
Abstract
N6-methyladenosine (m6A) is the most abundant modification of eukaryotic mRNA and is involved in almost every stage of RNA metabolism. The m6A modification on RNA has been demonstrated to be a regulator of the occurrence and development of a substantial number of diseases, especially cancers. Increasing evidence has shown that metabolic reprogramming is a hallmark of cancer and is crucial for maintaining the homeostasis of malignant tumors. Cancer cells rely on altered metabolic pathways to support their growth, proliferation, invasion and metastasis in an extreme microenvironment. m6A regulates metabolic pathways mainly by either directly acting on metabolic enzymes and transporters or indirectly influencing metabolism-related molecules. This review discusses the functions of the m6A modification on RNAs, its role in cancer cell metabolic pathways, the possible underlying mechanisms of its effects and the implication of this modification in cancer therapy.
Similar content being viewed by others

Introduction
N6-methyladenosine (m6A), an epigenetic modification, has been intensely studied in recent years. m6A is the predominant chemical modification on eukaryotic mRNA [1] and is involved in almost every stage of the RNA life cycle, including RNA splicing, nuclear output, decay, folding and translation [2, 3]. Significantly, m6A RNA modification plays a critical role in a variety of physiological processes and human diseases, such as tissue development [4], cell differentiation and pluripotency [5, 6], DNA damage repair [7], obesity [8], infertility [9] and even cancer progression [10]. Many studies have confirmed that m6A regulates the function of various RNA classes, including messenger RNAs (mRNAs) [11], ribosomal RNAs (rRNAs) [12], transfer RNAs (tRNAs) [13], long noncoding RNAs (lncRNAs) [14], microRNAs (miRNAs) [15,16,17], and circular RNAs (circRNAs) [18]. Due to the diversity of RNA modification sites, m6A can either facilitate or suppress the progression of many diseases, including human cancers. Recently, the most widely employed m6A detection technique has been methylated RNA immunoprecipitation sequencing [19], which can be used to identify m6A methylation hypermethylated regions. In contrast to methylated RNA immunoprecipitation sequencing (MeRIP-Seq/m6A-Seq), liquid chromatography–mass spectrometry (LC‒MS) is used to measure the overall m6A level of mRNA. An increasing number of novel methods, such as m6A individual-nucleotide resolution crosslinking and immunoprecipitation (miCLIP) [20], site-specific cleavage and radioactive labeling followed by ligation-assisted extraction and thin-layer chromatography (SCARLET) [21], have been developed to identify the diverse functions affected by the m6A modification.
Metabolic reprogramming, associated with a variety of deregulated metabolic pathways [22] and metabolic enzymes, is a main feature of cancer [23]. The metabolic pathways in cancer cells with increased proliferation are better adapted to survival in the extremely hypoxic and nutrient-altered microenvironment. In addition to aerobic glycolysis, best known as the Warburg effect [24], altered fatty acid metabolism, aberrant amino acid metabolism and mitochondrial metabolism have recently gained increasing attention in the field of cancer research.
Our review describes recent studies on the various functions of the m6A modification in cancer metabolic pathways, together with the pivotal biological roles of m6A in eukaryote cells. It then presents a discussion on the underlying mechanisms in cancer metabolism. These findings provide fresh insights into early cancer diagnosis and clinical therapies.
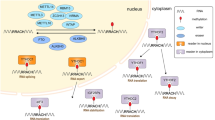
The m6A
Studies have revealed that approximately 0.1–0.4% of adenylate residues in mammals are modified with the m6A, accounting for approximately one-half of all methylated ribonucleosides [1, 25]. m6A, mostly deposited on the common DRm6ACH (D = A/G/U, R = G/A, H = A/C/U) motif [26,27,28,29], is enriched at the beginning of 3’-untranslated regions (3’-UTRs), proximal to translation termination codons and within internal long exons [19, 30]. The m6A modification is deposited, removed and identified by several writers, erasers and readers, respectively. The methyltransferase complex, termed the “writer”, catalyzes the addition of the m6A mark to target RNAs [31]. In contrast, demethylases, namely, “erasers”, remove m6A RNA methyl groups. To perform various downstream biological functions, “readers”, a group of specific proteins, bind to m6A methylation sites. Readers are proteins that specifically recognize and bind to m6A-modified sites, performing different downstream biological functions. Altogether, these enzymes modulate an integrated m6A network and are involved in cancer progression.
Methyltransferases mediating the m6A modification
m6A is installed by a multicomponent methyltransferase complex that mainly comprises the catalytic subunit METTL3 and many other components, such as METTL14 [32], METTL16 [33, 34], WTAP [35], VIRMA (KIAA1429) [36, 37], RBM15/15B [14] and ZC3H13 [38, 39]. In the 1990s, methyltransferase-like 3 (METTL3), a 70 kD protein, was discovered to be an important element of the m6A methyltransferase complex in human HeLa cells, and it primarily exhibited catalytic activity in eukaryotes ranging from yeast to humans [40, 41]. Homologous to METTL3, methyltransferase-like 14 (METTL14) was found to be located in nuclear plaques, and the two writers together formed a heterodimer. METTL14 is catalytically inactive but plays a significant role in facilitating METTL3 recognition of target RNAs [32, 42]. Methyltransferase-like 16 (METTL16), another m6A methyltransferase, has been reported to alter S-adenosylmethionine (SAM) levels and target precursor mRNAs and noncoding RNAs, including U6 small nuclear RNA [33, 34]. In addition, Wilms’ tumor 1-associated protein (WTAP) is a significant regulatory element in the m6A methyltransferase complex. Structurally, WTAP harbors no catalytic subunit and thus can neither catalyze the m6A modification nor positively affect the METTL3–METTL14 complex in vitro [32]. It has been demonstrated that WTAP is a regulator of METTL3-METTL14 complex accumulation in vivo. In other words, WTAP recruits METTL3 as well as METTL14 to constitute a stable dimer to ensure precise complex localization to nuclear spots [35].
Vir-like m6A methyltransferase associated (VIRMA), also KIAA1429, has been verified to facilitate mRNA methylation in mammalian cells [36]. Specifically, VIRMA preferentially recruits the METTL3/METTL14/WTAP component to 3’-UTRs and near a termination codon and is related to the cleavage factors CPSF5 and CPSF6. Moreover, RNA-binding motif protein 15 (RBM15) and its paralog RBM15B participate in m6A installation by guiding the m6A complex to specific RNA sites [14]. Zinc finger CCCH domain-containing protein 13 (ZC3H13) is important for m6A modification and required for nuclear localization of m6A by linking WTAP with Nito, an mRNA-binding factor [39].
Demethylases removing the m6A modification
Fat mass and obesity-associated protein (FTO), AlkB Homolog 5 (ALKBH5) and AlkB Homolog 3 (ALKBH3) are erasers that can dynamically reverse m6A RNA modification deposition. Among these three demethylases, FTO and ALKBH5 play major roles in removing the methyl group from the m6A mark. The first discovered demethylase of m6A modification was FTO. Previous studies have shown that FTO is associated with obesity and regulates energy homeostasis [43,44,45]. As a member of the dioxygenase AlkB family of proteins, FTO depends on Fe(ii) and α-ketoglutarate to catalyze the m6A modification [46]. Similar to FTO, iron(II)/α-ketoglutarate-dependent dioxygenase homolog 5 (ALKBH5) was later confirmed to be the second discovered m6A RNA eraser. Decreased levels of ALKBH5 augment m6A abundance on nuclear RNA, suggesting the reversibility of the m6A modification [47]. In 2017, another study confirmed that mammalian AlkB homolog 3 (ALKBH3) effectively promoted RNA demethylation and that the knockdown of ALKBH3 contributed to the accumulation of methylated RNAs. This research also identified tRNA as a new ALKBH3 substrate [48].
