutes Antigen-induced IL-2 Production. J Exp Med. 2003;198:947–55. https://doi.org/10.1084/jem.20021288.
Yu P, Pan J, Guo Z, Yang C, Mao L. CART cell therapy for prostate cancer: Status and promise. Onco Targets Ther. 2019;12:391–5. https://doi.org/10.2147/OTT.S185556.
Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12:6106–15. https://doi.org/10.1158/1078-0432.CCR-06-11833.
Hombach AA, Abken H. Costimulation by chimeric antigen receptors revisited the T cell antitumor response benefits from combined CD28-OX40 signalling. Int J Cancer. 2011;129:2935–44.
Zhang T, Cao L, Xie J, Shi N, Zhang Z, Luo Z, et al. Efficiency of CD19 chimeric antigen receptor-modified T cells for treatment of B cell malignancies in phase I clinical trials: a meta-analysis. Oncotarget. 2015;6:33961–71. https://doi.org/10.18632/oncotarget.5582.
Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci. 2009;106:3360–5. https://doi.org/10.1073/pnas.0813101106.
Chmielewski M, Kopecky C, Hombach AA, Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011;71:5697–706. https://doi.org/10.1158/0008-5472.CAN-11-0103.
Hillerdal V, Essand M. Chimeric antigen receptor-engineered t cells for the treatment of metastatic prostate cancer. BioDrugs. 2015;29:75–89. https://doi.org/10.1007/s40259-015-0122-9.
Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. 2015;15:1145–54. https://doi.org/10.1517/14712598.2015.1046430.
Krenciute G, Prinzing BL, Yi Z, Wu M-F, Liu H, Dotti G, et al. Transgenic expression of IL15 improves antiglioma activity of IL13Rα2-CAR T cells but results in antigen loss variants. Cancer Immunol Res. 2017;5:571–81. https://doi.org/10.1158/2326-6066.
Avanzi MP, Yeku O, Li X, Wijewarnasuriya DP, van Leeuwen DG, Cheung K, et al. Engineered Tumor-targeted t cells mediate enhanced anti-tumor efficacy both directly and through activation of the endogenous immune system. Cell Rep. 2018;23:2130–41. https://doi.org/10.1016/j.celrep.2018.04.051.
Shum T, Omer B, Tashiro H, Kruse RL, Wagner DL, Parikh K, et al. Constitutive signaling from an engineered IL7 receptor promotes durable tumor elimination by tumor-redirected T cells. Cancer Discov. 2017;7:1238–47. https://doi.org/10.1158/2159-8290.CD-17-0538.
Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res. 2017;23:2255–66. https://doi.org/10.1158/1078-0432.CCR-16-1300.
Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016;126:3130–44. https://doi.org/10.1172/JCI83092.
Petersen CT, Krenciute G. Next Generation CAR T Cells for the Immunotherapy of High-Grade Glioma. Front Oncol. 2019;9:69. https://doi.org/10.3389/fonc.2019.00069.
Hinrichs CS, Restifo NP. Reassessing target antigens for adoptive T-cell therapy. Nat Biotechnol. 2013;31:999–1008. https://doi.org/10.1038/nbt.2725.
Mirzaei HR, Rodriguez A, Badie B, Shepphird J, Brown CE. Chimeric antigen receptors T cell therapy in solid tumor: Challenges and clinical applications. Front Immunol. 2017;8:1850. https://doi.org/10.3389/fimmu.2017.01850.
Kiessling A, Wehner R, Füssel S, Bachmann M, Wirth MP, Schmitz M. Tumor-associated antigens for specific immunotherapy of prostate cancer. Cancers. 2012;4:193–217. https://doi.org/10.3390/cancers401019.
Westdorp H, Sköld AE, Snijer BA, Franik S, Mulder SF, Major PP, et al. Immunotherapy for prostate cancer: Lessons from responses to tumor-associated antigens. Front Immunol. 2014;5:191. https://doi.org/10.3389/fimmu.2014.00191.
Arredouani MS, Tseng-Rogenski SS, Hollenbeck BK, Escara-Wilke J, Leander KR, Defeo-Jones D, et al. Androgen ablation augments human HLA2.1-restricted T cell responses to PSA self-antigen in transgenic mice. Prostate. 2010;70:1002–11. https://doi.org/10.1002/pros.21134.
Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N Engl J Med. 2010;363:411–22.
Dannull J, Diener PA, Prikler L, Furstenberger G, Cerny T, Schmid U, et al. Prostate stem cell antigen is a promising candidate for immunotherapy of advanced prostate cancer. Cancer Res. 2000;60:5522–8.
Kiessling A, Schmitz M, Stevanovic S, Weigle B, Hölig K, Füssel M, et al. Prostate stem cell antigen: Identification of immunogenic peptides and assessment of reactive CD8+ T cells in prostate cancer patients. Int J Cancer. 2002;102:390–7.
Matsueda S, Kobayashi K, Nonaka Y, Noguchi M, Itoh K, Harada M. Identification of new prostate stem cell antigen-derived peptides immunogenic in HLA-A2+ patients with hormone-refractory prostate cancer. Cancer Immunol Immunother. 2004;53:479–89.
Garcia-Hernandez MDLL, Gray A, Hubby B, Klinger OJ, Kast WM. Prostate stem cell antigen vaccination induces a long-term protective immune response against prostate cancer in the absence of autoimmunity. Cancer Res. 2008;68(3):861–9.
Krupa M, Canamero M, Gomez CE, Najera JL, Gil J, Esteban M. Immunization with recombinant DNA and modified vaccinia virus Ankara (MVA) vectors delivering PSCA and STEAP1 antigens inhibits prostate cancer progression. Vaccine. 2011;29:1504–13.
Ross S, Spencer SD, Holcomb I, Tan C, Hongo JA, Devaux B, et al. Prostate stem cell antigen as therapy target: Tissue expression and in vivo efficacy of an immunoconjugate. Cancer Res. 2002;62(9):2546–53.
Saffran DC, Raitano AB, Hubert RS, Witte ON, Reiter RE, Jakobovits A. Anti-PSCA mAbs inhibit tumor growth and metastasis formation and prolong the survival of mice bearing human prostate cancer xenografts. Proc Natl Acad Sci. 2001;98:2658–63.
Olafsen T, Gu Z, Sherman MA, Leyton JV, Witkosky ME, Shively JE, et al. Targeting, imaging, and therapy using a humanized antiprostate stem cell antigen (PSCA) antibody. J Immunother. 2007;30:396–405.
Morgenroth A, Cartellieri M, Schmitz M, Günes S, Weigle B, Bachmann M, et al. Targeting of tumor cells expressing the prostate stem cell antigen (PSCA) using genetically engineered T-cells. Prostate. 2007;67:1121–31. https://doi.org/10.1002/pros.20608.
Rajasekaran AK, Anilkumar G, Christiansen JJ. Is prostate-specific membrane antigen a multifunctional protein? Am J Physiol Physiol. 2005;288:C975–81. https://doi.org/10.1152/ajpcell.00506.2004.
Ananias HJK, Van Den Heuvel MC, Helfrich W, De Jong IJ. Expression of the gastrin-releasing peptide receptor, the prostate stem cell antigen and the prostate-specific membrane antigen in lymph node and bone metastases of prostate cancer. Prostate. 2009;69:1101–8.
Caroli P, Sandler I, Matteucci F, De Giorgi U, Uccelli L, Celli M, et al. 68 Ga-PSMA PET/CT in patients with recurrent prostate cancer after radical treatment: prospective results in 314 patients. Eur J Nucl Med Mol Imaging. 2018;45(12):2035–44. https://doi.org/10.1007/s00259-018-4067-3.
Lu J, Celis E. Recognition of prostate tumor cells by cytotoxic T lymphocytes specific for prostate-specific membrane antigen. Cancer Res. 2002;62:5807–12.
Harada M, Matsueda S, Yao A, Ogata R, Noguchi M, Itoh K. Prostate-related antigen-derived new peptides having the capacity of inducing prostate cancer-reactive CTLs in HLA-A2+ prostate cancer patients. Oncol Rep. 2004;12:601–7.
Schroers R, Shen L, Rollins L, Rooney CM, Slawin K, Sonderstrup G, et al. Human Telomerase Reverse Transcriptase-Specific T-Helper Responses Induced by Promiscuous Major Histocompatibility Complex Class II-Restricted Epitopes. Clin Cancer Res. 2003;9:4743–55.
