
Abstract
The most common kind of acute leukemia in adults is acute myeloid leukemia (AML), which is often treated with induction chemotherapy regimens followed by consolidation or allogeneic hematopoietic stem cell transplantation (HSCT). However, some patients continue to develop relapsed or refractory AML (R/R-AML). Small molecular targeted drugs require long-time administration. Not all the patients hold molecular targets. Novel medicines are therefore needed to enhance treatment outcomes. T cells and natural killer (NK) cells engineered with chimeric antigen receptors (CARs) that target antigens associated with AML have recently been produced and are currently being tested in both pre-clinical and clinical settings. This review provides an overview of CAR-T/NK treatments for AML.
Similar content being viewed by others

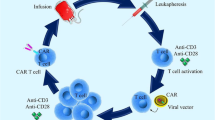
Background
Blocking differentiation and boosting proliferation are hallmarks of acute myeloid leukemia (AML), a clonal malignant hematologic illness. The "3 + 7" regimen of anthracycline and cytarabine is the gold standard treatment for AML, after which high-dose cytarabine or hematopoietic stem cell transplantation (HSCT) may be considered [1]. The 5-year overall survival (OS) is low for individuals with relapsed or refractory (R/R) AML or those who are unsuited for HSCT [2]. Novel small molecular targeting drugs, such as gilteritinib (FLT3 inhibitor) [3], enasidenib (IDH2 inhibitor) [4], ivosidenib (IDH1 inhibitor) [5], or venetoclax (BCL-2 inhibitor) [6], are promising for these patients. In spite of this, many cases of R/R AML persist that do not have a mutation in FLT3, IDH1, or IDH2. Nevertheless, most patients ultimately die from recurrence or disease progression due to treatment resistance or harmful side effects. Because of this, research into potential novel therapies for AML is essential.
Chimeric antigen receptor (CAR)T cells and natural killer (NK) cells are engineered cell treatments created to identify and kill tumor cells. The ability of both types of cells to recognize and bind tumor targets is based on the same principle of receptor-ligand binding, but the effector functions they exert upon binding are different.
CAR-T cells are engineered to express a chimeric antigen receptor and other specialized molecules that allow them to identify, bind to, and destroy their intended targets. When CAR-T cells are infused directly into a patient's bloodstream, they kill cancer cells without causing as many systemic, non-specific adverse effects as traditional anti-tumor treatments [7].
NK cells, just like T cells, can also be engineered to express the same CAR. CAR-NK cells, just like CAR-T cells, can recognize and target cancerous cells [8]. The natural characteristics of CAR-NK cells, however, give them a number of benefits over CAR-T cells, including their immediate availability, inducible proliferation, longer lifespan, and capacity to generate cytokines and chemokines which increase during infection [9]. Additionally, when compared to CAR-T cells, the safety profile of CAR-NK cells is much more reassuring. Nonetheless, there is still a small chance of developing graft-versus-host disease (GVHD) when employing donor NK cells [10].
In addition to Kymriah and Yescarta, two more CD19 CAR-T cell products, Tecartus (also known as Brexucabtagene Autoleucel or KTE-X19) from Gilead and Breyanzi (also known as lisocabtagene maraleucel, liso-cel) from Bristol-Myers Squibb, have been approved by the FDA for the treatment of B-cell malignant hematological disorders [11]. Two BCMA CAR-T cell products, Abecma (idecabtagene vicleucel) and Carvykti (ciltacabtagene autoleucel) also are approved, recently. This establishes a benchmark for the development of novel AML therapeutics. If targets as successful as CD19, or BCMA are found, AML immunotherapy with CAR may achieve a comparable outcome. The efficacy of CAR-T and CAR-NK cells in the treatment of AML has been demonstrated in a number of in-vitro and in-vivo investigations [7, 9].
This review primarily focuses on target selection and the corresponding research status of CAR-T therapy for AML, while also taking into account current interest in NK cells in relation to those targets. The detailed properties and distributions of the targets are described in Table 1.
Targets that have entered clinical trials
As technology advances, more targets, for instance, CD123, CD33, CD70, FLT3, CD38, and many others, have entered clinical trials. In order to induce cell death in AML cells, these targets are selected based on their capacity to bind to surface receptors. As a result of promising findings in preclinical trials, these targets are now the subject of extensive research into their clinical pharmacology, human safety, and therapeutic efficacy.
CD123
CD123, a high-affinity receptor for stem cell factor, is the α subunit of the interleukin 3 receptor (IL-3R). When its expression rises, AML patients' overall survival (OS) and progression-free survival (PFS) drop [15]. CD123 forms a heterodimer with β subunit of IL-3R to activate the JAK/STAT signaling which is required for hematopoiesis [80]. Furthermore, CD123 expression has been linked to a greater risk of treatment failure [81].
In AML patients, CD123 expression levels are correlated with the number of leukemic stem cells (LSCs) which are CD34+CD38− quiescent cells and demonstrate chemotherapy resistance [82,83,84]. CD123 is a considerably more promising target than CD33 since it is highly expressed by AML and relatively less expressed by hematopoietic stem and progenitor cells [16]. Anti-CD123 CAR-T cells efficiently reduce the burden of leukemia in vivo with minimal damage to normal hematopoietic stem and progenitor cells (HSPCs) [85]. Despite its specificity, off-target still brings significant hematopietic toxicities, especially when adjuvants that increase CD123 expression on both AML and normal HSCs are added to improve targeted killing [86].
Using a panel of CD123-specific monoclonal antibodies, Thokala et al. engineered CD123 CAR-T cells with variable VL and VH chains (mAb) [87]. This type of CD123 CAR T cell exhibits potent cytotoxicity toward CD123+AML cells but much lower toxicity toward normal hematopoietic stem cells. Several researchers have proposed CAR-T123 as a new pretreatment regimen for producing remission and bridge to HSCT [86]. However, its low remission rate and the major complications, such as infection and hemorrhage when prolonging the transplantation interval, limit its usage. In this context, subsequent studies have sought to optimize protocols for CD123 CAR-T treatment. For instance, because CD123 is significantly expressed on endothelial cells as well as hematopoietic cells, CD123 CAR-T can accidentally injure these normal tissue cells, leading to endothelial cell damage and hematopoietic toxicity. In order to overcome this shortcoming, Nadia EI Khawanky et al. developed a third-generation CD123 CAR-T cell based on single-chain variable fragments (scFvs) and the CD28-OX40-CD3 intracellular signaling domains of humanized CSL362, which demonstrated anti-AML activity in xenograft mice without damaging epithelial tissue [17]. Furthermore, it was discovered that 5'-azacytidine therapy increased CD123 expression on leukemic cells, leading to the expansion of CD123 CAR-T cells and a boost in effector capacity, in addition to increased levels of tumor necrosis factor (TNF) production and elevated downstream phosphorylation of a key T cell activation molecule. This demonstrates the superior efficacy of combining co-stimulatory domains with the induction of antigen expression on leukemic cells, thus maximizing the killing effect while ensuring safe treatment.
Budde reported that, after CD123 CAR-T cell treatment, 3/6 individuals with R/R AML had complete remission (CR), and two obtained partial remission, while 66% of these patients subsequently receiving allo-HSCT [88] (NCT02159495). Despite the fact that CD123 CAR-T has been shown to be clinically effective, early trials showed that its efficacy was significantly lower than CD 19 CAR-T, likely due to low targeting specificity [85]. Whereas CAR-T123 with HSCT has shown promise for the treatment of AML, more study is needed to determine the regimen's long-term anti-leukemic efficacy, the ideal transplantation interval, and the prevention of graft-versus-host disease (GVHD).
CD123 CAR-T cell treatment caused long-term myelosuppression in individuals who did not receive subsequent allo-HSCT, so it is still an open question as to how to enhance CR rates and minimize potential hematopoietic toxicity. This highlights the need for novel clinical strategies that put the power of CAR-T technology under a dependable and quick “ON/OFF” switch that regulates when it is active and when it is turned off in order to lessen both short-term and long-term side effects. For instance, the Herpes simplex virus (HSV)-thymidine kinase suicide gene was one of the original safety switches that transformed the prodrug into a toxic compound, preventing further replication of cellular DNA [89]. As a result, cells expressing the suicide gene died after the prodrug was administered, but the immunogenicity of the enzyme after gene expression and the restrictions of the activation latency prevented rapid termination of the targeted killing effect. Walid Warda et al. used an inducible caspase 9 suicide gene safety switch to circumvent the masked resistance to target epitopes that can emerge on low antigen-expressing AML primary cells after interleukin-1 receptor (IL-1RAP) CAR-T treatment [90].