Proteins binding to RNA m6A sites
The m6A modification of RNA is regulated by the mutual effect between m6A methyltransferases and demethylases. However, subsequent biological processes require the identification of various proteins that bind to specific modification sites. These proteins are “readers,” which recognize m6A modifications alone or in combination. The most noted m6A readers are in the YT521-B homolog.
(YTH) domain family and insulin-like growth factor 2 mRNA-binding protein (IGF2BP) family [49]. The former set includes YTH domain family protein 1/2/3 (YTHDF1/2/3) and YTH domain-containing 1/2 (YTHDC1/2), while the latter family is composed of IGF2BP1/2/3. The m6A binding sites of YTHDF1 cluster around the stop codons, and then YTHDF1 recognizes m6A, interacts with the translation initiation complex and accelerates the translation process of mRNA in mammalian species [50]. In contrast, YTHDF2 promotes mRNA decay. The C-terminus of YTHDF2 selectively identifies m6A-modified mRNA, while the N-terminus recruits the carbon catabolite repression 4-negative on TATA-less (CCR4-NOT) complex and forms bridges between the mRNA and the processing body, thus mediating the decay of select transcripts [51]. YTHDF3 plays a synergistic role in RNA metabolism by interacting with other YTH domain family members. It reinforces the translation of targeted RNAs by cooperating with YTHDF1 [52] and facilitates the degradation of m6A-modified mRNA in the presence of YTHDF2 [53]. Another m6A reader, YTHDC1, was initially shown to facilitate exon inclusion and regulate mRNA splicing by recruiting serine- and arginine-rich splicing factor 3 (SRSF3) or restricting SRSF10 recruitment, bridging nuclear m6A-modified RNA to the nuclear export adaptor protein SRSF3, nuclear RNA export factor 1 (NXF1) and the three-component prime repair exonuclease (TREX) complex, mediating nuclear efflux [54,55,56]. Additionally, YTHDC1 preferentially identifies m6A residues on X-inactive specific transcript (XIST), which is a long noncoding RNA, indirectly repressing transcriptional [14]. YTHDC2 has been reported to function together with meiosis-specific protein (MEIOC) after binding m6A inside the consensus GGACU motif, reducing mRNA abundance while promoting the translation efficiency of target mRNAs [57]. An increasing number of studies have shown that insulin-like growth factor 2 mRNA-binding protein 1/2/3 (IGF2BP1/2/3) are required for the m6A reading process. The K homology (KH) domains are critical to the capability of these readers to recognize the m6A modification. In contrast to YTHDF2, IGF2BPs are promoters of the stability, storage and translation of mRNAs, highlighting their significant biological roles in gene regulation [49, 58, 59].
Several members of the heterogeneous nuclear ribonucleoprotein (HNRNP) family are also m6A readers, and they are critical to pre-mRNA processing [60]. Binding to m6A sites in certain RNA transcripts, heterogeneous nuclear ribonucleoprotein A2B1 (HNRNPA2B1) induces alternative splicing effects. The interaction between HNRNPA2B1 and the primary microRNA microprocessor complex protein DiGeorge syndrome critical region 8 (DGCR8) enhances the processing of primary miRNAs [61]. This action is called “m6A switching’’. Heterogeneous nuclear ribonucleoprotein C (HNRNPC) and heterogeneous nuclear ribonucleoprotein G (HNRNPG) binding can exert obvious effects on the splicing of m6A-modified mRNAs, whereas m6A can remodel RNA structure to facilitate the binding of HNRNPC and HNRNPG to mediate mRNA abundance and splicing. “m6A-switching’’ is a possible underlying mechanism of m6A wide-ranging physiological functions [2, 62]. Eukaryotic initiation factor 3 (eIF3) is an essential component for initiating cap-independent translation under basal cellular conditions, and it is recruited by m6A in 5’-UTRs and subsequently recruits the 43S complex to promote mRNA translation [63]. During mRNA circularization, eIF3H physically and functionally interacts with the m6A writer METTL3 to facilitate cap-dependent translation [64]. (Table 1)
m6A in cancer metabolic pathways
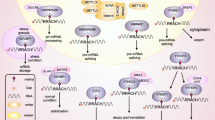
Increasing evidence shows that reprogrammed cellular metabolism is a main hallmark of cancer, in addition to the typical characteristics of tumor cell evasion of proliferation inhibitors, escape from immune attack, and capability of proliferation infinity; tumor-induced inflammation, invasion and metastasis; vascular leakage; genomic mutation; resistance to cell death and active proliferative signaling [65, 66]. A recent study reported that nonmutational epigenetic recombination, cellular senescence, phenotypic plasticity and polymorphic microbiomes are also hallmarks of cancer [67]. As one of the most noted characteristics in tumors, aberrant metabolism is, in part, critical for tumorigenesis and cancer progression. Various studies have verified that metabolic reprogramming is also a key factor in modulating resistance to chemotherapy [68, 69]. Tumor cells mainly take advantage of four metabolic pathways, aerobic glycolysis, altered fatty acid metabolism, glutamine-dependent anaplerosis and mitochondrial metabolism, to support biosynthesis and bioenergy metabolism [70, 71]. As the most prevalent RNA modification, m6A exerts widespread effects on cancer metabolism reprogramming by regulating a wide range of metabolic enzymes.
m6A in glucose metabolism pathways
In the 1950s, Warburg observed that glycolysis is highly activated in the presence of oxygen in cancer cells a process called aerobic glycolysis or the Warburg effect [24]. Depending on this typical glucose metabolism pathway, tumor cells can better adapt to hypoxic conditions to undergo malignant proliferation.
Glucose transporter 1 (GLUT1) is an important transporter in glycolysis, importing glucose into the cytoplasm. In gastric cancer (GC), the long noncoding RNA LINC00958 is highly expressed compared to normal gastric tissues and upregulated level of LINC00958 clinically indicates the poor survival of GC patients. The mechanism is that the m6A methyltransferase KIAA1429 catalyzes LINC00958 which stabilizes GLUT1 and promotes GLUT1 mRNA stability, thereby augmenting the effect of aerobic glycolysis of gastric cancer (GC) [72]. In colorectal cancer (CRC), METTL3 promotes GLUT1 translation to induce glucose uptake and lactate production and activates mammalian target of rapamycin complex 1(mTORC1) signaling, which results in cancer development [73]. High expression of ALKBH5 has been observed in breast cancer tissues from human epidermal growth factor receptor 2 (HER2)-therapy resistant patients. Mechanistic research has revealed that ALKBH5 targets GLUT4, ensuring its mRNA stability and facilitating glycolysis in breast cancer cells [74].