Kobayashi H, Omiya R, Sodey B, Yanai M, Oikawa K, Sato K, et al. Identification of Naturally Processed Helper T-Cell Epitopes from Prostate-Specific Membrane Antigen Using Peptide-Based in Vitro Stimulation. Clin Cancer Res. 2003;9:5386–93.
Kuroda K, Liu H, Kim S, Guo M, Navarro V, Bander NH. SaporinToxin-conjugated monoclonal antibody targeting prostate-specific membrane antigen has potent anticancer activity. Prostate. 2010;70:1286–94.
Wolf P, Alt K, Wetterauer D, Bühler P, Gierschner D, Katzenwadel A, et al. Preclinical evaluation of a recombinant anti-prostate specific membrane antigen single-chain immunotoxin against prostate cancer. J Immunother. 2010;33:262–71.
Bühler P, Wolf P, Gierschner D, Schaber I, Katzenwadel A, Schultze-Seemann W, et al. A bispecific diabody directed against prostate-specific membrane antigen and CD3 induces T-cell mediated lysis of prostate cancer cells. Cancer Immunol Immunother. 2008;57:43–52.
Bühler P, Molnar E, Dopfer EP, Wolf P, Gierschner D, Wetterauer U, et al. Target-dependent T-cell activation by coligation with a PSMA×CD3 diabody induces lysis of prostate cancer cells. J Immunother. 2009;32:565–73.
Friedmann RS, Spies AG, Kalos M. Identification of naturally processed CD8 T cell epitopes from prostein, a prostate tissue-specific vaccine candidate. Eur J Immunol. 2004;34:1091–101.
Carlsson B, Tötterman TH, Essand M. Generation of cytotoxic T lymphocytes specific for the prostate and breast tissue antigen TARP. Prostate. 2004;61:161–70.
Epel M, Carmi I, Soueid-Baumgarten S, Oh SK, Bera T, Pastan I, et al. Targeting TARP, a novel breast and prostate tumor-associated antigen, with T cell receptor-like human recombinant antibodies. Eur J Immunol. 2008;38:1706–20.
Tsavaler L, Shapero MH, Morkowski S, Laus R. Trp-p8, a novel prostate-specific gene, is up-regulated in prostate cancer and other malignancies and shares high homology with transient receptor potential calcium channel proteins. Cancer Res. 2001;61:3760–9.
Gade TPF, Hassen W, Santos E, Gunset G, Saudemont A, Gong MC, et al. Targeted elimination of prostate cancer by genetically directed human T lymphocytes. Cancer Res. 2005;65:9080–8. https://doi.org/10.1158/0008-5472.CAN-05-0436.
Maher J, Brentjens RJ, Gunset G, Rivière I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ/CD28 receptor. Nat Biotechnol. 2002;20:70–5. https://doi.org/10.1038/nbt0102-70.
Zuccolotto G, Fracasso G, Merlo A, Montagner IM, Rondina M, Bobisse S, et al. PSMA-Specific CAR-Engineered T Cells Eradicate Disseminated Prostate Cancer in Preclinical Models. PLoS One. 2014;9:e109427. https://doi.org/10.1371/journal.pone.0109427.
Ma Q, Gomes EM, Lo ASY, Junghans RP. Advanced generation anti-prostate specific membrane antigen designer T Cells for prostate cancer immunotherapy. Prostate. 2014;74:286–96. https://doi.org/10.1002/pros.22749.
Slovin SF, Wang X, Hullings M, Arauz G, Bartido S, Lewis JS, et al. Chimeric antigen receptor (CAR + ) modified T cells targeting prostate-specific membrane antigen (PSMA) in patients (pts) with castrate metastatic prostate cancer (CMPC). J Clin Oncol. 2013;31:72. https://doi.org/10.1200/jco.2013.31.6_suppl.72.
Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, et al. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol Ther. 2018;26:1855–66. https://doi.org/10.1016/j.ymthe.2018.05.003.
Zhang Q, Helfand BT, Carneiro BA, Qin W, Yang XJ, Lee C, et al. Efficacy Against Human Prostate Cancer by Prostate-specific Membrane Antigen-specific, Transforming Growth Factor-β Insensitive Genetically Targeted CD8+ T-cells Derived from Patients with Metastatic Castrate-resistant Disease. Eur Urol. 2018;73(5):648–52. https://doi.org/10.1016/j.eururo.2017.12.008.