Other optimization techniques also can be used to generate controlled CAR-T effects. In 2020, the University of Pennsylvania ran a clinical experiment with mRNA electroporated CD123 CAR-T cells [91] (NCT02623582). Because of the potential for CAR-T cells to persist and cause myelosuppression if left in the body for too long, mRNA electroporation was favored over lentiviral transduction. In vivo activation of CAR-T cells was reported to be transient during the clinical trial, which was consistent with expectations. In 2021, Jan-Erik Meyer et al. developed UniCAR, a fast switchable universal CAR-T platform [92]. Specifically, the first element of the system is a universal CAR that does not interact with any human surface antigens but is recognized by a targeting module (TM) in the second part of the system. TM is the central component of such a platform, providing specificity towards predetermined cancer antigens. Due to the adaptability of the tumor-binding domain, it is able to target a wide variety of antigens in both solid tumors and hematological malignancies. The antigen-specific TM, such as TM123, can connect CAR to antigen, such as CD123, on the surface of myeloblastic cells, thus starting the killing. Notably, the short half-life of these TMs, which is below 30 min, allows for rapid deactivation of the UniCAR system by TM withdrawal, consequently avoiding the long-term harmful effects associated with the ongoing activation of CAR-T cells. The “ON/OFF” switch depends on TM administration or not. Moreover, preclinical data demonstrated that UniCAR-T cells lyse cancer cells at a lower TM dose compared to cytokine release induction, indicating a greater therapeutic window for clinical application. Preliminary results suggest CD123 UniCAR-T is well-tolerated and fast convertible with promising outcomes even at the lowest dose levels. The Phase 1A study is still recruiting patients (NCT04230265).
Targeting CD123 for anti-AML CAR treatment has seen the greatest research, with numerous ongoing clinical trials investigating its use in UniCARs [92](NCT04230265) and bispecific CARs [16](NCT04678336, NCT03766126, NCT02623582). It has been suggested that CD33 and CD123 can be targeted jointly in AML therapy because 70% of AML initial cells show both of these markers [16]. Several clinical trials exploring multiple antigens have been launched (NCT04010877, NCT04033302).
CD33
Myeloid-specific transmembrane sialic acid-binding receptor CD33 is known to control leukocytes throughout the immune response due to its two extracellular Ig-like structural domains.
Initial feasibility studies showed that cytokine-induced killer cells could be used to deliver CD33-directed CAR T cells [93]. Myeloid-specific transmembrane sialic acid-binding receptor CD33 is known to control leukocytes throughout the immune response due to its two extracellular Ig-like structural domains. Almost all AML cells and leukemic stem cells express CD33 at high levels, whereas normal cells express it at low levels, even at low effector-to-target ratios of < 1:20, making CD33 a useful diagnostic marker and therapeutic target for AML. Recent research, however, has shown that CD33 is especially important since its loss from the surface of AML primitive cells as antigen-negative escape [93] is associated with a poorer prognosis for AML patients.
Ten individuals with R/R AML were enrolled in a phase I clinical research assessing the safety and efficacy of CD33 CAR-T cells [94] (NCT03126864). Three of these patients got cell treatment. Participants had adverse events such as cytokine release syndrome (CRS), immunological effector cell-associated neurotoxicity syndrome, tumor lysis syndrome, respiratory distress syndrome of grade 3, and infectious shock. Although no hepatotoxicity was recorded, all three recipients succumbed to the progression of the disease. In contrast, a case report of a 41-year-old male shows that CD33 CAR-T temporarily reduced the number of AML primitive cells in his bone marrow (NCT01864902), prior to disease progression 9 weeks after initiation of the treatment [95]. The therapy also caused CRS, indicating a partial response to the AML.
Preclinical and clinical research have frequently highlighted CD33’s potential as a target for the treatment of AML, but its use has not been standardized in AML patients until very recently. This is because its potent anti-AML activity is coupled with obvious side effects which may result from its expression on normal HSCs, albeit at low levels. Preclinical models have illustrated the typical hematopoietic toxicity associated with CD33-targeted treatments, such as hemocytopenia and decreased myeloid progenitor cells [96]. Severe hematopoietic suppression may lead to neutropenia and thrombocytopenia causing fatal infection and bleeding. This defect can be addressed in numerous ways. The first method is a combination of CD33 CAR-T treatment with CD33 knockout (KO) HSCs transplantation. In a recent study, Florence Borot et al. successfully used CRISPR/Cas9 to delete the CD33 antigen, and found no effect on the ability of HSCs to repopulate [96]. The dual-targeted guide RNA editing has also been proposed as an alternate method of CD33 modification. CD33 CAR-T cell treatment in combination with CD33 KO HSCs transplantation is likely to achieve precise targeting of AML cells and reduced myelotoxicity. This novel form of immunotherapy is attractive, but further research and clinical assessment are necessary prior to its application in humans. Furthermore, this approach may be extensible to other malignancies and antigens exhibiting analogous characteristics. The second strategy employs a "AND" logical gating strategy, whereby one CAR molecule is used for activation and another CAR molecule is used to provide co-stimulatory sites in the same T cell, optimizing the combination of antigen recognition to reduce "on-target/off-tumor" toxicity, and the killing effect of CAR-T can only be activated when at least two markers are expressed on the target cell [97]. While "OR" gating only requires the expression of one antigen in cells, "NOT" gating requires no other markers. BisCAR T cells which contain co-stimulatory domains of Nb 157/CD3ς and antiTIM3 scFv/CD28/4-1BB specifically killed CD13 + TIM3 + double positive tumors, but with reduced and likely acceptable toxicity to HSCs and other normal cells with only CD13 expression [97].
The third way is to select immortalized immune cell lines for CAR-T construction. Barsov et al. used these cells to produce CAR-T cells [98]. This approach is expected to lead to a commercially available cell therapy product, while avoiding manufacturing failure. However, irradiation of the immortalized cell line is essential before injection to avoid its malignant potential, compromising the in vivo durability of the cells. Additionally, to enhance on-target killing and reduce off-target toxicity, “AND” gating techniques may be employed to regulate CAR-T activity. Like the above mentioned CD123 UniCAR-T, logical gating solutions involve the employment of only one CAR molecule for activation and a second for co-stimulation. Additionally, other approaches, such as “ON” switch, “OFF” switch [99], SUPRA (split, universal, and programmable) CAR [100], RevCAR (Reverse CAR) [101], and STOP-CAR platform are also available [102]. Finally, maximizing the efficacy of CAR therapy requires optimizing techniques to enhance the production of CD33 targeted cells and decrease the time between single-cell collection and treatment [102].
The potential of bispecific CD33-CLL1 CARs [103] in clinical studies has brought into focus the necessity for rapid and efficient production methods to fully achieve this treatment's potential [103].
CD7
The glycoprotein CD7 is a kind of transmembrane protein. CD7 is mostly expressed in T cells, NK cells, and their precursor cells in healthy people because it serves as a co-stimulatory receptor for T and B cell contacts throughout lymphocyte maturation [21, 104, 105]. Some myeloid progenitor cells in cord blood express it as well, though it is not clear what they are used for. CD7 is expressed in both normal and leukemic bone marrow precursor cells, suggesting that it may be a useful therapeutic target for AML treatment.
The first preclinical evidence released by the lab of Gomes-Silva on CD7 CAR-T indicates its therapeutic efficacy in AML [21]. However, CAR-T targeting CD7 may attack other T and NK cells, causing “fratricide”, and depleting the CAR-T cells after infusion. Conversely, CD7-deficient animals showed largely unchanged peripheral T cell function [21], which suggests that CD7’s role in mature T cells may be redundant. In light of these two considerations, it is possible that eliminating the CD7 gene from CAR-T cells may allow them to develop to their full potential, so achieving the intended therapeutic effect while reducing the risk of T-cell “self-mutilation” and maximizing the efficiency of CAR-anti-leukemia T cells. After using CRISPR-Cas9 technology to deplete CD7 levels from T cell, Silva et al. discovered that CD7 CAR-T cells had potent anti-AML efficacy in mice [105]. Eighty-five percent to ninety percent of CAR-T cells can have CD7 expression deleted using the CRISPR-Cas9 technique, with no discernible off-target effect in the therapy of AML. It is also possible to remove CD7 from the surface of cells in ways other than genome editing. CD7 can be eliminated by attaching to an scFv that is linked to the cell’s endoplasmic reticulum [106]. Thus, inhibiting CD7’s trafficking to the cell surface reduces its ability to “self-flagellate” CD7 CAR-T cells. However, substantial clinical trial comparisons of the long-term efficacy and feasibility of these two methods are required (NCT04762485, NCT04033302).
CD70
CD70, a member of the tumor necrosis factor (TNF) family of type II transmembrane glycoproteins, is a ligand for the TNF receptor, CD27. It is significantly upregulated on AML cells, including LSCs. This makes CD70 an attractive therapeutic target for AML.
The anti-tumor benefits of modified CD70 CAR-T cells were demonstrated by Maus et al. at the 2021 annual meeting of the American Society of Hematology (ASH 2021) [106]. Moreover, CAR-T cells containing a truncated CD27, CD8 hinge and transmembrane region were found to be highly efficacious against AML. Additionally, LSCs can render more susceptible to CAR-T therapy by modulating their CD70 expression with hypomethylating agents and this was confirmed by Riether et al [22].
Patients with R/R AML are the focus of ongoing clinical trials with CD70 CAR-T cells (NCT04662294) and CD70 NK cells producing IL-15 (NCT05667155).