Hexokinase 2 (HK2) is an important enzyme in aerobic glycolysis, controlling the rate of glycolysis pathway activation. The m6A methyltransferase METTL3 links to the 3’-untranslated region of HK2 mRNA and recruits YTHDF1 to ensure HK2 stability, ultimately boosting aerobic glycolysis in cervical cancer (CC) cells, contributing to their proliferation and leading to poor prognosis in cervical cancer [75]. Glutamate also plays a role in pancreatic ductal adenocarcinoma by upregulating METTL3 activity and further promoting HK2 in an m6A dependent manner, ultimately enhancing the glycolysis rate [76]. The writer WTAP, playing functioning as an oncogene in gastric cancer and showing mechanistic action similar to that of METTL3, can bind the 3’-UTR m6A site of HK2, enhance its stability and augment the Warburg effect [77]. In lung cancer, sevoflurane inhibits tumor progression and aerobic glycolysis by indirectly reducing HK2 stability in a manner dependent on m6A [78]. In CRC cells, METTL3 is an oncogene that stabilizes HK2 as well as GLUT1 expression via IGF2BP2- or IGF2BP2/3-related mechanisms and activates the subsequent aerobic glycolysis pathway to boost colorectal cancer progression [79]. Additionally, in CRC cells, KIAA1429 increases HK2 levels to boost— aerobic glycolysis rate [80]. In addition to m6A writers, one of the m6A readers, YTHDF1, is closely related to cancer glucose metabolism. Based on the analysis of lncRNA expression data from gene expression omnibus (GEO) databases, it was discovered that Human Recombinant Protein 5 (HCP5) is significantly upregulated in esophageal squamous cell carcinoma (ESCC) and indicates unfavorable survival of ESCC patients. In mechanism, HCP5 directly interacts with YTHDF1, which subsequently promotes the binding of YTHDF1 to HK2 mRNA, enhances HK2 stability and thereby promotes ESCC progression [81].
Enolase 1 (ENO1), which catalyzes the synthesis of phosphoenolpyruvate (PEP) from 2-phosphoglycerate, plays a key role in promoting aerobic glycolysis. C5aR1-positive neutrophils reinforce aerobic glycolysis to drive breast cancer progression via m6A methylation of ENO1. The cascade involves C5aR1-positive neutrophils secreting IL-1β and TNF-α, thereby activating ERK1/2 signaling, leading to the phosphorylation of the m6A writer WTAP at serine341, which stabilizes it, thereby augmenting the abundance of ENO1 m6A methylation and facilitating glycolysis [82]. In lung adenocarcinoma (LUAD), the antagonism between the m6A writer METTL3 and eraser ALKBH5 is correlated with ENO1-dependent glycolysis. Due to the upregulation of METTL3 and downregulation of ALKBH5, an elevated m6A level on ENO1 suggests a bleak prognosis for LUAD patients because it stimulates glycolysis [83].
Similar to HK2, PKM2 catalyzes one of the rate-determining steps (RDSs) in glycolysis, producing pyruvate derived from phosphoenolpyruvate via substrate-level phosphorylation. The m6A eraser FTO has been demonstrated to show tumorigenic features in hepatocellular carcinoma (HCC). The Cancer Genome Atlas (TCGA) dataset shows that FTO is overexpressed in HCC, which is consistent with clinical data. In addition, the prognosis analysis reveals that high level of FTO indicates the poor survival of HCC patients. Mechanistically, FTO induces the demethylation of PKM2 mRNA to fuel translation, ultimately resulting in HCC [84]. In addition, the m6A writer YTHDF2 enhances PKM2 and subsequently augments aerobic glycolysis in breast cancer [85].
Lactate dehydrogenase A (LDHA) is a critical glycolytic enzyme that accelerates the dehydrogenation reaction of pyruvate conversion to lactic acid, while LDHB can reverse this process because of its higher affinity for lactate [86]. Recently, it was reported that METTL3 can upregulate the level of LDHA to trigger aerobic glycolysis in CRC, especially 5-FU-resistant CRC cells [87]. The landscape analysis based on TCGA shows that IGF2BP1 level is increased in clear cell renal cell carcinoma (ccRCC) and high expression of IGF2BP1 is closely correlated with the lower survival of ccRCC patients. IGF2BP1 participates in clear cell renal cell carcinoma (ccRCC) by recognizing sites on LDHA mRNA modified with m6A marks, increasing LDHA mRNA stability and accelerating aerobic glycolysis[88]. Moreover, R-2-hydroxyglutarate (R-2HG) specifically abrogates the upregulation of LDHB gene expression through m6A eraser FTO demethylation and thereby suppresses glycolysis in leukemia, indicating a function for m6A in cancer metabolic pathways [89] (Fig. 1).

The function of m6A in cancer glucose metabolic pathways
m6A can directly influence glucose transporters or metabolic enzymes to modulate glucose uptake and glycolysis in various cancers. Moreover, other enzymes that regulate glucose metabolism in tumors may be targets of m6A modification. For instance, it is demonstrated that ALKBH5 is downregulated in bladder cancer cells and predicts poor prognosis of bladder cancer individuals. ALKBH5 mediates glycolysis by attenuating the stability of casein kinase 2α (CK2α) in an m6A-dependent manner and thus suppressing the role of CK2α in the glycolytic pathway. The weakened function of CK2α can inhibit the multiplication of bladder cells and thus suppress tumor development [90]. Moreover, a series of studies confirmed that pyruvate dehydrogenase kinase 1 (PDK1) [91], pyruvate dehydrogenase kinase 4 (PDK4) [92], bromodomain PHD finger transcription factor (BPTF) [93], MYC proto-oncogene (MYC) [94,95,96,97] and hypoxia-inducible factor (HIF) [98,99,100] promoted glycolysis in tumor progression in an m6A-dependent manner. The detailed mechanisms are presented in Table 2.
In addition to aerobic glycolysis, branching glucose metabolic pathways, such as the pentose phosphate pathway (PPP), play important roles in the proliferation of tumor cells. m6A exerts an impact on the PPP to indirectly affect cancer metabolism. Numerous studies have shown that circ-0003215 inhibits tumor progression and metastasis. In CRC cells, YTHDF2 degrades circ-0003215 and further modulates the level of discs large MAGUK scaffold protein 4 (DLG4), which blocks the PPP via the ubiquitination of glucose-6-phosphate dehydrogenase (G6PD) [101]. Under serum starvation conditions, autophagy-induced METTL3 degradation promotes the stability of LINC01615. Thus, the overexpression of LINC01615 upregulates the level of G6PD by enhancing G6PD pre-mRNA splicing and further activates the PPP [102]. ALKBH5 expression is significantly increased in glioma cells and is involved in glioma cell proliferation. In the PPP, ALKBH5 facilitates the mRNA stability of G6PD, enhances its translation and ultimately stimulates cell proliferation [103]. In addition to G6PD, m6A targets 6-phosphogluconate dehydrogenase (6PGD), a cytosolic enzyme in the PPP, to influence cancer glucose metabolism. By recognizing the m6A site on 6PGD, YTHDF2 directly binds with 6PGD, promotes its expression and facilitates the progression of lung cancer [104].
Tricarboxylic acid cycle (TCA cycle), as the downstream pathways of glycolysis, is largely inhibited in primary solid tumor, which is one of the hallmarks of tumor. As shown in the previous studies, m6A modification plays an important role in glucose metabolism. In fact, the interaction between glucose metabolism and m6A is actually mutual. Although the study on the role of m6A in TCA cycle is limited, it is reported that the accumulation of intermediate products in TCA cycle can in turn regulate m6A modification. The lack of succinate dehydrogenase complex (SDH), an essential metabolic enzyme complex in TCA cycle, is quite common in gastrointestinal stromal tumors, which results in the abnormal accumulation of succinate, while succinate can effectively suppress α-ketoglutarate-dependent dioxygenase family enzymes which include m6A modification [105].
m6A in lipid metabolism pathways
Previous evidence has verified that lipid metabolism is deregulated in malignant tumors [106,107,108]. Elevated levels of de novo synthesis and altered fatty acid uptake and catabolism are the typical phenotypes acquired during lipid metabolic reprogramming, which satisfies the material needs for the proliferation of tumor cells and is conducive to cancer progression [109,110,111,112]. Mechanistically, the m6A modification exerts an effect on relevant metabolic enzymes and regulators to modulate lipid metabolism in cancer.