Hassani M, Hajari Taheri F, Sharifzadeh Z, Arashkia A, Hadjati J, van Weerden WM, et al. Construction of a chimeric antigen receptor bearing a nanobody against prostate a specific membrane antigen in prostate cancer. J Cell Biochem. 2019. https://doi.org/10.1002/jcb.28370.
Priceman SJ, Gerdts EA, Tilakawardane D, Kennewick KT, Murad JP, Park AK, et al. Co-stimulatory signaling determines tumor antigen sensitivity and persistence of CAR T cells targeting PSCA+ metastatic prostate cancer. Oncoimmunology. 2018;7:e1380764. https://doi.org/10.1080/2162402X.2017.1380764.
Hillerdal V, Ramachandran M, Leja J, Essand M. Systemic treatment with CAR-engineered T cells against PSCA delays subcutaneous tumor growth and prolongs survival of mice. BMC Cancer. 2014;14:30. https://doi.org/10.1186/1471-2407-14-30.
Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31:71–5. https://doi.org/10.1038/nbt.2459.
Feldmann A, Arndt C, Bergmann R, Loff S, Cartellieri M, Bachmann D, et al. Retargeting of T lymphocytes to PSCA- or PSMA positive prostate cancer cells using the novel modular chimeric antigen receptor platform technology “UniCAR”. Oncotarget. 2017;8:31368–85. https://doi.org/10.18632/oncotarget.15572.
Deng Z, Wu Y, Ma W, Zhang S, Zhang YQ. Adoptive T-cell therapy of prostate cancer targeting the cancer stem cell antigen EpCAM. BMC Immunol. 2015;16:1. https://doi.org/10.1186/s12865-014-0064-x.
Yang F, Wen W, Qin W. Bispecific antibodies as a development platform for new concepts and treatment strategies. Int J Mol Sci. 2017;18:48. https://doi.org/10.3390/ijms18010048.
Stieglmaier J, Benjamin J, Nagorsen D. Utilizing the BiTE (bispecific T-cell engager) platform for immunotherapy of cancer. Expert Opin Biol Ther. 2015;15(8):1093–9. https://doi.org/10.1517/14712598.2015.1041373.
Stone JD, Aggen DH, Schietinger A, Schreiber H, Kranz DM. A sensitivity scale for targeting t cells with chimeric antigen receptors (CARs) and bispecific t-cell engagers (BiTEs). Oncoimmunology. 2012;1(6):863–73.
Baum V, Bühler P, Gierschner D, Herchenbach D, Fiala GJ, Schamel WW, et al. Antitumor activities of PSMA×CD3 diabodies by redirected T-cell lysis of prostate cancer cells. Immunotherapy. 2013;5:27–38. https://doi.org/10.2217/imt.12.136.
Friedrich M, Raum T, Lutterbuese R, Voelkel M, Deegen P, Rau D, et al. Regression of Human Prostate Cancer Xenografts in Mice by AMG 212/BAY2010112, a Novel PSMA/CD3-Bispecific BiTE Antibody Cross-Reactive with Non-Human Primate Antigens. Mol Cancer Ther. 2012;11:2664–73. https://doi.org/10.1158/1535-7163.MCT-12-0042.
Hernandez-Hoyos G, Sewell T, Bader R, Bannink J, Chenault RA, Daugherty M, et al. MOR209/ES414, a novel bispecific antibody targeting PSMA for the treatment of metastatic castration-resistant prostate cancer. Mol Cancer Ther. 2016;15:2155–65. https://doi.org/10.1158/1535-7163.MCT-15-0242.
Bailis J, Deegen P, Thomas O, Bogner P, Wahl J, Liao M, et al. Preclinical evaluation of AMG 160, a next-generation bispecific T cell engager (BiTE) targeting the prostate-specific membrane antigen PSMA for metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. 2019;37(Suppl):301(Abstr.). https://doi.org/10.1200/JCO.2019.37.7_suppl.301.
Hummel H-D, Kufer P, Grüllich C, Deschler-Baier B, Chatterjee M, Goebeler M-E, et al. Phase 1 study of pasotuxizumab (BAY 2010112), a PSMA-targeting Bispecific T cell Engager (BiTE) immunotherapy for metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. 2019;37(Suppl):5034(Abstr. https://doi.org/10.1200/JCO.2019.37.15_suppl.5034.