ILT3 (LILRB4, CD85k)
ILT3, also known as leukocyte immunoglobulin-like receptor subfamily B member 4 (LILRB4), is an inhibitor of T cell activation and proliferation. Maintaining an immunosuppressive milieu for tumor cells, ILT3 may contribute to tumor’s evasion from the body’s immune system. ILT3 is expressed in myeloid cells and highly expressed in AML monocytes, therefore, representing an effective target for AML-M5.
In this regard, Samuel John et al. developed a ILT3 CAR-T therapy [24]. It was shown to trigger apoptosis specifically against monocytic AML in vitro and also decreased tumor burden in a xenograft model in vivo, with no observable adverse effects on normal hematopoiesis. To enhance ILT3 CAR therapeutic efficacy, it can be co-expressed with co-stimulatory domains or used in conjunction with other targeted therapies. Nevertheless, more work must be done to ensure the production of anti-ILT3 cells from scaled-up CAR-T to Good Manufacturing Practice grade in order to carefully address the various issues in production and application. These issues include the origin of the T cells, the limitations of the patient's own low lymphocyte count and the pollution of tumor cells when modifying autologous T cells, and the interference of HLA compatibility when infusing allogeneic T cells, etc. Clinical trials on ILT3 CAR-T are presently underway (NCT04803929).
NKG2DL
NKG2DL, or natural killer group 2 member D ligand, is a tetrameric protein complex that, when attached to its receptor, can trigger cell death. It is composed of transmembrane proteins that are linked via glycosylphosphatidyl alcohols. NKG2DL is increased in response to genotoxic stress, infection by some pathogens, and, most notably, malignant transformation.
NKG2DL overexpression has been seen in a variety of solid and hematological cancers, including AML and multiple myeloma (MM) [26]. Evidence suggests that its expression levels are associated with significant predictive prognosis in many AML patients. Therefore, therapeutic applications for NKG2DL-targeting CAR-T cells may be extensive.
Preclinical models of ovarian cancer and MM have displayed anti-tumor effects of NKG2DL CARs [27]. Nonetheless, clinical trials involving similar targeted CAR-T cells in AML patients achieved limited success. Baumeister et al. did not observe any notable impact after a single infusion [28], while Sallman et al. administered multiple infusions at higher doses and reported that one out of 22 patients experienced morphologic leukemia-free state [29]. Furthermore, activation of NKG2DL already presented in T cells can lead T cells to self-collapse. About this matter, NKG2DL CAR-T cells can be used in combination with a phosphatidylinositol 3-kinase (PI3K) inhibitor [30], and the cells can be further modified to express short hairpin RNA to decrease the amount of NKG2DL on the surface of CAR-T cells. Two AML patients showed a partial response to the treatment.
Patients with AML, myelodysplastic syndrome (MDS), and MM participated in the first human clinical trial evaluating the safety and feasibility of NKG2DL CAR-T cells [28] (NCT02203825). This Phase I dose-escalation trial obtained adequate number of NKG2DL CAR-T cells using special collecting bags for isolation products, and larger containers to provide a larger surface area for cells to grow (such as G-Rex). The manufactured NKG2D-CAR T cells demonstrated functional activity against autologous tumor cells in vitro. However, the extent of T cell expansion, the proportion of CD4 to CD8 cells, and the transfection efficiency of CD8 + cells were highly variable after transduction, which might be related to the clinical heterogeneity of the patients, including ages, stage of diseases, and previous treatment regimens. Nonetheless, this clinical trial offers circumstantial evidence that it is possible to produce sufficient number and effects of NKG2D CAR-T cells in a short amount of time.
Increased ligand expression may also cause toxic side effects in ordinarily functioning tissues. In clinical trials, however, no dose-limiting toxicity, cytokine release syndrome, or CAR T cell-related neurotoxicity was observed, nor were there any significant autoimmune reactions or adverse events of grade 3 attributable to NKG2D-CAR T cells. NKG2DL CAR-NK and CAR-T cell therapies are now being tested in a number of active clinical trials (NCT05734898).
FLT3
The FMS-like tyrosine kinase-3 (FLT3) receptor is one type of tyrosine kinase receptor. FLT3 ligand (FLT3Lg) is a natural ligand for FLT3. Upon FLT3Lg binds to FLT3, cells are activated. Furthermore, FLT3 mutations, which can be seen in 30% of AML patients, are extremely prevalent, making it one of the most frequently mutated genes [33]. The most prevalent FLT3 mutations are found in 24% of AML patients with internal tandem duplication (ITD) mutations and 7% of AML patients with tyrosine kinase structural domain (TKD) mutations [65, 100]. The PI3K/Akt, Raf/MEK/ERK, and JAK/STAT5 signaling pathways, which drive the progression of AML, are mediated by FLT3-ITD and FLT3-TKD. As a result, both FLT3-ITD and FLT3-TKD are linked to illness development and progression, and both indicate a bad prognosis.
Although FLT3 inhibitors, including gilteritinib [3] and midostaurin [107], have currently been used to improve clinical outcomes, allo-HSCT remains the only viable treatment option for patients with FLT3-mutated AML. Additionally, crenolanib, an FLT3 inhibitor, can upregulate FLT3-ITD expression on the cell surface, thus enhancing the targeting efficiency of FLT3 CAR-T [108].
Targeting a receptor can be accomplished in two ways, depending on its nature as a receptor. The first is to utilize anti-receptor antibodies, such as scFvs derived from anti-human FLT3 antibodies. Cesar Sommer et al. generated a kind of scFv-containing FLT3-CAR-T and examined its effects in a preclinical setting [109], and discovered that they removed primary AML primitive cells, as well as hematopoietic stem and progenitor cells, which bring certain bone marrow toxicity. This toxicity can be controlled by a way of rituximab-activated off-switches which deplete circulating CAR-T cells. Replication of natural binding ligands, FLT3L, is another approach for reducing immunogenicity and off-target toxic effects. Wang et al. developed FLT3L-4-1BB-CD3-CAR-T cells and evaluated their efficacy against FLT3+ leukemia cells [34]. FLT3L CAR-T cells demonstrated a greater lethality towards ITD-type mutations compared to wild-type FLT3, thereby protecting normal cells and hematopoiesis.
Several parallel clinical trials are currently enrolling patients (NCT05023707, NCT05445011, NCT05017883, and NCT05432401) to investigate the efficacy of FLT3 CAR-T cell therapy. Second-generation FLT3 CAR-T studies targeting FLT3 extracellular structural domain epitopes are in phase I clinical trials. One such study is using a kind of FLT3 CAR-T cells, named Amg553 (NCT03904069) which contains a scFv structural domain that binds extracellular epitopes of the FLT3, a CD28 co-stimulatory structure domain, and a CD3 zeta chain subunit activation structural domain. The preliminary results show that Amg553’s CAR-T cells target FLT3-expressing leukemia cells but spare normal hematopoietic stem cells, reducing therapeutic side effects. Additionally, Amg553’s CAR-T cells have a shorter lifespan, which allows for faster clearance and fewer side effects. Finally, Amg553’s CAR-T cells can be manufactured faster than other therapies, reducing treatment time. In summary, Amg553’s greater selectivity, safety, therapeutic efficacy, and shorter production time make it a promising treatment for acute myeloid leukemia.
CLL-1 (CLEC12A, MICL, KLRL1, DCAL-2)
Despite being regarded as a type II transmembrane glycoprotein with an inhibitory receptor function, the ligand for C-type lectin-like molecule 1 (CLL-1), also known as CLEC12A, MICL, KLRL1, or DCAL-2, has not been determined. As CLL-1 is not expressed in healthy tissues, it presents a promising therapeutic target.
Zhang et al. reported the first successful treatment using CLL-1 CAR-T cells. Following the administration of CAR-T, the patient had grade I–II cytokine release syndrome (CRS), which was characterized by an increase in body temperature and brief hypotension [40]. On day 29, the patient experienced morphological CR and MRD negative, which persisted for almost 9 months (NCT00846703). Multiple preclinical investigations have supported the potent anti-leukemic action of CLL-1 CAR-T without interfering with normal HSC. Subsequent modifications focused on enhancing the effect of the therapy, including improvement of the CAR-T construct as well as simultaneous transgenic expression of cytokines. However, these adjustments can lead to increased cytotoxicity. To address this challenge, Tashiro et al. developed a Caspase-9 suicide gene system [41] to provide targeted killing with the potential to modify its action.
Numerous CAR-T cell clinical trials targeting CLL-1 are currently recruiting participants (NCT05252572, NCT4219163, NCT04884984).
CD38
CD38 is a glycoprotein that is overexpressed in hematological malignant cells and broadly expressed on immune cells (especially plasma cells) and erythrocytes but not on HSCs [42, 110]. This expression feature allows CD38 as a therapeutic target. Myeloma patients have responded favorably to daratumumab, an anti-CD38 monoclonal antibody [111].
CD38 has a particular significance in AML, given that up to 83% of AML cells express CD38. Efforts must, therefore, be made to maximize the level of CD38 expression and binding intensity for these CAR-T treatments. To further enhance the efficacy of CAR-T therapy in AML, a study by Kazuyoshi Yoshida showed that all-trans retinoic acid (ATRA) could raise CD38 expression on AML cells [43].