In lipid metabolism, ATP citrate lyase (ACLY) catalyzes the conversion of cytoplasmic citrate to acetyl-CoA. Subsequently, acetyl-CoA carboxylase-alpha (ACC1) catalyzes the ATP-dependent carboxylation of acetyl-CoA and then produces malonyl-CoA, which is consumed during fatty acid synthesis. Omics research shows that HNRNPA2B1 is substantially increased in esophageal cancer (ESCA) and the prognosis is worse in patients with a high level of HNRNPA2B1. HNRNPA2B1 upregulates ACLY and ACC1 gene expression during fatty acid metabolism to promote esophageal cancer (ESCA) progression, while the knockdown of HNRNPA2B1 suppresses the growth, metastasis and invasion of tumor cells [113]. In addition, the dysregulation of the m6A writers METTL3 and METTL14 is closely connected with HCC development. Mechanistically, METTL3/14 target ACLY and stearoyl-CoA desaturase 1 (SCD1), increase their expression, promote lipid metabolism and ultimately lead to HCC [114].
In HepG2 cells, the first m6A eraser to be identified, FTO, regulates lipogenesis via fatty acid synthase (FASN) in the final step of de novo synthesis in an m6A-dependent manner. By knocking down FTO, the expression of the m6A reader YTHDF2 and the abundance of m6A on FASN mRNA were markedly increased, leading to the instability and ultimate decay of FASN mRNA. As FASN is positively correlated with de novo lipogenesis, the reduced level off FASN eventually results in deficient lipid accumulation [115]. In addition, FTO modulates lipid metabolism in HepG2 cells by acting on the transcription factor sterol regulatory element-binding protein (SREBP) 1 C to affect the downstream effectors FASN, stearoyl-CoA desaturase 1 (SCD1), ACC1, diacylglycerol acyltransferase 2 (DGAT2), cell death-inducing DFFA (DNA fragmentation factor-α)-like effector c (CIDEC) and carnitine palmitoyl transferase 1 (CPT1), eventually promoting lipid synthesis, lipid storage and fatty acid oxidation [116,117,118,119,120].
In HCC, METTL5 has been confirmed to promote tumorigenesis by targeting 18 S rRNA, impairing the 80 S ribosome, and reducing the levels of proteins related to fatty acid metabolism. Moreover, acyl-CoA synthetase long-chain family member (ACSL4) regulates the function of METTL5 in fatty acid metabolism and thus facilitates cancer progression[121].
In cervical squamous cell carcinoma (CESC), ALKBH5 and IGF2BP1 target silent mating type information regulation 2 homolog 3 (SIRT3), reducing its stability, subsequently inhibiting ACC1 deacetylation and lipid metabolism and ultimately repressing CESC[122].
m6A in amino acid metabolism pathways
An increasing number of studies have documented that cancer cells exhibit a higher demand for amino acids to maintain their vitality and proliferation. Amino acids are, on the one hand, substrates for biosynthesis, and on the other hand, they can be converted into α-ketoglutarate (α-KG) and other metabolites, which together constitute the basic process of amino acid metabolism in tumors. Studies showing that the m6A modification can exert a direct impact on relevant metabolic enzymes or metabolites in amino acid metabolism pathways are relatively rare. However, m6A acts on other significant factors, such as MYC [123], to mediate metabolic reprogramming in cancer.
SLC1A5 is an important transporter in amino metabolism pathways that imports glutamine into the cytoplasm [124]. After glutamine is transported, glutaminase (GLS) converts it to glutamate in the first step of glutamine catabolism [125]. The m6A RNA eraser FTO and von Hippel‒Lindau (VHL), a tumor-suppressing factor that is not expressed in ccRCC cells, are synthetic lethality-inducing partners that indirectly target SLC1A5 downstream to promote metabolic recombination [126]. The m6A reader YTHDF1 targets the putative binding motif in the 3’ UTR of GLS1 to boost the function of GLS1, facilitate glutamine metabolism and contribute to the progression of cisplatin-resistant colon cancer [127]. By analyzing TCGA datasets, IGF2BP2 is overexpressed in AML and acts an adverse prognostic factor for AML patients. IGF2BP2 is an adverse prognostic factor for AML patients. IGF2BP2 promotes AML progression by targeting several significant factors in the glutamine metabolism pathways, including MYC, glutamic-pyruvic transaminase 2 (GPT2), and SLC1A5 [128]. Recently, METTL16 was found to affect branched-chain amino acid metabolism in AML by upregulating the expression of branched-chain amino acid transaminase 1 (BCAT1) and BCAT2 mediated via m6A modification [129]. (Fig. 2)

The function of m6A in cancer lipid metabolic and amino acid metabolic pathways
m6A in mitochondrial metabolism
Glycolysis is one of the most notable and widely studied metabolic processes in all kinds of malignant tumors. Nevertheless, mitochondria are also key elements providing energy for the proliferation of cancer cells in addition to oncogenesis, regulating tumor anabolism, redox and calcium homeostasis, etc. [70]. Therefore, the role of mitochondrial metabolism in cancer cannot be ignored.
Peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α) is a transcription coactivator in mitochondrial metabolism. FTO plays an indispensable antitumorigenic role in ccRCC by reducing the abundance of m6A on PGC-1α mRNA, increasing transcript levels and leading to the recovery of mitochondrial activity [130].
Adenylate kinase 4 (AK4) has been demonstrated to indirectly modulate mitochondrial metabolism probably via ADP/ATP translocase, thereby enhancing energy homeostasis [131]. In tamoxifen-resistant breast cancer cells, METTL3 selectively targets the 5’ UTR of AK4 mRNA, which ultimately inhibits mitochondrial apoptosis and enhances tamoxifen resistance [132].
Toll-like receptor 4 (TLR4) signaling plays an important role in multiple myeloma (MM) by maintaining cancer mitochondrial metabolism. Clinical data show that HNRNPA2B1 indicates poor prognosis in MM patients. Mechanistically, HNRNPA2B1 can enhance this process by enriching at the m6A sites of TLR4, thus facilitating cancer progression[133].
m6A in cancer targeted therapy
Under normal physiological conditions, the m6A modification is a homeostatic process, while m6A dysregulation is involved in cancer cell metabolism, indicating that m6A-targeted therapy may play an important role in inhibiting tumor progression.
m6A-targeted anti-tumor treatment
Previous studies demonstrated that m6A enzymes were differently expressed in numerous cancers and the abnormal level of m6A in metabolic pathways predicted the poor prognosis of cancer patients. Based on mechanisms of m6A in cancer metabolism, m6A-targeted therapy may provide an insight into the study of targeted anti-tumor treatment. Strategies include: establishing m6A gene-editing system to achieve the knockdown or knockout of oncogenes and the overexpression of tumor suppressor genes, for example clustered regularly interspaced short palindromic repeats associated protein 9 (CRISPR-Cas9) or short hairpin RNA (shRNA) encapsulated by adeno-associated virus (AAV); using nanoparticles to deliver small interfering RNA for targeting m6A enzymes; screening small molecule compounds that specifically regulate m6A; and developing specific and efficient m6A-targeted inhibitors.
