
Abstract
Background
Adoptive transfer of T cells genetically engineered with a chimeric antigen receptor (CAR) has successfully been used to treat both chronic and acute lymphocytic leukemia as well as other hematological cancers. Experimental therapy with CAR-engineered T cells has also shown promising results on solid tumors. The prostate stem cell antigen (PSCA) is a protein expressed on the surface of prostate epithelial cells as well as in primary and metastatic prostate cancer cells and therefore a promising target for immunotherapy of prostate cancer.
Methods
We developed a third-generation CAR against PSCA including the CD28, OX-40 and CD3 ζ signaling domains. T cells were transduced with a lentivirus encoding the PSCA-CAR and evaluated for cytokine production (paired Student’s t-test), proliferation (paired Student’s t-test), CD107a expression (paired Student’s t-test) and target cell killing in vitro and tumor growth and survival in vivo (Log-rank test comparing Kaplan-Meier survival curves).
Results
PSCA-CAR T cells exhibit specific interferon (IFN)-γ and interleukin (IL)-2 secretion and specific proliferation in response to PSCA-expressing target cells. Furthermore, the PSCA-CAR-engineered T cells efficiently kill PSCA-expressing tumor cells in vitro and systemic treatment with PSCA-CAR-engineered T cells significantly delays subcutaneous tumor growth and prolongs survival of mice.
Conclusions
Our data confirms that PSCA-CAR T cells may be developed for treatment of prostate cancer.
Similar content being viewed by others

Background
Adoptive transfer of ex vivo-expanded tumor infiltrating lymphocytes (TILs) has shown promising results as a treatment of human cancers [1]. However, since it is not possible to isolate and expand TILs from all patients and tumor types, an attractive alternative technology is to isolate T cells from peripheral blood of a cancer patient and genetically engineer them with a novel T cell receptor (TCR), recognizing a tumor-associated antigen in the context of human leukocyte antigen (HLA) presentation, or a chimeric antigen receptor (CAR), recognizing a tumor-associated antigen on the surface of tumor cells. The engineered T cells are expanded and adoptively transferred back to the patient. Engineered T cells with specificities for a variety of tumor-associated antigens have been developed [2]. The first successful attempt to treat cancer patients with TCR-engineered T cells was reported from the Surgery Branch at the National Cancer Institute in 2006, where 2 patients out of 15 (13%) demonstrated objective regression of metastatic melanoma lesions when treated with MART-1-TCR-engineered autologous T cells [3]. CARs are artificial single chain antibody fragment (ScFv)-based receptors linked to a signaling domain for T cell activation [4]. First-generation CARs contain the CD3 ζ chain signaling domain from the TCR complex for T cell activation, whereas second-generation CARs include also a second co-stimulatory signaling domain from CD28 [5], 4-1BB [6], OX-40 [7] or CD27 [8]. Third-generation CARs contain two co-stimulatory signaling domains along with the CD3 ζ chain [9]. A successful report with complete remission of two out of three B-cell chronic lymphocytic leukemia (CLL) patients using CD19-CAR T cells was reported from University of Pennsylvania in 2011 [10]. This was followed up by successful treatment also of B-cell acute lymphocytic leukemia (ALL) in 2013 [11].
Prostate cancer is one of the most common cancers in the developed world. Curative treatment is not possible when the tumor has spread beyond the prostate gland. Since the prostate is a dispensable organ, T cell immunotherapy is an attractive approach for treatment of prostate cancer as it allows for targeting of tissue-specific antigens that are also expressed on malignant prostatic cells. Prostate stem cell antigen (PSCA) is a prostate tissue-restricted antigen highly expressed on primary and metastatic prostate cancer cells [12]. PSCA has been evaluated as a DNA vaccine in an experimental model for prostate cancer [13] and T cell epitopes from PSCA have been identified [14]. Furthermore, HLA-A2-positive prostate cancer patients have been found to have circulating T cells against PSCA [15]. Positive results have been reported in a study using a bi-specific antibody against PSCA and CD3, thereby re-directing T cells towards PSCA-expressing cells [16]. Humanized anti-PSCA antibodies have entered clinical trials [17, 18]. Herein, we use a third-generation CAR targeting PSCA, which besides the CD3 ζ chain contains the signaling domains of CD28 and OX-40. We evaluate whether primary T cells from peripheral blood of healthy volunteers transduced with a lentiviral vector encoding the PSCA-CAR molecule are able to recognize and kill cancer cells expressing PSCA both in vitro and in vivo.
Methods
Lentivirus vector design and lentivirus production
Lentivirus for target cell modification: A number of third-generation self-inactivating lentiviral plasmids expressing two transgenes separated by the sequence for the Thosea asigna virus 2A (T2A) peptide were constructed using pGreenPuro (SBI System Biosciences, Mountain View, CA). The plasmids are denoted pBMN(TurboRFP-Luc2), pBMN(copGFP-PSCA) and pBMN(copGFP-TARP), where TurboRFP encodes turbo red fluorescent protein, Luc2 encodes codon-optimized firefly luciferase, copGFP encodes copepod green fluorescent protein, PSCA encodes the human prostate stem cell antigen and TARP encodes human T cell receptor γ-chain alternate reading frame protein.