In order to prevent CAR-T/NK cell depletion, CD38 expression should be knocked down from the T or NK cells using, for example, the CRISPR-Cas9 technology [44]. CD38 is expressed in immune cells such as T, B, and NK cells. Anti-CD38 CAR-NK cells with CD38 knockdown have shown significant benefits in preclinical studies [42]. Six AML patients who suffered from recurrent AML following allo-HSCT have been enrolled in CD38 CAR-T clinical studies (NCT04351022). Four weeks after the initial CD38 CAR-T cell infusion, four out of the six patients achieved CR or CR with incomplete count recovery (CRi). One case experienced a relapse 117 days after the first CD38 CAR-T cell infusion and went into remission after the second infusion. All six of the patients had clinically manageable side effects, including grade I–II CRS and grade III hepatotoxicity. The results of the preliminary studies indicated that CD38 was a viable CAR-T target for the therapy of AML. And there are a number of clinical trials under recruitment (NCT05239689, NCT04351022).
Targets that have entered pre-clinical trials
A number of new targets have entered preclinical trials in addition to those that have already been the subject of clinical trials. These targets, such as CD117, FRβ, CD93, TIM3, and others, are currently being examined in animal models to determine their efficacy and safety.
CD117
CD117 (c-kit) plays an important role in HSC homeostasis [32, 112]. Its level of expression correlates with the prognosis of AML.
Preclinical research on CD117 is currently underway. Because CD117 is highly expressed in HSPC, CAR treatments targeting CD117 can cause severe suppression of hematopoiesis. Therefore, a bridge to transplantation is necessary. To this end, Myburgh et al. proposed a three-step immunotherapy technique consisting of the eradication of AML cells and original HSPCs via CD117 CAR-T cells, cessation of clearance effects, and finally, transplantation of healthy HSPC [32]. Complete elimination of healthy and leukemic cells was shown in a xenograft mouse model with CAR-T treatment, and subsequent hematopoiesis recovery after depletion of CAR-T cells with anti-thymocyte globulin and rituximab, followed by HSCT. Several in vitro investigations showed that, despite promising findings, low target antigen expression and loss of this antigen under selective therapeutic pressure can lead to CAR-T cells not fully exerting their effects [32]. Therefore, more investigation into the benefits and downsides of CD117 CAR-T cell therapy is required, along with an examination of how antigen loss can affect clinical trials in the future.
CD4
CD4, a T cell membrane glycoprotein that binds to major histocompatibility complex class II antigens, is expressed at significantly high levels in some AML subtypes. For instance, CD4 is expressed in 65.0% and 78.3% of the acute myeloid leukemia and acute monocytic leukemia, respectively, but only in 30–40% of other subtypes. It is neither expressed on HSPCs, nor on non-hematopoietic cells. CD4 is a promising target for CAR-T treatment in AML because of its consistent, albeit low, expression.
The initial CD4 CAR treatment preclinical trial, assessing its safety and efficacy, was undertaken by Huda Salman et al. The result showed that it was effective at killing CD4 + AML cells in culture and in animals [112]. It inhibited the growth of leukemia cells in a xenograft mice model and improved survival time. Transient CD4 + T cell reduction and off-target toxicity were still detected since CD4 is also expressed in T cells; however, hematopoiesis was unaffected. These results provide credence to CD4 CAR-T cells’ feasibility as a bridging therapy preceding HSCT for tumor burden reduction or remission induction. Additionally, a safety switch based on alemtuzumab (anti-CD52 antibody) could be utilized to rapidly and completely reduce CD4 + CAR-T cell levels in patients who are ineligible for HSCT transplantation, optimizing killing efficacy whilst minimizing toxicity.
FRβ
A total of four receptors—FRα, FRβ, FRγ, and FRδ—make up the folate receptor (FR) family and are found in varying degrees of expression throughout different organs and tissues. Epithelial cells are the only ones that express FRα, although myeloid hematopoietic cells predominately express FRβ. In addition, FRβ can also be induced to be up-regulated during macrophage activation, which is a possible mechanism for off-target toxicity of FRβ CAR-T cells [113].
Preclinical testing shows that FR CAR-T has anti-leukemic activity both in vitro and in vivo. However, the treatment’s potential efficacy in people with low FRβ expression is concerned, because the degree of CAR activation is correlated with the expression level of FR. Researchers have found that ATRA can increase FRβ expression in primary AML cells. Further stimulation of FR expression on AML in vitro was observed when ATRA was combined with histone deacetylase (HDAC) inhibitors [114]. Optimizing the use of ATRA in conjunction with other FR-inducing treatments, represented by HDAC, may not only extend the therapeutic reach of ATRA in AML, but also the efficacy of FRβ-targeted therapy.
FRβ CAR-T cell treatment has entered preclinical trials but not clinical trials.
CD93
CD93 is a transmembrane antibody of the C-type lectin family that has a role in both cell–cell adhesion and host immune defense.
Richards et al. collaborated with Dr. Majeti to investigate a CD93 CAR-T preclinical trial in 2021 [63]. In vivo, the study showed that although damage to HSPCs was minimal, there was severe “on-target, off-tumor” injury due to the ubiquitous expression of CD93 on endothelial cells. The team developed a non-gated CAR-T design optimization strategy, as a result, allowing non-gated CAR T cells to avoid the off-tumor toxicity. These non-gated CD93 CAR T cells also expressed a second inhibitory CD19 CAR (iCAR), which was chosen for this investigation because it selectively targeted CD19, an antigen that is present on healthy cells but absent from AML cells. A CD19-specific inhibitory CAR consists of a CD19 scFv, a CD8α transmembrane signaling internal domain, and an immunoreceptor tyrosine-based inhibitory motif which encodes proteins, including PD-1 and TIGIT. This non-gated CAR-T demonstrated “on-target, on-tumor” cytotoxicity to CD93 + AML cells, while “on-target, off-tumor” effect to CD93 + immortalized human umbilical vein endothelial cells which was engineered to stably express truncated CD19, thus killing the AML cells but avoiding the toxicity to endothelial cell.
MSLN
Mesothelin (MSLN), which is completely absent during normal hematopoiesis, is highly expressed in AML cells. This differential expression suggests a new target for CAR-T therapy with the goal of protecting hematopoiesis.
Preclinical testing of MSLN CAR-T for the treatment of AML was undertaken by Quy Le et al [115]. Primary AML cells and a CD34+CD38− cell subpopulation were shown to express MSLN at the cell surface. To prove that MSLN is a therapeutically realistic target for CAR-T cell therapy in AML, the researchers used a xenograft model to show that MSLN CAR-T successfully killed MSLN+ AML cells while also targeting and eliminating CD34+CD38− cells without interfering with the function of normal HSCs. Additionally, the team found that suppression of ADAM17 metalloproteinase, a protease that promotes MSLN shedding, could help to sustain CAR-T function.
IL-10R
Interleukin receptor 10 receptor (IL-10R) includes two α and two β molecules.
Overexpression of IL-10R was found in the vast majority of AML cells, and IL-10 was found to promote cell proliferation in these cells by activating the IL-10R/PI3K/AKT/OCT4 signaling axis [116]. To take advantage of this, a new CAR-T cell was developed using IL-10 as its natural ligand as the antigen-binding structural domain; this CAR-T cell showed a considerable killing capability on AML cells both in vitro and in vivo. Additionally, the expression of IL-10 on the surface of CAR-T cells had no impact on their survival, biological activity, or ability to proliferate, and normal hematopoietic cells, like HSPCs, appeared to sustain no off-target damage. These findings imply that IL-10R is a promising AML CAR-T treatment target.
WT1
Wilms Tumor 1 (WT1) is an intracellular protein, and a zinc-finger transcription factor frequently associated with cancer. It plays a role in many different biological processes, such as organ development, differentiation, proliferation, and apoptosis [59]. Its expression is considerably elevated in various types of solid tumors and hematological malignant cells but relatively modest in healthy tissues. A poorer prognosis is seen in AML when WT1 is overexpressed.
Intracellular localization of WT1 prevents antibodies and antibody-derived CAR-T cells from recognizing its antigens [60]. So, it is difficult in generating effective WT1 CAR-T cells. Researchers have discovered that peptides produced from intracellular WT1 can be expressed in the context of human leukocyte antigen on the surface of tumor cells (HLA). WT1 CAR can be designed based on the monoclonal antibody, such as ESK1 [60], which can recognize WT1/HLA A2 complexes. Moreover, if IL-12 gene is integrated into CAR, it can improve T-cell activity and amplify the therapeutic benefit. This approach was applied by Rafiq S. et al., who discovered that co-administration of IL-12 with T cell receptor-mimic CAR-T improves the efficiency of T cells against tumors and has the potential to boost anti-tumor treatment responses.
GRP78 (HSPA5)
Glucose regulating protein 78 (GRP78) is typically found in the endoplasmic reticulum (ER) and plays a crucial role in controlling the highly conserved unfolded protein response during species evolution. Because the ER has so little space, GRP78 is shuttled out to the cell surface in a cancer-specific form. This process has been observed in many types of cancer, both solid and blood-related. Despite its potential as a target for CAR-T treatment in AML due to its expression properties, its efficacy has been the subject of relatively few investigations.