In CRC, METTL3 is significantly upregulated and promotes glucose uptake via targeting at metabolic enzymes in glycolysis. It is found that METTL3 knockout by using lentiviral-based CRISPR gene editing system markedly inhibits tumorigenesis in mice, and this may be due to decreased HK2 and GLUT1 level and the reduced hexokinase activity [79]. In vivo experiments confirm that METTL3 knockdown via shRNA has anti-tumor effect in liver cancer. The knockdown of METTL3 significantly attenuates PDK4 mRNA stability, reduces glucose uptake and lactate production and accelerates mitochondrial oxidative respiration [92]. Similarly, METTL3 is significantly upregulated in cervical cancer and promotes glucose uptake via targeting at metabolic enzymes in glycolysis. The knockdown of METTL3 via shRNA induces reduced lactate production and ATP level in vivo and suppresses tumorigenesis [75]. After METTL5 knockout, treatment with ACSL4 knockdown by using siRNA significantly represses fatty acid metabolism of HCC in mice. To be specific, METTL5 knockout and ACSL4 knockdown synergistically reduce the levels of free fatty acids, triglycerides and intracellular lipid droplets and then blocks tumor initiation in HCC tissues [121]. In AML in vivo experiments, the lentiviral vector-based shRNA system was applied to knock down METTL16 in mice, and it was demonstrated that METTL16 knockdown largely inhibited tumorigenesis and development. Consistently, METTL16 knockout also exhibited anti-tumor effect by suppressing the expression of BCAT1 and BCAT2 in amino acid metabolism [129]. KIAA1429 overexpression is associated with the malignancy maintenance of CRC cells. Conversely, KIAA1429 knockdown via shRNA in xenograft model represses tumor growth, which leads to reduced glucose uptake and lack of ATP production [80].
In addition to m6A writers, targeting m6A erasers can also inhibit cancer progression by regulating metabolic pathways. In papillary thyroid cancer (PTC), the overexpression of FTO via lentiviruses containing complete FTO coding sequence suppresses tumor growth in xenograft model. GLUT1, HK2 and LDHA levels are markedly reduced, which ultimately leads to weakened aerobic glycolysis [134]. High expressions of FTO and glutamine transporter SLC1A5 are closely correlated with glutamine metabolism and the poor prognosis of ccRCC patient, while FTO knockdown decreases the survival of ccRCC cells, therefore targeting FTO is a possible anti-tumor therapy. Using shRNA to knockdown FTO, it is found that the growth of ccRCC tumor is significantly decreased in xenograft model. The levels of SLC1A5 and glutamine uptake are reduced upon FTO knockdown [126]. There are various m6A enzymes that participate in cancer metabolism, which forms a complicated regulating network, but it is clear that the abnormal global level of m6A is closely correlated with tumor initiation and progression. In LUAD, high global m6A level is observed due to METTL3 upregulation and ALKBH5 downregulation, and this leads to elevated PEP, pyruvate and ATP levels, while METTL3 knockout and ALKBH5 overexpression exhibit synergistic effect on tumor inhibition in LUAD mouse models [83].
After bone marrow transplantation, IGF2BP2 knockdown via shRNA dramatically decreased the level of immature blast cells in peripheral blood, substantially delayed leukemogenesis and development, and obviously prolonged the lifespan of mice. Moreover, CWI1-2 was identified as a small-molecule IGF2BP2 inhibitor. Treatment with CWI1-2 effectively inhibited AML initiation in vivo and this inhibitor exhibited synergistic effects with other AML chemotherapy, including daunorubicin and homoharringtonine. IGF2BP2 knockdown resulted in the reduced stability of GPT2 and SLC1A5, suppressed amino acid metabolism and decreased levels of metabolites in glycolysis and TCA cycle [128].
Recently, siRNA encapsulated by small extracellular vesicles (sEV) was used to deplete YTHDF1 in GC. It was revealed that YTHDF1 depletion suppressed tumor development and metastasis in vivo. YTHDF1 loss modulated immune regulation in GC by increasing interferon (IFN)-γ receptor 1 expression to promote IFN-γ response and upregulating major histocompatibility complex class I to enhance cytotoxic T lymphocyte reaction [135]. (Table 3)
Development of m6A-targeted inhibitors
Since FTO is involved in the occurrence and poor prognosis of various cancers, such as glioblastoma (GBM) and acute myeloid leukemia (AML), FTO inhibitors are thought to be a promising cancer targeted therapy. In 2011, rhein was first discovered to exert inhibitory effects on FTO and ALKBH5 in vitro. Molecular modeling showed that rhein competitively bound to 3-methylthymine (m3T) and 2-oxoglutarate (2OG), active sites of the FTO enzyme, and Fe2+, effectively preventing m6A recognition. Therefore, rhein increased the abundance of m6A modification inside cells[136]. Meclofenamic acid (MA) was later found to be another in vitro FTO inhibitor demonstrated to preferentially act on FTO over ALKBH5[137]. Inspired by this discovery, researchers have reported an increasing number of FTO inhibitors, confirming that FTO is a drug-treatable cancer target. Several fluorescein derivatives, with the base name FL1-11, have been designed and can selectively inhibit FTO demethylation in vitro neither by mimicking 2-OG nor by forming an Fe2+ chelate[138]. In 2019, a new derivative of MA named FB23-2 was confirmed to upregulate the abundance of methylation marks on genes essential to leukemia, increasing the level of a tumor suppressor protein, and reducing the abundance of a tumor promoting protein, thereby inhibiting the proliferation of AML cells in vitro and in vivo[139]. Compared to FB23-2, two new small-molecule FTO inhibitors, CS1 and CS2, showed higher efficacy in inhibiting the activity of AML cells. CS1 and CS2 bind to the enzymatic reaction center of FTO, blocking its binding to a target gene and thus inhibiting its methyltransferase activity. In addition, CS1 and CS2 inhibit the immune escape of AML cells by targeting leukocyte immunoglobulin-like receptor subfamily B4 (LILRB4), an immune checkpoint protein [140]. Recently, the oxetane-based compounds were shown to inhibit FTO. Among these chemical compounds, FTO-43 N has been reported to significantly inhibit GBM, AML and GC progression and show the an antiproliferative effect similar to that of 5-fluorouracil in GC cells [141].
As mentioned earlier, m6A is a double-edged sword in tumorigenesis and tumor development. An increase or decrease in m6A levels can promote the progression of different tumors. Hence, in addition to inhibitors of the m6A eraser FTO, small-molecule inhibitors of the m6A writers METTL3/14 have garnered increasing attention in recent years. The first selective small-molecule inhibitors of METTT3 to be identified were adenine derivatives, which exhibited high ligand-binding efficiency, shedding light on the study of selective METTL3/14 inhibitors[142]. Based on adenine library screening, UZH1a was designed as a nanomolar inhibitor of METTL3 and showed cell permeability. Importantly, UZH1a lowered the m6A levels in AML cells, osteosarcoma cells and embryonic kidney cells by binding SAM[143]. UZH2, established via optimization of UZH1a, was later confirmed to be another nanomolar inhibitor of METTL3 in AML cells and prostate cancer cells, showing better metabolic stability than UZH1a [144]. STM2457 is a highly specific and selective inhibitor of METTL3/14 that can directly bind to the SAM-binding site in METTL3 and inhibit the activity of METTL3 methyltransferase and its translation, thereby reducing the m6A level in AML cells. STM2457 effectively restrains the expansion of AML cells in vivo, significantly prolonging the survival of mice and exerting no obvious toxic effect [145]. STM2457 is now in the first stage of a clinical trial. Obviously, most of the METTL3/14 inhibitors to date are competitive inhibitors.