Lentivirus for T cell engineering: An anti-PSCA CAR-expressing lentiviral plasmid, pBMN(PSCA-CAR), was generated by fusing a PSCA-recognizing single chain antibody fragment, obtained through reversed genetics [19] with the signaling moieties of CD28, OX-40 and CD3 ζ chain, from a plasmid obtained from M Brenner, Baylor College of Medicine, Houston, TX [20].
Lentiviruses were produced in HEK-293 T cells using polyethyleneimine (Sigma-Aldrich, St Louis, MO) transfection. The pBMN-based lentiviral plasmid and the packaging plasmids pLP1, pLP2 and pVSV-G (Invitrogen) were used at a ratio of 2:1:1:1. The supernatant was harvested 48 and 72 hours post-transfection, concentrated through ultracentrifugation at 75,000 × g for 90 minutes and stored at -80°C. Mock lentivirus was produced using an empty pRRL lentiviral plasmid (Addgene, Cambridge, MA).
Target cell lines
The mel526 cell line was obtained from T Boon, Ludwig Institute for Cancer Research, Brussels, Belgium and cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA). Mel526-based target cells were produced through lentiviral transduction followed by sorting using a FACS Aria III sorter (BD Biosciences, Franklin Lakes, NJ). Mel526 cells co-expressing TARP, copGFP, Luc2 and turboRFP will be referred to in the text as mel526(TARP), and mel526 cells co-expressing PSCA, copGFP, Luc2 and turboRFP will be referred to as mel526(PSCA).
T cells from activated and lentivirus transducted of PBMCs
Peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats from healthy donors using Ficoll-Paque (GE Healthcare, Uppsala, Sweden) and cultured in RPMI-1640 supplemented with 10% human AB serum (our own production), 2 mM L-glutamine, 10 mM HEPES, 20 μM β-mercaptoethanol and 1% penicillin/streptomycin. The PBMCs were activated with 100 ng/ml OKT-3 (Nordic Biosite, Täby, Sweden) and 100 IU/ml IL-2 (Proleukin, Novartis, Basel, Switzerland) for 2 days to selectively stimulate T cells. Activated cells were transduced with 50 μl concentrated PSCA-CAR-encoding lentivirus or Mock lentivirus for 4 hours at 37°C in the presence of 10 μg/ml protamine sulphate and 100 IU IL-2 (Sigma-Aldrich). Transduction was repeated 24 hours later and the cells were cultured and expanded for 2-4 weeks before analysis. For analysis of PSCA-CAR expression, cells were stained with biotinylated protein-L (Genscript, Piscataway, NJ) [21], washed 3 times with PBS containing 4% BSA, followed by labeling with phycoerythrin (PE)-conjugated streptavidin (BD Biosciences) or stained with Alexa Fluor® 647 F(ab')2 Fragment of Goat Anti-Mouse IgG (H + L) (Invitrogen) and stained with an allophycocyanin (APC)-conjugated anti-CD3 or fluorescein isothiocyanate (FITC)-conjugated anti-CD3 antibody (Nordic Biosite). Flow cytometry analysis was performed using FACSCanto II or LSRII (BD Biosciences).
IFN-γ and IL-2 ELISA
Activated and PSCA-CAR-transduced or Mock-transduced T cells (105 cells) were co-cultured with mel526(PSCA) or mel526(TARP) cells at a 1:1 ratio in 96-well plates. Supernatants were collected after 16 hours. ELISA (Mabtech, Nacka Strand, Sweden) was used to detect IFN-γ and IL-2 secretion.
Proliferation assay
Activated and PSCA-CAR-transduced or Mock-transduced T cells (105 cells) were labeled for 20 minutes at 37°C with 5 μM Cell Trace Violet (Invitrogen) in PBS and then washed with cold cell culture medium containing 10% human serum to stop the labeling reaction. Labeled PBMCs were co-cultured with irradiated (50 Gy) mel526(PSCA) or mel526(TARP) cells at a 1:1 ratio in 96-well plates for 5 days. The T cells received a low dose of IL-2 (10 IU/ml) on day 1. The labeled cells were then collected and stained with an APC-conjugated anti-CD3 antibody followed by flow cytometry analysis.
CD107a degranulation flow cytometry analysis
Activated and PSCA-CAR-transduced or Mock-transduced T cells (105 cells) were co-cultured with mel526(PSCA) or mel526(TARP) cells at a 1:1 ratio in 96-well plates for 16 hours. Cells were stained with a FITC-conjugated anti-CD107a antibody and an APC-conjugated anti-CD3 antibody followed by flow cytometry analysis.