To determine whether or not GRP78 CAR-T cells could be used to treat AML, Nikhil Hebbar et al. conducted in vitro and in vivo tests, finding that the CAR-T cells successfully recognized and eradicated GRP78+ AML, but HSPCs did not elicit a substantial toxic response [117]. Blocking differentiation with dasatinib may also increase the target-killing impact of CAR-T because antigen-dependent T cell differentiation may limit CAR-efficacy.
Future directions
For the therapy of AML, researchers are looking into a wide variety of avenues. Though some offer more promise than others, the development of any of them could lead to useful therapeutics. The biochemical pathways that these prospective targets cover need to be thoroughly explored in order to guarantee safe and effective therapeutics.
Transmembrane proteins during CAR exploration phase include the well-known programmed death 1 (PD1) protein. Despite the critical role played by the PD-1/PD-L1 axis in immunosuppression in AML, it is not known if PD-L1 is constitutively expressed in AML primary cells. The expression of PD-L1 may be upregulated in response to inflammatory stimuli such as toll-like receptor (TLR) ligands and interferon. A poorer prognosis may be associated with AML cells expressing PD-L1 and/or PD-L2 [64]. Monoclonal antibodies and blockers targeting PD-1 can effectively induce anti-tumor immune responses, reduce tumor burden, and increase OS in AML patients. Studies have shown that PD-1-deficient CLL-1 CAR-T cells are superior at killing off AML cells. The PD-1-deficient CLL-1 CAR-T cells were effective in treating two individuals with R/R AML, resulting in molecular complete remission and incomplete hematological recovery for both.
Just like WT1 mentioned above, unique intracellular tumor-specific mutational epitopes can also be employed for CAR-T cell treatment, and studies targeting intracellular antigens have the potential to expand CAR recognition beyond extracellular antigens. To do this, innovative strategies, including TCRm CAR, have been designed and are currently being successfully evaluated in scientific cases [60]. Studies on IDH1, IDH2, RHAMM, Survivin, hTERT, and many other topics have been published [118, 119]. Additionally, the sequentially tumor-selected antibody and antigen retrieval (STAR) system can be used to screen for nanobodies that preferentially bind to AML cells [97], and small conditional RNA (scRNA)-seq can provide information on immunotherapeutic target screening [120].
More cancer patients may benefit from CAR-T cell treatment if it is expanded to the targeting of intracellular antigens.
Conclusions
The treatment of AML includes traditional chemotherapy, targeted drug and hematopoietic stem cell transplantation, as well as CAR-T and CAR-NK cell therapies. Every therapeutic way has its unique advantages and challenges.
Traditional chemotherapy regimens for AML have been developed so mature until now and are the first-line treatment for newly diagnosed AML. While the organ damage and hematopoietic inhibition caused by chemotherapeutics may be fatal to patients, especially to the elders. Novel small molecular targeting drugs, such as gilteritinib (FLT3 inhibitor) [3], enasidenib (IDH2 inhibitor) [4], ivosidenib (IDH1 inhibitor) [5], or venetoclax (BCL-2 inhibitor) [6], have shown promising results. However, the cost of these drugs can be a challenge, and they may not be accessible to all patients. Stem cell transplantation remains a viable option for AML, particularly in patients who are young and have a suitable donor. However, this treatment can also cause severe side effects, such as graft-versus-host disease and infection. CAR-T and CAR-NK cell therapies have shown promising results in the treatment of AML, particularly in patients who are relapsed or refractory after standard therapies. CAR-T and CAR-NK cell therapies can target specific antigens on cancer cells and kill the tumor. CAR-NK cell therapy has the potential to target a wider range of tumor cells, including those that are resistant to T cell therapy. However, the use of CAR-T cell therapy can also cause severe side effects, such as cytokine release syndrome and neurotoxicity, which can be life-threatening. Furthermore, the effectiveness of CAR-NK cell therapy may be limited by the availability of suitable NK cells for engineering.
The treatment approach for AML should be tailored to the individual patient’s condition and disease severity. For the younger, or those in good physical condition, traditional chemotherapy followed by stem cell transplantation may be the preferred treatment option, as it can provide long-term therapeutic benefits. For the elder, or those with comorbidities, targeted drug therapy may be the more suitable option, as it can offer a gentler treatment and can be administered at home. For the patients who have already undergone traditional treatments, but have not achieved sustained remission, CAR-T and CAR-NK cell therapies may be the best option.
In general, the treatment sequence for AML should be determined based on the individual patient’s condition and disease severity. Stem cell transplantation following chemotherapy may be the first-line treatment option for the younger or fit patients. Targeted drug therapy may be a more suitable option for older or frail patients. CAR-T and CAR-NK cell therapies may be the best option for patients with relapsed and/or refractory AML. When selecting a treatment approach, besides individual patient’s condition and comorbidities, the communication and collaboration with their family are also important.
Availability of data and materials
Not applicable.
Abbreviations
- Allo-HSCT:
-
Allogenic hematopoietic stem cell transplantation
- AML:
-
Acute myeloid leukemia
- ATRA:
-
All-trans retinoic acid
- CAR-T:
-
Chimeric antigen receptor T cell
- CLL-1:
-
C-type lectin-like molecule-1
- CR:
-
Complete remission
- CRS:
-
Cytokine release syndrome
- ER:
-
Endoplasmic reticulum
- FDA:
-
U.S. Food and Drug Administration
- FLT3:
-
Fms-like tyrosine kinase 3
- FLT3Lg:
-
FLT3 ligament
- FR:
-
Folate Receptor
- GRP78:
-
Glucose regulated protein 78
- GVHD:
-
Graft-versus-host disease
- HDAC:
-
Histone deacetylase
- HSPC:
-
Hematopoietic stem and progenitor cells
- IL:
-
Interleukin; ITD: Internal tandem duplication
- KDEL:
-
Lys-Asp-Glu-Leu, a peptide sequence
- LILRB:
-
Leukocyte immunoglobulin-like receptor B
- LSC:
-
Leukemic stem cells
- mAb:
-
Monoclonal antibodies
- MDS:
-
Myelodysplastic syndromes
- MLFS:
-
Morphological leukemia-free state
- MM:
-
Multiple myeloma
- MSLN:
-
Mesothelin
- NK:
-
Natural killer cell
- NKG2DL:
-
Natural killer group 2, member D ligands
- OS:
-
Overall survival
- PD1:
-
Programmed cell death protein 1
- PI3K:
-
Phosphoinositide 3-kinases
- R/R:
-
Relapsed or refractory
- RevCAR:
-
Reversible chimeric antigen receptor
- scFv:
-
Single-chain variable fragment
- SUPRA:
-
Split, universal, and programmable CAR
- TCRm:
-
T-cell receptor mimic
- TKD:
-
Tyrosine kinase domain
- TM:
-
Targeting module
- TNF:
-
Tumor necrosis factor
- Treg:
-
Regulatory T cells
- UniCAR:
-
Universal CAR
- 5’-AZA:
-
5’-Azacytidine
References
Appelbaum FR. Effectiveness of allogeneic hematopoietic cell transplantation for older patients with acute myeloid leukemia. Best Pract Res Clin Haematol. 2021;34(4): 101320.
Kreidieh F, Abou Dalle I, Moukalled N, El-Cheikh J, Brissot E, Mohty M, et al. Relapse after allogeneic hematopoietic stem cell transplantation in acute myeloid leukemia: an overview of prevention and treatment. Int J Hematol. 2022;116(3):330–40.
Perl AE, Martinelli G, Cortes JE, Neubauer A, Berman E, Paolini S, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N Engl J Med. 2019;381(18):1728–40.
Pollyea DA, Tallman MS, de Botton S, Kantarjian HM, Collins R, Stein AS, et al. Enasidenib, an inhibitor of mutant IDH2 proteins, induces durable remissions in older patients with newly diagnosed acute myeloid leukemia. Leukemia. 2019;33(11):2575–84.
Martelli MP, Martino G, Cardinali V, Falini B, Martinelli G, Cerchione C. Enasidenib and ivosidenib in AML. Minerva Med. 2020;111(5):411–26.
Pollyea DA, Amaya M, Strati P, Konopleva MY. Venetoclax for AML: changing the treatment paradigm. Blood Adv. 2019;3(24):4326–35.
Marofi F, Rahman HS, Al-Obaidi ZMJ, Jalil AT, Abdelbasset WK, Suksatan W, et al. Novel CAR T therapy is a ray of hope in the treatment of seriously ill AML patients. Stem Cell Res Ther. 2021;12(1):465.
Xie G, Dong H, Liang Y, Ham JD, Rizwan R, Chen J. CAR-NK cells: a promising cellular immunotherapy for cancer. EBioMedicine. 2020;59: 102975.
Marofi F, Saleh MM, Rahman HS, Suksatan W, Al-Gazally ME, Abdelbasset WK, et al. CAR-engineered NK cells; a promising therapeutic option for treatment of hematological malignancies. Stem Cell Res Ther. 2021;12(1):1–18.
Lu H, Zhao X, Li Z, Hu Y, Wang H. From CAR-T cells to CAR-NK cells: a developing immunotherapy method for hematological malignancies. Front Oncol. 2021;11: 720501.