Recently, eltrombopag was demonstrated to be a noncompetitive inhibitor that may bind to the allosteric site of METTL3/14, inhibit METTL3/14 complex activity and thus reduce m6A levels in AML cells[146]. Moreover, a previous study verified that eltrombopag prolonged the lifespan of mouse models of AML via antiproliferative and differentiation-inducing effects in vivo[147]. Hence, further optimization of eltrombopag may facilitate new drug development for AML. 4-[2-[5-Chloro-1-(diphenylmethyl)-2-methyl-1 H-indol-3-yl]-ethoxy] benzoic acid (CDIBA), an allosteric inhibitor of METTL3/14, was optimized by combining the best substituents in different regions, and it showed a higher METTL3/14 inhibitory effect than other inhibitors [148]. Although the development of small-molecular drugs targeting METTL3 and METTL14 has slower than that of FTO inhibitors, the study of METTL3/14 inhibitors shows great promise.
m6A and therapeutic resistance
In fact, m6A enzymes are not only closely correlated with cancer initiation and progression, but also responsible for the therapeutic resistance and altered metabolic pathways in various cancers [149], which may expand the application of m6A-targeted inhibitors. Combination of m6A-targeted inhibitors and chemotherapy may contribute to anti-tumor treatment.
In primary leukemia, FTO increases the stability of a series of proliferation and survival mRNAs, for example B-cell lymphoma-2 (BCL-2) and tyrosine-protein kinase Mer (MERTK), and subsequently promotes protein synthesis. Leukemia cells with the overexpression of FTO demonstrate higher tyrosine kinase inhibitor (TKI) tolerance, while cells exposed to the FTO inhibitor rhein reverses TKI resistance [150]. Interestingly, leukemia cells with the upregulation of FTO are more sensitive to R-2HG by FTO-enhanced stability of MYC transcripts [151]. In CRC, FTO promotes the resistance to 5-fluorouracil (5-FU) and cisplatin. Mechanistically, FTO regulates the expression of G6PD, modulates NADPH and (reactive oxygen species) ROS levels and consequently affects DNA damage. Meanwhile, FTO also regulates the mRNA stability of poly ADP-ribose polymerase 1 (PARP1) to mediate DNA damage repair, which ultimately leads to the reduced chemotherapy sensitivity [152]. In GBM, JPX modulates the expression of PDK1 by interacting with FTO, which prevents PDK1 mRNA from degradation and results in enhanced aerobic glycolysis and the resistance to temozolomide [91]. In addition to chemotherapy resistance, FTO also participates in immunotherapy resistance by regulating glycolysis-associated genes. FTO enhances the expression of c-Jun, JunB, and CCAAT/enhancer binding protein β (C/EBPβ), and then rewires glycolysis metabolism, which impairs the function of T cells and leads to poor immunotherapy response in melanoma. Therefore, the inhibition of FTO is a potential strategy for improving immunotherapy effects [153].
In addition to FTO, other m6A enzymes are also involved in cancer drug resistance via regulating metabolism. METTL3-mediated m6A methylation induces the splicing of estrogen receptor related receptor γ (ERRγ) precursor mRNA, and then ERRγ binds to ATP binding cassette subfamily B member 1 (ABCB1), reinforces its transcription and lowers the sensitivity of cancer cells to numerous anticancer agents. Moreover, ERRγ interacts with the rate-limiting enzyme CPT1B to promote fatty acid metabolism and leads to chemoresistance [120]. In bone marrow mesenchymal stem cells of AML, however, the expression METTL3 is reduced, which enhances adipogenesis and mediates the resistance to penicillin and streptomycin via upregulating AKT protein level [154].
It was revealed that an increased level of ALKBH5 was closely correlated with the glycolysis of breast cancer and the resistance to trastuzumab and lapatinib via the demethylation of GLUT4 and suppression of GLUT4 re-sensitized the resistant cells, indicating that the inhibition of ALKBH5/GLUT4 axis may contribute to breast cancer targeted therapy [74].
YTHDF1 interacts with the binding motif of GLS1 and increases glutamine uptake to enhance glutamine metabolism in cisplatin-resistant CRC cells. The inhibition of GLS1 leads to an increased sensitivity to cisplatin-induced cell death and YTHDF1 is also a potential target in overcoming resistant colon cancers [127]. In gastric cancer, YTHDF2 reduces the stability of cystathionine β-synthase (CBS) mRNA, subsequently decreases the methylation of ACSL4, an important enzyme in lipid metabolism, and results in ACSL4 degradation and chemoresistance. Patients with low CBS levels have a worse prognosis and are less sensitive to chemotherapy [155]. As mentioned above, m6A enzymes participate in chemoresistance via altered metabolic pathways in many different cancers. Hence, m6A inhibitors have great development perspective.
Conclusion
In the past few years, numerous studies have demonstrated that m6A fuels cancer progression by regulating tumor metabolism. Owing to the development of methods for detecting the m6A modification, numerous studies have revealed a possible role for m6A in aerobic glycolysis, the deregulated metabolism of fatty acids and amino acids and aberrant mitochondrial metabolism. This review analyzes several confirmed functions of the m6A modification in cancer metabolic pathways as well as the underlying mechanisms. In fact, the mutual effects of m6A and cancer metabolic pathways are very complicated. Signaling pathways, transcription factors and noncoding RNAs can exert large impacts on metabolic enzymes in a wide range of cancers, forming a complex network involved in metabolic reprogramming. m6A exerts different influences on cancer metabolism by targeting upstream molecules or downstream metabolic enzymes and transporters, thereby extensively modulating metabolic recombination. Conversely, cancer metabolic reprogramming regulates the abnormal m6A modification. Take glucose metabolism for example, numerous studies confirm that m6A participate in cancer glycolysis, while the accumulation of succinate, an intermediate product in glucose metabolism, can in turn regulate m6A modification. Studies on metabolic enzymes in amino acid metabolic pathways and mitochondrial metabolism are relatively rare, and in-depth investigation is needed to further explain and elucidate the systematic molecular mechanisms involved. There is still a long way to go in the study on the mutual effect of cancer metabolism on m6A modification.
To date, an increasing number of m6A inhibitors have been designed and a series of experiments in vivo and in vitro have confirmed that they have a good application prospect. However, relevant clinical evidence is quite insufficient and clinical trials are urgently needed.
Since dysregulated m6A is closely correlated with tumor development, m6A may also function as specific biomarkers involved in early diagnosis. For instance, multi-omics data suggests that METTL7A is differently expressed in ccRCC, renal mesothelioma and sarcoma, and shows high accuracy in predicting tumorigenesis. Therefore, METTL7A is a potential diagnostic biomarker in certain cancer types [156]. Although accumulating evidence shows that abnormal m6A level may play an important role in the development and progression of cancer, most of m6A-related proteins are located in the nucleus, which cannot be detected in body fluids unless invasive puncture examination is performed. Serological marker, including secretory proteins, exosomes, cytoplasmic tRNAs (ctRNAs), cell-free RNAs (cfRNAs) and some non-coding RNAs, is a better choice for early diagnosis. Actin filament-associated protein 1-antisense RNA1 (AFAP1-AS1) is a lncRNA that encodes AFAP1-AS1 translated mitochondrial-localized peptide (ATMLP) under the control of m6A. The serum level of ATMLP is upregulated in NSCLC, which indicates that ATMLP is a diagnostic marker for lung cancer [157]. In HCC, m6A-modulated circRNAs also function as biomarkers which can be detected in serum, for example circular cleavage and polyadenylation specific factor 6 (circCPSF6), circular mitogen-activated protein kinase kinase kinase 4 (circMAP3K4) and circular fucosyltransferase 8 (circFUT8) [158,159,160].
In conclusion, m6A can regulate cancer metabolic reprogramming in distinct ways, providing a more comprehensive knowledge of epigenetics and cancer metabolism to better carry out clinical experiments. These studies may be particularly helpful in early diagnosis, disease control and therapeutic approaches to cancer.