Bioluminescence in vitrokilling assay
Activated and PSCA-CAR-transduced or Mock-transduced T cells were co-cultured with luciferase-expressing mel526(PSCA) or mel526(TARP) (15000 cells) in various effector to target cell ratios (0.4:1–50:1) in flat-bottomed 96-well plates. Co-cultures were harvested 48 hours later and analyzed for luciferase expression using Steady-Glo® Luciferase Assay System (Promega Corporation, Madison, WI), according to the manufacturer’s instruction, and the luminescence was measured in a luminometer (Wallac Victor 2 Multi-label Counter, Perkin Elmer, Waltham, MA). Luciferase activity from target cells not exposed to T cells was set as 100% cell viability (survival).
Animal model for T cell treatment
Nude NMRI mice (Harlan, Netherlands) were injected subcutaneously (hind flank) with 3 × 106 mel526(PSCA) cells. One, seven and fourteen days later the mice received intravenous injection of 1 × 107 PSCA-CAR-transduced T cells or Mock-transduced T cells. Twelve mice per group were used. The tumors were measured by caliper and tumor volume was calculated using the equation (length × width2)/2. Animals were sacrificed, when tumors reached over 1000 mm3. The Uppsala Animal Ethics Committee has approved the animal studies (ID numbers C319/9 and C195/11).
Statistics
Statistics were performed using GraphPad prism software version 5.04 (La Jolla, CA, USA). Statistical analysis for IFN-γ and IL-2 secretion, cell proliferation and CD107a degranulation were performed using paired Student’s t-test. Log-rank test was used to compare survival curves created by the Kaplan-Meier method. Values of p < 0.05 were considered statistically significant.
Results
Transduced T cells efficiently express the PSCA-CAR molecule
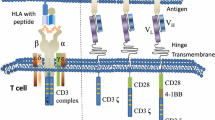
The ScFv nucleotide sequence of the anti-PSCA molecule was synthesized by reverse genetics from the amino acid sequence presented in the reference by Reiter and co-workers [19] and cloned together with the rest of the CAR molecule into a pGreenPuro-derived lentiviral vector under transcriptional control of the spleen-focus forming virus (SFFV) promoter (Figure 1A). To improve signal transduction through the CAR molecule, the intracellular domains of CD28 and OX-40 were included [20] and fused to the CD3 ζ chain. The leader sequence from the immunoglobulin kappa light chain was included for efficient expression of the CAR molecule on the cell surface of transduced cells. The anti-PSCA ScFv sequence was separated from the signaling part of the CAR molecule with a hinge region from an IgG heavy chain to allow for better flexibility.

Lentivirus cassette and surface expression of PSCA-CAR on T cells. (A) The design of the PSCA-CAR-encoding lentiviral vector is shown. (B) Expression of the PSCA-CAR molecule on the surface CD3+ T cells after transduction with the lentiviral vector was analyzed by flow cytometry using Alexa-647 F(ab')2 Fragment of Goat Anti-Mouse IgG (H + L). The solid filled histogram represents PSCA-CAR expression of transduced T cells, complex histogram represents CAR expression on Mock lentivirus-transduced T cells and the dashed histogram represents untransduced control T cells.
Peripheral blood lymphocytes isolated from healthy donors were activated for 24 hours and transduced with the PSCA-CAR-encoding lentivirus, or Mock lentivirus followed by two weeks of culture. The expression of PSCA-CAR was verified using Alexa-647 F(ab')2 Fragment of Goat Anti-Mouse IgG (H + L) (Invitrogen), which labels the heavy and light chain of mouse IgG and analyzed by flow cytometry analysis. T cells were efficiently transduced and expressed significant levels of PSCA-CAR when compared to Mock lentivirus-transduced or untransduced PBMCs (Figure 1B).
PSCA-CAR T cells specifically secrete IFN-γ and IL-2 and proliferate when exposed to target cells expressing the PSCA antigen
We first wanted to evaluate the PSCA-CAR T cells, generated from peripheral blood, against target cells in vitro. We could not make use of prostate cancer target cell lines with endogenous PSCA as they have been reported to down-regulate expression of PSCA during in vitro culture [22]. We also screen a large number of prostate cancer cell line as well as primary prostate epithelial cells at different passages for PSCA expression by flow cytometry (Additional file 1: Methods) but were unable to detect any PSCA expression (Additional file 2: Figure S1A). Immunohistochemistry analysis has detected PSCA expression in pancreatic cancer cell lines [23]. However, we were unable to detect PSCA expression on the surface of pancreatic cancer cell lines by flow cytometry (Additional file 2: Figure S1B). There are reports that suggest that xenografted pancreatic cancer cell lines regain PSCA expression in vivo. We therefore, transplanted two human pancreatic cancer cell lines to NMRI nude mice, excised the grafts after 3–4 weeks, made single cell suspension and examined the cell-surface expression of PSCA by flow cytometry (Additional file 1: Methods). These cell lines did not regain PSCA cell surface expression (Additional file 3: Figure S2). Therefore, we took the approach to lentivirally transduce target cells (mel526) to express the relevant antigen, PSCA, or an irrelevant control antigen, TARP. Thereby we establish a target cell line with a stable and intermediate strong PSCA expression. PSCA expression levels on mel526(PSCA) is shown in Additional file 2: Figure S1C.