Watanabe N, Mo F, McKenna MK. Impact of Manufacturing Procedures on CAR T Cell Functionality. Front Immunol. 2022;13:876339.
Kenderian SS, Ruella M, Shestova O, Klichinsky M, Aikawa V, Morrissette JJD, et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia. 2015;29(8):1637–47.
Liu Y, Wang S, Schubert ML, Lauk A, Yao H, Blank MF, et al. CD33-directed immunotherapy with third-generation chimeric antigen receptor T cells and gemtuzumab ozogamicin in intact and CD33-edited acute myeloid leukemia and hematopoietic stem and progenitor cells. Int J Cancer. 2022;150(7):1141–55.
Rafiq S, Purdon TJ, Schultz LM, Brentjens RJ. CD33-Directed Chimeric Antigen Receptor (CAR) T Cells for the Treatment of Acute Myeloid Leukemia (AML). Blood. 2016;128(22):2825-.
Lamble AJ, Eidenschink Brodersen L, Alonzo TA, Wang J, Pardo L, Sung L, et al. CD123 expression is associated with high-risk disease characteristics in childhood acute myeloid leukemia: a report from the children’s oncology group. J Clin Oncol. 2022;40(3):252–61.
Boucher JC, Shrestha B, Vishwasrao P, Leick MB, Tu N, Ghafoor T, et al. Bispecific CD33/CD123 targeted chimeric antigen receptor T cells for the treatment of acute myeloid leukemia. Blood. 2022;140(Supplement 1):10275–6.
El Khawanky N, Hughes A, Yu W, Myburgh R, Matschulla T, Taromi S, et al. Demethylating therapy increases anti-CD123 CAR T cell cytotoxicity against acute myeloid leukemia. Nat Commun. 2021;12(1):6436.
Ehninger A, Kramer M, Röllig C, Thiede C, Bornhäuser M, von Bonin M, et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. 2014;4(6): e218.
Jetani H, Navarro-Bailón A, Maucher M, Frenz S, Verbruggen C, Yeguas A, et al. Siglec-6 is a novel target for CAR T-cell therapy in acute myeloid leukemia. Blood. 2021;138(19):1830–42.
Li C, Chen X, Yu X, Zhu Y, Ma C, Xia R, et al. Tim-3 is highly expressed in T cells in acute myeloid leukemia and associated with clinicopathological prognostic stratification. Int J Clin Exp Pathol. 2014;7(10):6880–8.
Gomes-Silva D, Atilla E, Atilla PA, Mo F, Tashiro H, Srinivasan M, et al. CD7 CAR T cells for the therapy of acute myeloid leukemia. Mol Ther. 2019;27(1):272–80.
Riether C, Pabst T, Höpner S, Bacher U, Hinterbrandner M, Banz Y, et al. Targeting CD70 with cusatuzumab eliminates acute myeloid leukemia stem cells in patients treated with hypomethylating agents. Nat Med. 2020;26(9):1459–67.
Mark BL, Harrison S, Irene S, Rebecca L, Bryan DC, Amanda AB, et al. Rational chemical and genetic modifications enhance avidity and function of CD70-directed CAR-T-cells for myeloid leukemia. Blood. 2021;138:405.
John S, Chen H, Deng M, Gui X, Wu G, Chen W, et al. A novel Anti-LILRB4 CAR-T Cell for the treatment of monocytic AML. Mol Ther. 2018;26(10):2487–95.
Deng M, Gui X, Kim J, Xie L, Chen W, Li Z, et al. LILRB4 signalling in leukaemia cells mediates T cell suppression and tumour infiltration. Nature. 2018;562(7728):605–9.
Frazao A, Rethacker L, Messaoudene M, Avril MF, Toubert A, Dulphy N, et al. NKG2D/NKG2-ligand pathway offers new opportunities in cancer treatment. Front Immunol. 2019;10:661.
Barber A, Meehan KR, Sentman CL. Treatment of multiple myeloma with adoptively transferred chimeric NKG2D receptor-expressing T cells. Gene Ther. 2011;18(5):509–16.
Baumeister SH, Murad J, Werner L, Daley H, Trebeden-Negre H, Gicobi JK, et al. Phase I trial of autologous CAR T cells targeting NKG2D ligands in patients with AML/MDS and multiple myeloma. Cancer Immunol Res. 2019;7(1):100–12.
David AS, Tessa K, Xavier P, Violaine H, Philippe L, Marco LD, et al. Remissions in relapse/refractory acute myeloid leukemia patients following treatment with NKG2D CAR-T therapy without a prior preconditioning chemotherapy. Blood. 2018;132:902.
Deeren D, Maertens JA, Lin T, Beguin Y, Demoulin B, Fontaine M, et al. First results from the dose escalation segment of the phase i clinical study evaluating Cyad-02, an optimized non gene-edited engineered NKG2D CAR T-cell product, in relapsed or refractory acute myeloid leukemia and myelodysplastic syndrome patients. Blood. 2020;136(Supplement 1):36-.
Lichtman EI, Du H, Shou P, Song F, Suzuki K, Ahn S, et al. Preclinical evaluation of B7-H3-specific chimeric antigen receptor T cells for the treatment of acute myeloid leukemia. Clin Cancer Res. 2021;27(11):3141–53.
Myburgh R, Kiefer JD, Russkamp NF, Magnani CF, Nuñez N, Simonis A, et al. Anti-human CD117 CAR T-cells efficiently eliminate healthy and malignant CD117-expressing hematopoietic cells. Leukemia. 2020;34(10):2688–703.
Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, Belton AA, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001;98(6):1752–9.
Wang Y, Xu Y, Li S, Liu J, Xing Y, Xing H, et al. Targeting FLT3 in acute myeloid leukemia using ligand-based chimeric antigen receptor-engineered T cells. J Hematol Oncol. 2018;11(1):60.
Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100(5):1532–42.
Wang B, Yang B, Ling Y, Zhang J, Hua X, Gu W, et al. Role of CD19 and specific KIT-D816 on risk stratification refinement in t(8;21) acute myeloid leukemia induced with different cytarabine intensities. Cancer Med. 2021;10(3):1091–102.
Tasian SK. Acute myeloid leukemia chimeric antigen receptor T-cell immunotherapy: how far up the road have we traveled? Ther Adv Hematol. 2018;9(6):135–48.
Peinert S, Prince HM, Guru PM, Kershaw MH, Smyth MJ, Trapani JA, et al. Gene-modified T cells as immunotherapy for multiple myeloma and acute myeloid leukemia expressing the Lewis Y antigen. Gene Ther. 2010;17(5):678–86.
Daver N, Alotaibi AS, Bucklein V, Subklewe M. T-cell-based immunotherapy of acute myeloid leukemia: current concepts and future developments. Leukemia. 2021;35(7):1843–63.
Zhang H, Gan W-T, Hao W-G, Wang P-F, Li Z-Y, Chang L-J. Successful Anti-CLL1 CAR T-Cell Therapy in Secondary Acute Myeloid Leukemia. Front Oncol. 2020;10:685.
Tashiro H, Sauer T, Shum T, Parikh K, Mamonkin M, Omer B, et al. Treatment of acute myeloid leukemia with T cells expressing chimeric antigen receptors directed to c-type lectin-like molecule 1. Mol Ther. 2017;25(9):2202–13.
Cui Q, Qian C, Xu N, Kang L, Dai H, Cui W, et al. CD38-directed CAR-T cell therapy: a novel immunotherapy strategy for relapsed acute myeloid leukemia after allogeneic hematopoietic stem cell transplantation. J Hematol Oncol. 2021;14(1):82.
Yoshida T, Mihara K, Takei Y, Yanagihara K, Kubo T, Bhattacharyya J, et al. All-trans retinoic acid enhances cytotoxic effect of T cells with an anti-CD38 chimeric antigen receptor in acute myeloid leukemia. Clin Transl Immunol. 2016;5(12):e116.
Gurney M, Stikvoort A, Nolan E, Kirkham-McCarthy L, Khoruzhenko S, Shivakumar R, et al. CD38 knockout natural killer cells expressing an affinity optimized CD38 chimeric antigen receptor successfully target acute myeloid leukemia with reduced effector cell fratricide. Haematologica. 2020;107(2):437–45.
Casucci M, Nicolis di Robilant B, Falcone L, Camisa B, Norelli M, Genovese P, et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood. 2013;122(20):3461–72.
Ghamari A, Pakzad P, Majd A, Ebrahimi M, Hamidieh AA. Design and production an effective bispecific tandem chimeric antigen receptor on T cells against CD123 and folate receptor ss towards B-acute myeloid leukaemia blasts. Cell J. 2021;23(6):650–7.
Lynn RC, Poussin M, Kalota A, Feng Y, Low PS, Dimitrov DS, et al. Targeting of folate receptor beta on acute myeloid leukemia blasts with chimeric antigen receptor-expressing T cells. Blood. 2015;125(22):3466–76.
Hasegawa A, Saito S, Narimatsu S, Nakano S, Nagai M, Ohnota H, et al. Mutated GM-CSF-based CAR-T cells targeting CD116/CD131 complexes exhibit enhanced anti-tumor effects against acute myeloid leukaemia. Clin Transl Immunology. 2021;10(5): e1282.