Data Availability
Data sharing not applicable to this article as no data-sets were generated or analyzed during the current study.
References
Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger rna from novikoff hepatoma cells.Proc Natl Acad Sci U S a. 1974 1974Oct;71(10):3971–75. doi: https://doi.org/10.1073/pnas.71.10.3971
Liu N, Zhou KI, Parisien M, Dai Q, Diatchenko L, Pan T. N6-methyladenosine alters rna structure to regulate binding of a low-complexity protein.Nucleic Acids Res. 2017 2017 Jun2;45(10):6051–63. doi: https://doi.org/10.1093/nar/gkx141
Liu Q, Gregory RI. Rnamod: an integrated system for the annotation of mrna modifications. Nucleic Acids Res. 2019 2019 Jul 2;47(W1):W548-55. doi: https://doi.org/10.1093/nar/gkz479
Wang Y, Li Y, Yue M, Wang J, Kumar S, Wechsler-Reya RJ et al. N(6)-methyladenosine rna modification regulates embryonic neural stem cell self-renewal through histone modifications. Nat Neurosci. 2018 2018 Feb;21(2):195–206. doi: https://doi.org/10.1038/s41593-017-0057-1
Zhao BS, He C. Fate by rna methylation: m6a steers stem cell pluripotency.Genome Biol. 2015 2015 Feb22;16(1):43. doi: https://doi.org/10.1186/s13059-015-0609-1
Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M et al. Stem cells. M6a mrna methylation facilitates resolution of naïve pluripotency toward differentiation.Science. 2015 2015 Feb27;347(6225):1002–06. doi: https://doi.org/10.1126/science.1261417
Zhang X, Blumenthal RM, Cheng X. A role for n6-methyladenine in dna damage repair.Trends Biochem Sci. 2021 2021Mar;46(3):175–83. doi: https://doi.org/10.1016/j.tibs.2020.09.007
Church C, Moir L, McMurray F, Girard C, Banks GT, Teboul L et al. Overexpression of fto leads to increased food intake and results in obesity.Nat Genet. 2010 2010Dec;42(12):1086–92. doi: https://doi.org/10.1038/ng.713
Yang Y, Huang W, Huang JT, Shen F, Xiong J, Yuan EF et al. Increased n6-methyladenosine in human sperm rna as a risk factor for asthenozoospermia.Sci Rep. 2016 2016 Apr13;6:24345. doi: https://doi.org/10.1038/srep24345
Wang T, Kong S, Tao M, Ju S. The potential role of rna n6-methyladenosine in cancer progression. Mol Cancer. 2020 2020 May 12;19(1):88. doi: https://doi.org/10.1186/s12943-020-01204-7
Covelo-Molares H, Bartosovic M, Vanacova S. Rna methylation in nuclear pre-mrna processing. Wiley Interdiscip Rev Rna. 2018 2018 Nov;9(6):e1489. doi: https://doi.org/10.1002/wrna.1489
Ignatova VV, Stolz P, Kaiser S, Gustafsson TH, Lastres PR, Sanz-Moreno A et al. The rrna m(6)a methyltransferase mettl5 is involved in pluripotency and developmental programs.Genes Dev. 2020 2020May 1;34(9–10):715–29. doi: https://doi.org/10.1101/gad.333369.119
Huang H, Li H, Pan R, Wang S, Liu X. Trna modifications and their potential roles in pancreatic cancer.Arch Biochem Biophys. 2021 2021 Dec15;714:109083. doi: https://doi.org/10.1016/j.abb.2021.109083
Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M et al. M(6)a rna methylation promotes xist-mediated transcriptional repression.Nature. 2016 2016 Sep15;537(7620):369–73. doi: https://doi.org/10.1038/nature19342
Zhang J, Bai R, Li M, Ye H, Wu C, Wang C et al. Excessive mir-25-3p maturation via n(6)-methyladenosine stimulated by cigarette smoke promotes pancreatic cancer progression. Nat Commun. 2019 2019 Apr 23;10(1):1858. doi: https://doi.org/10.1038/s41467-019-09712-x
Han J, Wang JZ, Yang X, Yu H, Zhou R, Lu HC et al. Mettl3 promote tumor proliferation of bladder cancer by accelerating pri-mir221/222 maturation in m6a-dependent manner.Mol Cancer. 2019 2019 Jun22;18(1):110. doi: https://doi.org/10.1186/s12943-019-1036-9
Gu S, Sun D, Dai H, Zhang Z. N(6)-methyladenosine mediates the cellular proliferation and apoptosis via micrornas in arsenite-transformed cells.Toxicol Lett. 2018 2018Aug;292:1–11. doi: https://doi.org/10.1016/j.toxlet.2018.04.018
Di Timoteo G, Dattilo D, Centrón-Broco A, Colantoni A, Guarnacci M, Rossi F et al. Modulation of circrna metabolism by m(6)a modification. Cell Rep. 2020 2020 May 12;31(6):107641. doi: https://doi.org/10.1016/j.celrep.2020.107641
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mrna methylation reveals enrichment in 3’ utrs and near stop codons.Cell. 2012 2012 Jun22;149(7):1635–46. doi: https://doi.org/10.1016/j.cell.2012.05.003
Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. Single-nucleotide-resolution mapping of m6a and m6am throughout the transcriptome. Nat Methods. 2015 2015 Aug;12(8):767–72. doi: https://doi.org/10.1038/nmeth.3453
Liu N, Parisien M, Dai Q, Zheng G, He C, Pan T. Probing n6-methyladenosine rna modification status at single nucleotide resolution in mrna and long noncoding rna. Rna 2013. 2013 Dec;19(12):1848–56. https://doi.org/10.1261/rna.041178.113.
DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv 2016. 2016 May;2(5):e1600200. https://doi.org/10.1126/sciadv.1600200.
Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate.Cancer Cell. 2012 2012 Mar20;21(3):297–308. doi: https://doi.org/10.1016/j.ccr.2012.02.014
WARBURG O. On the origin of cancer cells. Science. 1956 1956 Feb 24;123(3191):309–14. doi: https://doi.org/10.1126/science.123.3191.309
Wei CM, Gershowitz A, Moss B. Methylated nucleotides block 5’ terminus of hela cell messenger rna.Cell. 1975 1975Apr;4(4):379–86. doi: https://doi.org/10.1016/0092-8674(75)90158-0
Narayan P, Ludwiczak RL, Goodwin EC, Rottman FM. Context effects on n6-adenosine methylation sites in prolactin mrna. Nucleic Acids Res. 1994 1994 Feb 11;22(3):419–26. doi: https://doi.org/10.1093/nar/22.3.419
Carroll SM, Narayan P, Rottman FM. N6-methyladenosine residues in an intron-specific region of prolactin pre-mrna.Mol Cell Biol. 1990 1990Sep;10(9):4456–65. doi: https://doi.org/10.1128/mcb.10.9.4456-4465.1990
Kane SE, Beemon K. Precise localization of m6a in rous sarcoma virus rna reveals clustering of methylation sites: implications for rna processing.Mol Cell Biol. 1985 1985Sep;5(9):2298–306. doi: https://doi.org/10.1128/mcb.5.9.2298-2306.1985
Csepany T, Lin A, Baldick CJ, Beemon K. Sequence specificity of mrna n6-adenosine methyltransferase.J Biol Chem. 1990 1990 Nov25;265(33):20117–22.