PSCA-CAR T cells released high levels of IFN-γ (Figure 2A) and IL-2 (Figure 2C) in response to relevant mel526(PSCA) target cell exposure but not in response to the irrelevant mel526(TARP) target cells. In another control experiment it was observed that when PSCA-CAR T cells or Mock T cells were exposed to relevant target mel526(TARP) only PSCA-CAR T cells secreted IFN-γ (Figure 2B) and IL-2 (Figure 2D). We next evaluated the proliferative capacity of the PSCA-CAR T cells by labeling them with CellTrace Violet and exposing them to target cells. The PSCA-CAR T cells proliferated significantly better upon stimulation with mel526(PSCA) target cells than with mel526(TARP) cells, as detected through dilution of the dye and a lower intensity of the fluorescent signal. The results on proliferation from a pool of four donors are shown in Figure 2E and a representative image of proliferation for T cells established from one donor is presented in Figure 2G. The same proliferation pattern was observed when PSCA-CAR T cells were compared to Mock T cells co-cultured with relevant target mel526(PSCA) with pooled results in Figure 2F and a representative image in Figure 2H. The experiments with PSCA-CAR T cells and Mock T cells (Figures 2F, 2H) were performed with PBMCs isolated from different donors when compared to the experiments with relevant and irrelevant targets (Figure 2E,G).

Specific IFN-γ and IL-2 release and proliferation of PSCA-CAR-engineered T cells. Lymphocytes were isolated from healthy donors and T cells were stimulated before being transduced with a lentiviral vector encoding the CAR against PSCA or Mock lentivirus. PSCA-CAR T cells were then co-cultured overnight with mel526(PSCA) or irrelevant mel526(TARP) target cells. In a separate experiment PSCA-CAR T cells and Mock T cells were co-cultured with relevant target mel526(PSCA) cells. ELISA was used to analyze IFN-γ release from (A) PSCA-CAR T cells against mel526(PSCA) or mel526(TARP) target cells (n = 4), (B) PSCA-CAR T cells and Mock T cells against mel526(PSCA) target cells (n = 4). ELISA was used to analyze IL-2 release from (C) PSCA-CAR T cells against mel526(PSCA) or mel526(TARP) target cells (n = 4), (D) PSCA-CAR T cells and Mock T cells against mel526(PSCA) target cells (n = 4). PSCA-CAR T cells and Mock T cells were labeled with a fluorescent dye and T cell proliferation, assessed as dilution of the dye for each cell division, was measured by flow cytometry after 5 days of co-culture with mel526(PSCA) target cells or mel526(TARP) control cells. Pooled T cell proliferation data for (E) PSCA-CAR T cells against mel526(PSCA) or mel526(TARP) target cells, (F) PSCA-CAR T cells and Mock T cells against mel526(PSCA) target cells (n = 4). (G) A representative histogram of proliferation for T cells established from one donor in the pool presented in E. (H) A representative histogram of proliferation for T cells established from one donor in the pool presented in F. Asterisks indicate significance (***p < 0.001, paired Student’s t-test). Error bars represent standard deviation from four individual donors run in triplicates.
PSCA-CAR T cells specifically degranulate upon specific antigen recognition and kill PSCA-expressing target cells
When an activated T cell recognizes its cognate antigen, it initiates killing of the target cell. During this process, the content of proteolytic granules are released from the cytoplasm of the T cell, creating pores within the cell membrane of target cells. The emptying of the granule vesicles in T cells is associated with translocation of molecules from the granules to the cell surface. We analyzed the cell surface expression of one such molecule, CD107a, which is also known as lysosomal-associated membrane protein (LAMP)-1. The PSCA-CAR-engineered T cells degranulated, as seen by CD107a surface expression, in response to mel526(PSCA) cells, but not in response to mel526(TARP) cells. When T cells from four donors were pooled we found that a significantly higher proportion of PSCA-CAR-engineered T cells degranulated when exposed to mel526(PSCA) target cells compared to mel526(TARP) cells (Figure 3A). We also observed that Mock T cells degranulated to a significantly lesser degree when exposed to relevant target mel526 (PSCA) when compared to PSCA-CAR T cells (Figure 3B). To evaluate whether the PSCA-CAR T cells can kill target cells in vitro, we used a luciferase-based killing assay where viable mel526(PSCA) and mel526(TARP) target cells express luciferase. Target cells were co-cultured with PSCA-CAR T cells or Mock T cells at different ratios for 2–4 days before evaluating the percentage of viable target cells by luminescence measurements. The PSCA-CAR T cells efficiently killed PSCA-expressing tumor cells from the three donors evaluated, but did not kill TARP-expressing tumor cells (Figure 3C). The Mock T cells did not exhibit any unspecific cytotoxic activity against the target cells when compared to PSCA-CAR T cells (Figure 3D). The experiments presented in Figure 3D were performed with PBMCs isolated from different donors when compared to the experiments with PSCA-CAR T cells against relevant or irrelevant targets (C).