Kageyama Y, Miwa H, Arakawa R, Tawara I, Ohishi K, Masuya M, et al. Expression of CD25 fluctuates in the leukemia-initiating cell population of CD25-positive AML. PLoS ONE. 2018;13(12): e0209295.
Cerny J, Yu H, Ramanathan M, Raffel GD, Walsh WV, Fortier N, et al. Expression of CD25 independently predicts early treatment failure of acute myeloid leukaemia (AML). Br J Haematol. 2013;160(2):262–6.
Dehbashi M, Hojati Z, Motovali-bashi M, Ganjalikhany MR, Cho WC, Shimosaka A, et al. A novel CAR expressing NK cell targeting CD25 with the prospect of overcoming immune escape mechanism in cancers. Front Oncol. 2021;11:649710.
Saito Y, Kitamura H, Hijikata A, Tomizawa-Murasawa M, Tanaka S, Takagi S, et al. Identification of therapeutic targets for quiescent, chemotherapy-resistant human leukemia stem cells. Sci Transl Med. 2010;2(17):17ra9-ra9.
Chao MP, Takimoto CH, Feng DD, McKenna K, Gip P, Liu J, et al. Therapeutic targeting of the macrophage immune checkpoint CD47 in myeloid malignancies. Front Oncol. 2019;9:1380.
Zhao J, Zhang HT. Effect of CD56 expression on prognosis of AML patients with AML/ETO mutation. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2020;28(1):63–7.
Buccisano F, Rossi FM, Venditti A, Del Poeta G, Cox MC, Abbruzzese E, et al. CD90/Thy-1 is preferentially expressed on blast cells of high risk acute myeloid leukaemias. Br J Haematol. 2004;125(2):203–12.
Hosen N, Park CY, Tatsumi N, Oji Y, Sugiyama H, Gramatzki M, et al. CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia. Proc Natl Acad Sci U S A. 2007;104(26):11008–13.
Ågerstam H, Karlsson C, Hansen N, Sandén C, Askmyr M, von Palffy S, et al. Antibodies targeting human IL1RAP (IL1R3) show therapeutic effects in xenograft models of acute myeloid leukemia. Proc Natl Acad Sci. 2015;112(34):10786–91.
Stroopinsky D, Rosenblatt J, Ito K, Mills H, Yin L, Rajabi H, et al. MUC1 is a potential target for the treatment of acute myeloid leukemia stem cells. Cancer Res. 2013;73(17):5569–79.
Sugiyama H. WT1 (Wilms’ tumor gene 1): biology and cancer immunotherapy. Jpn J Clin Oncol. 2010;40(5):377–87.
Rafiq S, Purdon TJ, Daniyan AF, Koneru M, Dao T, Liu C, et al. Optimized T-cell receptor-mimic chimeric antigen receptor T cells directed toward the intracellular Wilms Tumor 1 antigen. Leukemia. 2017;31(8):1788–97.
Steger B, Floro L, Amberger DC, Kroell T, Tischer J, Kolb HJ, et al. WT1, PRAME, and PR3 mRNA Expression in Acute Myeloid Leukemia (AML). J Immunother. 2020;43(6):204–15.
Ma Q, Garber HR, Lu S, He H, Tallis E, Ding X, et al. A novel TCR-like CAR with specificity for PR1/HLA-A2 effectively targets myeloid leukemia in vitro when expressed in human adult peripheral blood and cord blood T cells. Cytotherapy. 2016;18(8):985–94.
Richards RM, Zhao F, Freitas KA, Parker KR, Xu P, Fan A, et al. NOT-Gated CD93 CAR T cells effectively target AML with minimized endothelial cross-reactivity. Blood Cancer Discovery. 2021;2(6):648–65.
Salih HR, Wintterle S, Krusch M, Kroner A, Huang YH, Chen L, et al. The role of leukemia-derived B7–H1 (PD-L1) in tumor-T-cell interactions in humans. Exp Hematol. 2006;34(7):888–94.
Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer. 2003;3(8):582–91.
Megías-Vericat JE, Ballesta-López O, Barragán E, Montesinos P. IDH1-mutated relapsed or refractory AML: current challenges and future prospects. Blood Lymphat Cancer. 2019;9:19–32.
Stein EM. IDH2 inhibition in AML. Blood. 2023;141(2):124–5.
Ranieri R, Pianigiani G, Sciabolacci S, Perriello VM, Marra A, Cardinali V, et al. Current status and future perspectives in targeted therapy of NPM1-mutated AML. Leukemia. 2022;36(10):2351–67.
Dong H, Ham JD, Hu G, Xie G, Vergara J, Liang Y, et al. Memory-like NK cells armed with a neoepitope-specific CAR exhibit potent activity against NPM1 mutated acute myeloid leukemia. Proc Natl Acad Sci U S A. 2022;119(25): e2122379119.
Adamia S, Bar-Natan M, Haibe-Kains B, Pilarski PM, Bach C, Pevzner S, et al. NOTCH2 and FLT3 gene mis-splicings are common events in patients with acute myeloid leukemia (AML): new potential targets in AML. Blood. 2014;123(18):2816–25.
Schneider V, Zhang L, Bullinger L, Rojewski M, Hofmann S, Wiesneth M, et al. Leukemic stem cells of acute myeloid leukemia patients carrying NPM1 mutation are candidates for targeted immunotherapy. Leukemia. 2014;28(8):1759–62.
Zhang F, Liu X, Chen C, Zhu J, Yu Z, Xie J, et al. CD244 maintains the proliferation ability of leukemia initiating cells through SHP-2/p27(kip1) signaling. Haematologica. 2017;102(4):707–18.
Casalegno-Garduño R, Meier C, Schmitt A, Spitschak A, Hilgendorf I, Rohde S, et al. Immune responses to RHAMM in patients with acute myeloid leukemia after chemotherapy and allogeneic stem cell transplantation. Clin Dev Immunol. 2012;2012: 146463.
Huang J, Lyu H, Wang J, Liu B. Influence of survivin-targeted therapy on chemosensitivity in the treatment of acute myeloid leukemia. Cancer Lett. 2015;366(2):160–72.
Greiner J, Brown E, Bullinger L, Hills RK, Morris V, Döhner H, et al. Survivin’ acute myeloid leukaemia-a personalised target for inv(16) patients. Int J Mol Sci. 2021;22(19):10482.
Eid MM, Helmy NA, Omar IM, Mohamed AA, El Sewefy D, Fadel IM, et al. Clinical significance of telomerase genes (hTERC and hTERT) amplification in patients with acute myeloid leukemia. Gulf J Oncolog. 2013;1(13):51–60.
Tong Y, Xiang Y, Li B, Bao S, Zhou Y, Yuan W, et al. Association between TERT gene polymorphisms and acute myeloid leukemia susceptibility in a Chinese population: a case–control study. Cancer Cell Int. 2020;20(1):313.
Yik MY, Azlan A, Rajasegaran Y, Rosli A, Yusoff NM, Moses EJ. Mechanism of human telomerase reverse transcriptase (hTERT) regulation and clinical impacts in leukemia. Genes (Basel). 2021;12(8):1188.
Salman H, Pinz KG, Wada M, Shuai X, Yan LE, Petrov JC, et al. Preclinical targeting of human acute myeloid leukemia using CD4-specific chimeric antigen receptor (CAR) T Cells and NK cells. J Cancer. 2019;10(18):4408–19.
Wittwer NL, Brumatti G, Marchant C, Sandow JJ, Pudney MK, Dottore M, et al. High CD123 levels enhance proliferation in response to IL-3, but reduce chemotaxis by downregulating CXCR4 expression. Blood Adv. 2017;1(15):1067–79.
Testa U, Pelosi E, Frankel A. CD 123 is a membrane biomarker and a therapeutic target in hematologic malignancies. Biomark Res. 2014;2(1):4.
Moshaver B, Wouters RF, Kelder A, Ossenkoppele GJ, Westra GAH, Kwidama Z, et al. Relationship between CD34/CD38 and side population (SP) defined leukemia stem cell compartments in acute myeloid leukemia. Leuk Res. 2019;81:27–34.
Zeijlemaker W, Grob T, Meijer R, Hanekamp D, Kelder A, Carbaat-Ham JC, et al. CD34(+)CD38(-) leukemic stem cell frequency to predict outcome in acute myeloid leukemia. Leukemia. 2019;33(5):1102–12.
Herrmann H, Sadovnik I, Eisenwort G, Rülicke T, Blatt K, Herndlhofer S, et al. Delineation of target expression profiles in CD34+/CD38- and CD34+/CD38+ stem and progenitor cells in AML and CML. Blood Adv. 2020;4(20):5118–32.
Sun Y, Jianlin C, Yarong L, Li B, Qinghan W, Hongliang F, et al. Donor-derived CD123-targeted CAR T cell serves as a ric regimen for haploidentical transplantation in a patient with FUS-ERG+ AML. Front Oncol. 2019;9:1358.
Kovtun Y, Jones GE, Adams S, Harvey L, Audette CA, Wilhelm A, et al. A CD123-targeting antibody-drug conjugate, IMGN632, designed to eradicate AML while sparing normal bone marrow cells. Blood Adv. 2018;2(8):848–58.