Gires O, Klein CA, Baeuerle PA. On the abundance of EpCAM on cancer stem cells. Nat Rev Cancer. 2009;9:143. https://doi.org/10.1038/nrc2499-c1.
Ni J, Cozzi PJ, Duan W, Shigdar S, Graham PH, John KH, et al. Role of the EpCAM (CD326) in prostate cancer metastasis and progression. Cancer Metastasis Rev. 2012;31:779–91. https://doi.org/10.1007/s10555-012-9389-1.
Enblad G, Karlsson H, Loskog ASI. CAR T-Cell Therapy: The Role of Physical Barriers and Immunosuppression in Lymphoma. Hum Gene Ther. 2015;26:498–505. https://doi.org/10.1089/hum.2015.054.
Roberts E, Cossigny DAF, Quan GMY. The Role of Vascular Endothelial Growth Factor in Metastatic Prostate Cancer to the Skeleton. Prostate Cancer. 2013;2013:418340. https://doi.org/10.1155/2013/418340.
Shi S, Chen L, Huang G. Antiangiogenic therapy improves the antitumor effect of adoptive cell immunotherapy by normalizing tumor vasculature. Med Oncol. 2013;30:698. https://doi.org/10.1007/s12032-013-0698-1.
O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9:eaaa0984. https://doi.org/10.1126/scitranslmed.aaa0984.
Wolf D, Sopper S, Pircher A, Gastl G, Wolf AM. Treg(s) in Cancer: Friends or Foe? J Cell Physiol. 2015;230:2598–605. https://doi.org/10.1002/jcp.25016.
Chrétien S, Zerdes I, Bergh J, Matikas A, Foukakis T. Beyond PD-1/PD-L1 inhibition: What the future holds for breast cancer immunotherapy. Cancers (Basel). 2019;11. https://doi.org/10.3390/cancers11050628.
Gomes B, Driessens G, Bartlett D, Cai D, Cauwenberghs S, Crosignani S, et al. Characterization of the selective indoleamine 2,3-dioxygenase-1 (IDO1) catalytic inhibitor EOS200271/PF-06840003 supports IDO1 as a critical resistance mechanism to PD-(L)1 blockade therapy. Mol Cancer Ther. 2018;17:2530–42. https://doi.org/10.1158/1535-7163.MCT-17-1104.
Zhang X, Zhu S, Li T, Liu Y-J, Chen W, Chen J. Targeting immune checkpoints in malignant glioma. Oncotarget. 2017;8:7157–74. https://doi.org/10.18632/oncotarget.12702.
Villanueva N, Bazhenova L. New strategies in immunotherapy for lung cancer: beyond PD-1/PD-L1. Ther Adv Respir Dis. 2018;12:1753466618794133. https://doi.org/10.1177/1753466618794133.
Moon YW, Hajjar J, Hwu P, Naing A. Targeting the indoleamine 2,3-dioxygenase pathway in cancer. J ImmunoTher Cancer. 2015;3:51. https://doi.org/10.1186/s40425-015-0094-9.
Bollard CM, Tripic T, Cruz CR, Dotti G, Gottschalk S, Torrano V, et al. Tumor-specific t-cells engineered to overcome tumor immune evasion induce clinical responses in patients with relapsed hodgkin lymphoma. J Clin Oncol. 2018;36:1128–39. https://doi.org/10.1200/JCO.2017.74.3179.
Pinthus JH, Waks T, Malina V, Kaufman-Francis K, Harmelin A, Aizenberg I, et al. Adoptive immunotherapy of prostate cancer bone lesions using redirected effector lymphocytes. J Clin Invest. 2004;114:1774–81. https://doi.org/10.1172/JCI22284.
Kantele JM, Kurk S, Jutila MA. Effects of Continuous Exposure to Stromal Cell-Derived Factor-1α on T Cell Rolling and Tight Adhesion to Monolayers of Activated Endothelial Cells. J Immunol. 2000;164:5035–40. https://doi.org/10.4049/jimmunol.164.10.5035.