PSCA-CAR-engineered T cells specifically degranulate upon antigen recognition and kill PSCA-expressing tumor cells. PSCA-CAR T cells were co-cultured with mel526(PSCA) target cells or mel526(TARP) control cells. In a separate experiment PSCA-CAR T cells and Mock T cells were co-cultured with relevant target mel526(PSCA) cells. T cells (CD3+) were analyzed for CD107a expression (degranulation) by flow cytometry after 16 hours. Pooled data of CD107a expression on T cells from (A) PSCA-CAR T cells against mel526(PSCA) or mel526(TARP) target cells (n = 4), (B) PSCA-CAR T cells and Mock T cells against mel526(PSCA) target cells (n = 4). Asterisks indicate significance (*** p < 0.001, paired Student’s t-test). Error bars represent standard deviation from four individual donors run in triplicates. (C) PSCA-CAR T cells from three donors (#1, #2, #3) were co-cultured with luciferase-expressing mel526(PSCA) or mel526(TARP) target cells for 2 days. (D) PSCA-CAR T cells or Mock T cells from three donors (#4, #5, #6) were co-cultured with luciferase-expressing mel526(PSCA) for 4 days. Luciferase expression in target cells was then measured. Target cell viability was related to the luciferase signal for target cells not exposed to T cells. Error bars represent standard deviation from triplicate samples.
Systemic administration of PSCA-CAR T cells delay tumor growth and prolong survival of mice with subcutaneous PSCA-expressing tumors
Next we wanted to evaluate the ability of the PSCA-CAR T cells to control tumor growth in vivo. Mel526(PSCA) tumor cells were implanted subcutaneously in nude mice and PSCA-CAR-transduced T cells or Mock lentivirus-transduced T cells were infused systemically three times, one week apart, by intravenous injections. The tumor sizes of twelve individual mice treated with PSCA-CAR T cells are shown in Figure 4A and the tumor sizes of twelve individual mice treated with Mock T cells are shown in Figure 4B. As an example, by day 38, two out of the twelve mice treated with PSCA-CAR T cells had to be sacrificed while at the same day eleven out of twelve mice treated by Mock T cells had already been sacrificed. The pooled data showed significantly smaller tumor volumes for mice treated with PSCA-CAR T cells compared to mice treated with Mock T cells (Figure 4C). Furthermore, the survival analysis showed significantly prolonged survival for mice treated with PSCA-CAR T cells compared to mice treated with Mock T cells (Figure 4D).

Systemic treatment of mice with subcutaneous tumors with PSCA-CAR-engineered T cells leads to delayed tumor growth and prolonged survival. Nude NMRI mice were injected subcutaneously with 3 × 106 mel526(PSCA) cells. One, seven and fourteen days later the mice received intravenous injection of 1 × 107 PSCA-CAR-transduced T cells (CAR T) or Mock-transduced T cells (Mock T). Twelve mice per group were used and tumor volumes were measured by caliper every second day. One experiment out of two is presented. (A) Tumor volume for individual mice treated with CAR T cells (n = 12). (B) Tumor volume for individual mice treated with Mock T cells (n = 12). (C) Pooled data on tumor volume for PSCA-CAR T cell-treated and Mock T cell-treated mice shows significant difference in tumor volume (p < 0.01, paired t-test). (D) Kaplan-Meier survival analysis shows significant difference in survival for mice treated with PSCA-CAR T cells and Mock T cells (p < 0.01, log rank test).
Discussion
PSCA is a tissue-restricted antigen highly expressed on primary and metastatic prostate cancer cells in vivo[12, 24]. It may therefore be an appropriate target for cancer immunotherapy [24]. Fully humanized antibodies against PSCA are now in clinical trial for prostate cancer but do not lead to cure [18]. The potential of therapeutic T cells to traffic to sites of disease, expand and persist remains a major advantage compared with antibodies. In fact, complete objective remissions have been observed for some cancer patients when autologous, engineered T cells have been used for treatment [3, 10, 11].
A number of CARs have recently been developed against PSCA [21, 23, 25, 26]. Most publications on PSCA-CAR T cells use PSCA-transfected target cell lines to show T cell activity and only one publication show reactivity against a pancreatic tumor cell line with endogenous PSCA expression [23]. It should be noted that we did not detect PSCA expression on the surface of any prostate cancer nor pancreatic cancer cell line in vitro. Neither did we detect any PSCA expression on cultured primary prostate epithelial cells at different passages. Furthermore, we did not detect any PSCA expression on xenografted pancreatic cancer cell lines that were examined. Therefore, we were limited to use transduced target cells for PSCA-CAR T cell evaluation. We chose to use stable lentiviral transduction instead of transcient transfection, which can give unnaturally high levels of transgene expression.