Thokala R, Olivares S, Mi T, Maiti S, Deniger D, Huls H, et al. Redirecting specificity of T cells using the sleeping beauty system to express chimeric antigen receptors by mix-and-matching of vl and vh domains targeting CD123+ tumors. PLoS ONE. 2016;11(8): e0159477.
Budde L, Song JY, Kim Y, Blanchard S, Wagner J, Stein AS, et al. Remissions of acute myeloid leukemia and blastic plasmacytoid dendritic cell neoplasm following treatment with CD123-specific CAR T cells: a first-in-human clinical trial. Blood. 2017;130(Supplement 1):811-.
Bordignon C, Bonini C, Verzeletti S, Nobili N, Maggioni D, Traversari C, et al. Transfer of the HSV-tk gene into donor peripheral blood lymphocytes for in vivo modulation of donor anti-tumor immunity after allogeneic bone marrow transplantation. Hum Gene Ther. 1995;6(6):813–9.
Warda W, Da Rocha MN, Trad R, Haderbache R, Salma Y, Bouquet L, et al. Overcoming target epitope masking resistance that can occur on low-antigen-expresser AML blasts after IL-1RAP chimeric antigen receptor T cell therapy using the inducible caspase 9 suicide gene safety switch. Cancer Gene Ther. 2021;28(12):1365–75.
Katherine DC, Noelle F, Anne Marie N, Aliza S, Selina L, Randi EI, et al. Treating Relapsed Refractory (RR) AML with biodegradable anti-CD123 CAR modified T cells. Blood. 2017;130:1359.
Meyer JE, Loff S, Dietrich J, Spehr J, Jurado Jiménez G, von Bonin M, et al. Evaluation of switch-mediated costimulation in trans on universal CAR-T cells (UniCAR) targeting CD123-positive AML. Oncoimmunology. 2021;10(1):1945804.
Dutour A, Marin V, Pizzitola I, Valsesia-Wittmann S, Lee D, Yvon E, et al. In vitro and in vivo antitumor effect of anti-CD33 chimeric receptor-expressing EBV-CTL against CD33 acute myeloid leukemia. Adv Hematol. 2012;2012: 683065.
Tambaro FP, Singh H, Jones E, Rytting M, Mahadeo KM, Thompson P, et al. Autologous CD33-CAR-T cells for treatment of relapsed/refractory acute myelogenous leukemia. Leukemia. 2021;35(11):3282–6.
Wang Q-S, Wang Y, Lv H-Y, Han Q-W, Fan H, Guo B, et al. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol Ther. 2015;23(1):184–91.
Borot F, Wang H, Ma Y, Jafarov T, Raza A, Ali AM, et al. Gene-edited stem cells enable CD33-directed immune therapy for myeloid malignancies. Proc Natl Acad Sci U S A. 2019;116(24):11978–87.
He X, Feng Z, Ma J, Ling S, Cao Y, Gurung B, et al. Bispecific and split CAR T cells targeting CD13 and TIM3 eradicate acute myeloid leukemia. Blood. 2020;135(10):713–23.
Barsov EV. Selective immortalization of tumor-specific T cells to establish long-term T-cell lines maintaining primary cell characteristics. Methods Mol Biol. 2009;511:143–58.
Mestermann K, Giavridis T, Weber J, Rydzek J, Frenz S, Nerreter T, et al. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci Transl Med. 2019;11(499):eaau5907.
Cho JH, Collins JJ, Wong WW. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell. 2018;173(6):1426-38.e11.
Kittel-Boselli E, Soto KEG, Loureiro LR, Hoffmann A, Bergmann R, Arndt C, et al. Targeting acute myeloid leukemia using the revcar platform: a programmable, switchable and combinatorial strategy. Cancers (Basel). 2021;13(19):4785.
Giordano-Attianese G, Gainza P, Gray-Gaillard E, Cribioli E, Shui S, Kim S, et al. A computationally designed chimeric antigen receptor provides a small-molecule safety switch for T-cell therapy. Nat Biotechnol. 2020;38(4):426–32.
Liu F, Cao Y, Pinz K, Ma Y, Wada M, Chen K, et al. First-in-Human CLL1-CD33 Compound CAR T cell therapy induces complete remission in patients with refractory acute myeloid leukemia: update on phase 1 clinical trial. Blood. 2018;132(Supplement1):901-.
Saxena A, Sheridan DP, Card RT, McPeek AM, Mewdell CC, Skinnider LF. Biologic and clinical significance of CD7 expression in acute myeloid leukemia. Am J Hematol. 1998;58(4):278–84.
Silva D, Tashiro H, Srinivasan M, Brenner MK, Mamonkin M. CD7 CAR for the Treatment of Acute Myeloid and Lymphoid Leukemia. Blood. 2016;128(22):4555-.
Yi Tian P, Natasha V, Takahiro K, Noriko S, Elaine C-S, Dario C. Blockade of CD7 expression in T cells for effective chimeric antigen receptor targeting of T-cell malignancies. Blood Adv. 2017;1(25):2348–60.
Larson RA, Mandrekar SJ, Huebner LJ, Sanford BL, Laumann K, Geyer S, et al. Midostaurin reduces relapse in FLT3-mutant acute myeloid leukemia: the Alliance CALGB 10603/RATIFY trial. Leukemia. 2021;35(9):2539–51.
Jetani H, Garcia-Cadenas I, Nerreter T, Thomas S, Rydzek J, Meijide JB, et al. CAR T-cells targeting FLT3 have potent activity against FLT3−ITD+ AML and act synergistically with the FLT3-inhibitor crenolanib. Leukemia. 2018;32(5):1168–79.
Sommer C, Cheng HY, Nguyen D, Dettling D, Yeung YA, Sutton J, et al. Allogeneic FLT3 CAR T cells with an off-switch exhibit potent activity against AML and can be depleted to expedite bone marrow recovery. Mol Ther. 2020;28(10):2237–51.
Drent E, Groen RW, Noort WA, Themeli M, Lammerts van Bueren JJ, Parren PW, et al. Pre-clinical evaluation of CD38 chimeric antigen receptor engineered T cells for the treatment of multiple myeloma. Haematologica. 2016;101(5):616–25.
Usmani SZ, Weiss BM, Plesner T, Bahlis NJ, Belch A, Lonial S, et al. Clinical efficacy of daratumumab monotherapy in patients with heavily pretreated relapsed or refractory multiple myeloma. Blood. 2016;128(1):37–44.
Escribano L, Ocqueteau M, Almeida J, Orfao A, San Miguel JF. Expression of the c-kit (CD117) molecule in normal and malignant hematopoiesis. Leuk Lymphoma. 1998;30(5–6):459–66.
Ross JF, Chaudhuri PK, Ratnam M. Differential regulation of folate receptor isoforms in normal and malignant tissues in vivo and in established cell lines. Physiologic Clin implications Cancer. 1994;73(9):2432–43.
Lynn RC, Poussin M, Kalota A, Feng Y, Low PS, Dimitrov DS, et al. Targeting of folate receptor β on acute myeloid leukemia blasts with chimeric antigen receptor–expressing T cells. Blood. 2015;125(22):3466–76.
Le Q, Castro S, Tang T, Loeb AM, Hylkema T, McKay CN, et al. Therapeutic targeting of mesothelin with chimeric antigen receptor T cells in acute myeloid leukemia. Clin Cancer Res. 2021;27(20):5718–30.
Chen N, Xu Y, Mou J, Rao Q, Xing H, Tian Z, et al. Targeting of IL-10R on acute myeloid leukemia blasts with chimeric antigen receptor-expressing T cells. Blood Cancer J. 2021;11(8):144.
Hebbar N, Epperly R, Vaidya A, Thanekar U, Moore SE, Umeda M, et al. CAR T cells redirected to cell surface GRP78 display robust anti-acute myeloid leukemia activity and do not target hematopoietic progenitor cells. Nat Commun. 2022;13(1):587.
Saygin C, Carraway HE. Emerging therapies for acute myeloid leukemia. J Hematol Oncol. 2017;10(1):93.
Lichtenegger FS, Krupka C, Haubner S, Köhnke T, Subklewe M. Recent developments in immunotherapy of acute myeloid leukemia. J Hematol Oncol. 2017;10(1):142.
Chen G, Ning B, Shi T. Single-cell RNA-Seq technologies and related computational data analysis. Front Genet. 2019;10:317.
Acknowledgements
Not applicable.
Funding
This review article was supported by the National Natural Science Foundation of China (Grant No. 81700130).
Author information
Authors and Affiliations
Contributions
X.S. and K.X. designed the outline of this manuscript. R.S. and Z.L. were the main contributor to writing the first draft. H.X. prepared the tables. S.J. and Y.Z. selected and organized the references. J.L. reviewed and revised the references. R.H. reviewed and edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shao, R., Li, Z., Xin, H. et al. Biomarkers as targets for CAR-T/NK cell therapy in AML. Biomark Res 11, 65 (2023). https://doi.org/10.1186/s40364-023-00501-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-023-00501-9