Hirbe AC, Morgan EA, Weilbaecher KN. The CXCR4/SDF-1 Chemokine Axis: A Potential Therapeutic Target for Bone Metastases? Curr Pharm Des. 2010;16:1284–90. https://doi.org/10.2174/138161210791034012.
Lee JY, Kang DH, Chung DY, Kwon JK, Lee H, Cho NH, et al. Meta-Analysis of the Relationship between CXCR4 Expression and Metastasis in Prostate Cancer. World J Mens Health. 2014;32:167–75. https://doi.org/10.5534/wjmh.2014.32.3.167.
Zhang J, Patel L, Pienta KJ. CC chemokine ligand 2 (CCL2) promotes prostate cancer tumorigenesis and metastasis. Cytokine Growth Factor Rev. 2010;21:41–8. https://doi.org/10.1016/j.cytogfr.2009.11.009.
John LB, Devaud C, Duong CPM, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. 2013;19:5636–46. https://doi.org/10.1158/1078-0432.CCR-13-0458.
Sanchez C, Chan R, Bajgain P, Rambally S, Palapattu G, Mims M, et al. Combining T-cell immunotherapy and anti-androgen therapy for prostate cancer. Prostate Cancer Prostatic Dis. 2013;16:123–31. https://doi.org/10.1038/pcan.2012.49.
Kochenderfer JN, Dudley ME, Carpenter RO, Kassim SH, Rose JJ, Telford WG, et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood. 2013;122:4129–39. https://doi.org/10.1182/blood-2013-08-519413.
Xu XJ, Tang YM. Cytokine release syndrome in cancer immunotherapy with chimeric antigen receptor engineered T cells. Cancer Lett. 2014;343:172–8. https://doi.org/10.1016/j.canlet.2013.10.004.
Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra38. https://doi.org/10.1126/scitranslmed.3005930.
Maus MV, Haas AR, Beatty GL, Albelda SM, Levine BL, Liu X, et al. T Cells Expressing Chimeric Antigen Receptors Can Cause Anaphylaxis in Humans. Cancer Immunol Res. 2013;1:26–31. https://doi.org/10.1158/2326-6066.CIR-13-0006.
Di Stasi A, Tey S-K, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible Apoptosis as a Safety Switch for Adoptive Cell Therapy. N Engl J Med. 2011;365:1673–83. https://doi.org/10.1056/NEJMoa1106152.
Deniger DC, Switzer K, Mi T, Maiti S, Hurton L, Singh H, et al. Bispecific T-cells expressing polyclonal repertoire of endogenous γδ T-cell receptors and introduced CD19-specific chimeric antigen receptor. Mol Ther. 2013;21:638–47. https://doi.org/10.1038/mt.2012.267.
Klingemann H. Are natural killer cells superior CAR drivers? Oncoimmunology. 2014;3:e28147. https://doi.org/10.4161/onci.28147.
Jakobczyk H, Sciortino F, Chevance S, Gauffre F, Troadec MB. Promises and limitations of nanoparticles in the era of cell therapy: Example with CD19-targeting chimeric antigen receptor (CAR)-modified T cells. Int J Pharm. 2017;532:813–24. https://doi.org/10.1016/j.ijpharm.2017.07.075.
Singh S, Asal R, Bhagat S. Multifunctional antioxidant nanoliposome-mediated delivery of PTEN plasmids restore the expression of tumor suppressor protein and induce apoptosis in prostate cancer cells. J Biomed Mater Res - Part A. 2018;106:3152–64. https://doi.org/10.1002/jbm.a.36510.
Yazdanifar M, Zhou R, Mukherjee P. Emerging immunotherapeutics in adenocarcinomas: A focus on CAR-T cells. Curr Trends Immunol. 2016;17:95–115.
Acknowledgements
The authors thank Gráinne Tierney and Cristiano Verna for editorial assistance.
Funding
No funding source.
Author information
Authors and Affiliations
Contributions
GS, MCC and UDG: conception and design. UDG: study supervision. All authors: analysis and interpretation of literature. All authors: writing, review, and/or revision of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Schepisi, G., Cursano, M.C., Casadei, C. et al. CAR-T cell therapy: a potential new strategy against prostate cancer. j. immunotherapy cancer 7, 258 (2019). https://doi.org/10.1186/s40425-019-0741-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40425-019-0741-7