There is only one report were PSCA-CAR-engineered T cells has been used in an in vivo model, in that case highly immunodeficient NSG mice with transplanted human tumors transduced to express PSCA [26]. Significant reduction in tumor growth rate was observed when the authors transferred T cells engineered with both a CAR that provides suboptimal activation upon binding of one antigen, PSCA, and a chimeric costimulatory receptor that recognizes a second antigen, PSMA, or vice versa. The authors further showed that co-transduced T cells destroy tumors that express both antigens but do not affect tumors expressing either antigen alone [26].
Herein, we used a third generation CAR against PSCA and we also show significant delay in tumor growth rate and significantly prolonged survival of nude mice. However, adoptive transfer of PSCA-CAR T cells alone did not cure any tumor-bearing mice. Whole-body irradiation as a preconditioning treatment before adoptive T cell transfer together with supportive administration of IL-2 have shown significantly improved results in mice [27]. It is therefore promising that our PSCA-CAR-engineered T cells are able to delay tumor growth in vivo without irradiation preconditioning or IL-2 support, though it may be beneficial to combine those treatments in the future for better effects. More experiments are needed to determine how long PSCA-CAR-engineered T cells persist or whether they proliferate at the tumor site. For example, T cells with longer telomeres that have high capacity to proliferate have been correlated with better prognosis for the patients receiving adoptive T cell transfer [28]. It may therefore be important to analyze the telomeres as well the phenotype of T cells and possibly select an optimal T cell subpopulation for genetic engineering and transfer. The method of T cell activation before transduction as well as the condition for in vitro culture of engineered T cells may also affect the performance of adoptively transferred CAR T cells.
Conclusions
We confirm others finding that adoptive transfer of PSCA-CAR T cells is a potentially promising approach to treat prostate cancer. Although the expression of PSCA-CAR on the surface of the transduced T cells was intermediate high, almost all T cells were expressing the CAR, Figure 1B. Our experiments therefore indicate that even low level of expression of the CAR may be sufficient for T cell activation and T cell-mediated killing. Although in adoptive T cell transfer only highly reactive clones are selected (secreting more than 200 pg/ml IFN-γ after co-culture with targets), no correlation between IFN-γ secretion and persistence and efficacy of the cells in vivo has been found [29].
Prostate cancer has, like most cancers, an immunosuppressive tumor microenvironment [30] and it is important to have highly active T cells that will be able to proliferate and kill tumors also in this harsh environment. Therefore, our future focus will be on enhancing the resistance of the PSCA-CAR T cells to immunosuppressive factors.
Authors’ information
Mohanraj Ramachandran and Justyna Leja shared authorship.
Abbreviations
- CAR:
-
Chimeric antigen receptor
- PSCA:
-
Prostate stem cell antigen
- CD:
-
Cluster of differentiation
- IFN:
-
Interferon
- IgG (H + L):
-
Immunoglobulin heavy and light chain
- IL:
-
Interleukin
- TIL:
-
Tumor infiltrating lymphocyte
- TCR:
-
T cell receptor
- HLA:
-
Human leukocyte antigen
- ScFv:
-
Single chain antibody fragment
- CLL:
-
Chronic lymphocytic leukemia
- ALL:
-
Acute lymphocytic leukemia
- T2A:
-
Thosea asigna virus 2A
- Luc:
-
Firefly luciferase
- copGFP:
-
Copepod green fluorescent protein
- TARP:
-
T cell receptor γ-chain alternate reading frame protein
- PE:
-
Phycoerythrin
- APC:
-
Allophycocyanin
- FITC:
-
Fluorescein isothiocyanate
- SFFV:
-
Spleen-focus forming virus
- LAMP:
-
Lysosomal-associated membrane protein.
References
Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME: Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008, 8 (4): 299-308. 10.1038/nrc2355.
Essand M, Loskog AS: Genetically engineered T cells for the treatment of cancer. J Intern Med. 2013, 273 (2): 166-181. 10.1111/joim.12020.
Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, Zheng Z, Nahvi A, de Vries CR, Rogers-Freezer LJ, Mavroukakis SA, Rosenberg SA: Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006, 314 (5796): 126-129. 10.1126/science.1129003.
Sadelain M, Brentjens R, Riviere I: The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. 2009, 21 (2): 215-223. 10.1016/j.coi.2009.02.009.
Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Olivares S, Gonzalez N, Smith DD, Forman SJ, Jensen MC, Cooper LJN: CD28 Costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 2006, 66 (22): 10995-11004. 10.1158/0008-5472.CAN-06-0160.
Song D-G, Ye Q, Carpenito C, Poussin M, Wang L-P, Ji C, Figini M, June CH, Coukos G, Powell DJ: In vivo persistence, tumor localization, and antitumor activity of CAR-engineered T cells is enhanced by costimulatory signaling through CD137 (4-1BB). Cancer Res. 2011, 71 (13): 4617-4627. 10.1158/0008-5472.CAN-11-0422.
Pule MA, Straathof KC, Dotti G, Heslop HE, Rooney CM, Brenner MK: A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther. 2005, 12 (5): 933-941. 10.1016/j.ymthe.2005.04.016.
Song DG, Ye Q, Poussin M, Harms GM, Figini M, Powell DJ: CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood. 2012, 119 (3): 696-706. 10.1182/blood-2011-03-344275.
Wang J, Jensen M, Lin Y, Sui X, Chen E, Lindgren CG, Till B, Raubitschek A, Forman SJ, Qian X, James S, Greenberg P, Riddell S, Press OW: Optimizing adoptive polyclonal T cell immunotherapy of lymphomas, using a chimeric T cell receptor possessing CD28 and CD137 costimulatory domains. Hum Gene Ther. 2007, 18 (8): 712-725. 10.1089/hum.2007.028.
Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH: T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011, 3 (95): 95ra73-
Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, Milone MC, Levine BL, June CH: Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. The New England J of Med. 2013, 368 (16): 1509-1518. 10.1056/NEJMoa1215134.
Gu Z, Thomas G, Yamashiro J, Shintaku IP, Dorey F, Raitano A, Witte ON, Said JW, Loda M, Reiter RE: Prostate stem cell antigen (PSCA) expression increases with high gleason score, advanced stage and bone metastasis in prostate cancer. Oncogene. 2000, 19 (10): 1288-1296. 10.1038/sj.onc.1203426.
Zhang KQ, Yang F, Ye J, Jiang M, Liu Y, Jin FS, Wu YZ: A novel DNA/peptide combined vaccine induces PSCA-specific cytotoxic T-lymphocyte responses and suppresses tumor growth in experimental prostate cancer. Urology. 2012, 79 (6): 1410 e1417-1413-
Kiessling A, Schmitz M, Stevanovic S, Weigle B, Holig K, Fussel M, Fussel S, Meye A, Wirth MP, Rieber EP: Prostate stem cell antigen: identification of immunogenic peptides and assessment of reactive CD8+ T cells in prostate cancer patients. Int J Cancer. 2002, 102 (4): 390-397. 10.1002/ijc.10713.
Forsberg O, Carlsson B, Malmstrom PU, Ullenhag G, Totterman TH, Essand M: High frequency of prostate antigen-directed T cells in cancer patients compared to healthy age-matched individuals. Prostate. 2009, 69 (1): 70-81. 10.1002/pros.20858.
Leyton JV, Olafsen T, Lepin EJ, Hahm S, Bauer KB, Reiter RE, Wu AM: Humanized radioiodinated minibody for imaging of prostate stem cell antigen–expressing tumors. Clin Cancer Res. 2008, 14 (22): 7488-7496. 10.1158/1078-0432.CCR-07-5093.
Antonarakis E, Carducci M, Eisenberger M, Denmeade S, Slovin S, Jelaca-Maxwell K, Vincent M, Scher H, Morris M: Phase I rapid dose-escalation study of AGS-1C4D4, a human anti-PSCA (prostate stem cell antigen) monoclonal antibody, in patients with castration-resistant prostate cancer: a PCCTC trial. Cancer Chemother Pharmacol. 2012, 69 (3): 763-771. 10.1007/s00280-011-1759-9.
Morris MJ, Eisenberger MA, Pili R, Denmeade SR, Rathkopf D, Slovin SF, Farrelly J, Chudow JJ, Vincent M, Scher HI, Carducci MA: A phase I/IIA study of AGS-PSCA for castration-resistant prostate cancer. Ann Oncol. 2012, 23 (10): 2714-2719. 10.1093/annonc/mds078.
Olafsen T, Gu Z, Sherman MA, Leyton JV, Witkosky ME, Shively JE, Raubitschek AA, Morrison SL, Wu AM, Reiter RE: Targeting, imaging, and therapy using a humanized antiprostate stem cell antigen (PSCA) antibody. J Immunother. 2007, 30 (4): 396-405. 10.1097/CJI.0b013e318031b53b.
Yvon E, Del Vecchio M, Savoldo B, Hoyos V, Dutour A, Anichini A, Dotti G, Brenner MK: Immunotherapy of metastatic melanoma using genetically engineered GD2-specific T cells. Clin Cancer Res. 2009, 15 (18): 5852-5860. 10.1158/1078-0432.CCR-08-3163.
Zheng Z, Chinnasamy N, Morgan RA: Protein L: a novel reagent for the detection of chimeric antigen receptor (CAR) expression by flow cytometry. J Transl Med. 2012, 10: 29-10.1186/1479-5876-10-29.
Taylor RM, Severns V, Brown DC, Bisoffi M, Sillerud LO: Prostate cancer targeting motifs: expression of alphanu beta3, neurotensin receptor 1, prostate specific membrane antigen, and prostate stem cell antigen in human prostate cancer cell lines and xenografts. Prostate. 2012, 72 (5): 523-532. 10.1002/pros.21454.
Katari UL, Keirnan JM, Worth AC, Hodges SE, Leen AM, Fisher WE, Vera JF: Engineered T cells for pancreatic cancer treatment. HPB: the Off J of the Int Hepato Pancreato Biliary Assoc. 2011, 13 (9): 643-650. 10.1111/j.1477-2574.2011.00344.x.
Jalkut MW, Reiter RE: Role of prostate stem cell antigen in prostate cancer research. Curr Opin Urol. 2002, 12 (5): 401-406. 10.1097/00042307-200209000-00006.
Morgenroth A, Cartellieri M, Schmitz M, Gunes S, Weigle B, Bachmann M, Abken H, Rieber EP, Temme A: Targeting of tumor cells expressing the prostate stem cell antigen (PSCA) using genetically engineered T-cells. Prostate. 2007, 67 (10): 1121-1131. 10.1002/pros.20608.
Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M: Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013, 31 (1): 71-75.
Kochenderfer JN, Yu ZY, Frasheri D, Restifo NP, Rosenberg SA: Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood. 2010, 116 (19): 3875-3886. 10.1182/blood-2010-01-265041.
Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR, Morton KE, Laurencot CM, Steinberg SM, White DE, Dudley ME: Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011, 17 (13): 4550-4557. 10.1158/1078-0432.CCR-11-0116.
Besser MJ, Shapira-Frommer R, Treves AJ, Zippel D, Itzhaki O, Schallmach E, Kubi A, Shalmon B, Hardan I, Catane R, Segal E, Markel G, Apter S, Nun AB, Kuchuk I, Shimoni A, Nagler A, Schachter J: Minimally cultured or selected autologous tumor-infiltrating lymphocytes after a lympho-depleting chemotherapy regimen in metastatic melanoma patients. J Immunother. 2009, 32 (4): 415-423. 10.1097/CJI.0b013e31819c8bda.
Barach YS, Lee JS, Zang XX: T cell coinhibition in prostate cancer: new immune evasion pathways and emerging therapeutics. Trends Mol Med. 2011, 17 (1): 47-55. 10.1016/j.molmed.2010.09.006.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/14/30/prepub
Acknowledgements
The authors wish to thank Berith Nilsson, Di Yu, Chuan Jin, Fredrik Eriksson and Angelica Loskog for technical assistance and advices and the BioVis core facility at Uppsala University for cell sorting.
Grant support
The Swedish Cancer Society (12 0569), Gunnar Nilsson’s Cancer Foundation, the Swedish Children Cancer Foundation (PROJ10/027) and the Marcus and Marianne Wallenberg’s Foundation supported this work. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
VH, MR, JL and ME designed research; VH, MR and JL performed research and analyzed the data; VH, MR and ME wrote the paper. All authors read and approved the final manuscript.
Electronic supplementary material
12885_2013_4260_MOESM2_ESM.tiff
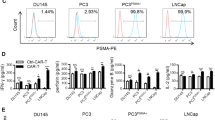
Additional file 2: Figure S1: PSCA expression was not detected on various prostate and pancreatic cancer cells. A) PSCA expression was analyzed by flow cytometry on the surface of the human prostate cancer cell lines LNCaP, DU145, PC3, VCaP and on primary prostate epithelial cells of different passage (p). B) PSCA expression was analyzed by flow cytometry on the surface of human pancreatic cancer cell lines AsPc1, CaPan1, CaPan2 and Panc1 C) PSCA expression was analyzed by flow cytometry on the surface of the transduced mel526(PSCA) target cells and transduced mel526(TARP) control cells. Grey filled histograms represent anti-PSCA-stained cells while white filled histograms represent isotype control antibody staining. (TIFF 18 MB)
12885_2013_4260_MOESM3_ESM.tiff
Additional file 3: Figure S2: PSCA expression was not detected on xenografted pancreatic cancer cells. PSCA expression was analyzed by flow cytometry on the surface of the pancreatic cell lines AsPc1 and CaPan2 after they have been grown subcutaneously in nude mice. Grey filled histograms represent anti-PSCA-stained cells while white filled histograms represent isotype control antibody staining. (TIFF 5 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Hillerdal, V., Ramachandran, M., Leja, J. et al. Systemic treatment with CAR-engineered T cells against PSCA delays subcutaneous tumor growth and prolongs survival of mice. BMC Cancer 14, 30 (2014). https://doi.org/10.1186/1471-2407-14-30
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-14-30