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S et al. Topology of the human and mouse m6a rna methylomes revealed by m6a-seq.Nature. 2012 2012 Apr29;485(7397):201–06. doi: https://doi.org/10.1038/nature11112
Bokar JA, Rath-Shambaugh ME, Ludwiczak R, Narayan P, Rottman F. Characterization and partial purification of mrna n6-adenosine methyltransferase from hela cell nuclei. Internal mrna methylation requires a multisubunit complex. J Biol Chem. 1994 1994 Jul 1;269(26):17697-704.
Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L et al. A mettl3-mettl14 complex mediates mammalian nuclear rna n6-adenosine methylation.Nat Chem Biol. 2014 2014Feb;10(2):93–95. doi: https://doi.org/10.1038/nchembio.1432
Warda AS, Kretschmer J, Hackert P, Lenz C, Urlaub H, Höbartner C et al. Human mettl16 is a n(6)-methyladenosine (m(6)a) methyltransferase that targets pre-mrnas and various non-coding rnas. Embo Rep. 2017 2017 Nov;18(11):2004-14. doi: https://doi.org/10.15252/embr.201744940
Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP et al. The u6 snrna m(6)a methyltransferase mettl16 regulates sam synthetase intron retention.Cell. 2017 2017 May18;169(5):824–35. doi: https://doi.org/10.1016/j.cell.2017.05.003
Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ et al. Mammalian wtap is a regulatory subunit of the rna n6-methyladenosine methyltransferase.Cell Res. 20142014 Feb;24(2):177–89. doi: https://doi.org/10.1038/cr.2014.3
Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG et al. Perturbation of m6a writers reveals two distinct classes of mrna methylation at internal and 5’ sites.Cell Rep. 2014 2014Jul 10;8(1):284–96. doi: https://doi.org/10.1016/j.celrep.2014.05.048
Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang Z, et al. Virma mediates preferential m(6)a mrna methylation in 3’utr and near stop codon and associates with alternative polyadenylation. Cell Discov 2018. 2018;4:10. https://doi.org/10.1038/s41421-018-0019-0.
Wen J, Lv R, Ma H, Shen H, He C, Wang J et al. Zc3h13 regulates nuclear rna m(6)a methylation and mouse embryonic stem cell self-renewal.Mol Cell. 2018 2018 Mar15;69(6):1028–38. doi: https://doi.org/10.1016/j.molcel.2018.02.015
Knuckles P, Lence T, Haussmann IU, Jacob D, Kreim N, Carl SH et al. Zc3h13/flacc is required for adenosine methylation by bridging the mrna-binding factor rbm15/spenito to the m(6)a machinery component wtap/fl(2)d. Genes Dev. 2018 2018 Mar 1;32(5–6):415–29. doi: https://doi.org/10.1101/gad.309146.117
Clancy MJ, Shambaugh ME, Timpte CS, Bokar JA. Induction of sporulation in saccharomyces cerevisiae leads to the formation of n6-methyladenosine in mrna: a potential mechanism for the activity of the ime4 gene.Nucleic Acids Res. 2002 2002Oct 15;30(20):4509–18. doi: https://doi.org/10.1093/nar/gkf573
Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM. Purification and cdna cloning of the adomet-binding subunit of the human mrna (n6-adenosine)-methyltransferase. Rna. 1997 1997 Nov;3(11):1233-47.
Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z et al. Structural basis of n(6)-adenosine methylation by the mettl3-mettl14 complex.Nature. 2016 2016 Jun23;534(7608):575–78. doi: https://doi.org/10.1038/nature18298
Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM et al. A common variant in the fto gene is associated with body mass index and predisposes to childhood and adult obesity.Science. 2007 2007May 11;316(5826):889–94. doi: https://doi.org/10.1126/science.1141634
Thorleifsson G, Walters GB, Gudbjartsson DF, Steinthorsdottir V, Sulem P, Helgadottir A et al. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity.Nat Genet. 2009 2009Jan;41(1):18–24. doi: https://doi.org/10.1038/ng.274
Dina C, Meyre D, Gallina S, Durand E, Körner A, Jacobson P et al. Variation in fto contributes to childhood obesity and severe adult obesity.Nat Genet. 2007 2007Jun;39(6):724–26. doi: https://doi.org/10.1038/ng2048
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y et al. N6-methyladenosine in nuclear rna is a major substrate of the obesity-associated fto.Nat Chem Biol. 2011 2011 Oct16;7(12):885–87. doi: https://doi.org/10.1038/nchembio.687
Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ et al. Alkbh5 is a mammalian rna demethylase that impacts rna metabolism and mouse fertility.Mol Cell. 2013 2013 Jan10;49(1):18–29. doi: https://doi.org/10.1016/j.molcel.2012.10.015
Ueda Y, Ooshio I, Fusamae Y, Kitae K, Kawaguchi M, Jingushi K et al. Alkb homolog 3-mediated trna demethylation promotes protein synthesis in cancer cells.Sci Rep. 2017 2017 Feb13;7:42271. doi: https://doi.org/10.1038/srep42271
Huang H, Weng H, Sun W, Qin X, Shi H, Wu H et al. Recognition of rna n(6)-methyladenosine by igf2bp proteins enhances mrna stability and translation. Nat Cell Biol. 2018 2018 Mar;20(3):285–95. doi: https://doi.org/10.1038/s41556-018-0045-z
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H et al. N(6)-methyladenosine modulates messenger rna translation efficiency.Cell. 2015 2015 Jun4;161(6):1388–99. doi: https://doi.org/10.1016/j.cell.2015.05.014
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D et al. N6-methyladenosine-dependent regulation of messenger rna stability.Nature. 2014 2014 Jan2;505(7481):117–20. doi: https://doi.org/10.1038/nature12730
Li A, Chen YS, Ping XL, Yang X, Xiao W, Yang Y et al. Cytoplasmic m(6)a reader ythdf3 promotes mrna translation.Cell Res. 2017 2017Mar;27(3):444–47. doi: https://doi.org/10.1038/cr.2017.10
Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ et al. Ythdf3 facilitates translation and decay of n(6)-methyladenosine-modified rna.Cell Res. 2017 2017Mar;27(3):315–28. doi: https://doi.org/10.1038/cr.2017.15
Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF et al. Nuclear m(6)a reader ythdc1 regulates mrna splicing.Mol Cell. 2016 2016 Feb18;61(4):507–19. doi: https://doi.org/10.1016/j.molcel.2016.01.012
Lesbirel S, Viphakone N, Parker M, Parker J, Heath C, Sudbery I et al. The m(6)a-methylase complex recruits trex and regulates mrna export. Sci Rep. 2018 2018 Sep 14;8(1):13827. doi: https://doi.org/10.1038/s41598-018-32310-8
Roundtree IA, Luo GZ, Zhang Z, Wang X, Zhou T, Cui Y et al. Ythdc1 mediates nuclear export of n(6)-methyladenosine methylated mrnas. Elife. 2017 2017 Oct 6;6. doi: https://doi.org/10.7554/eLife.31311
Hsu PJ, Zhu Y, Ma H, Guo Y, Shi X, Liu Y, et al. Ythdc2 is an n(6)-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res 2017. 2017 Sep;27(9):1115–27. https://doi.org/10.1038/cr.2017.99.
Wang S, Chim B, Su Y, Khil P, Wong M, Wang X et al. Enhancement of lin28b-induced hematopoietic reprogramming by igf2bp3.Genes Dev. 2019 2019 Aug1;33(15–16):1048–68. doi: https://doi.org/10.1101/gad.325100.119
Müller S, Glaß M, Singh AK, Haase J, Bley N, Fuchs T et al. Igf2bp1 promotes srf-dependent transcription in cancer in a m6a- and mirna-dependent manner.Nucleic Acids Res. 2019 2019 Jan10;47(1):375–90. doi: https://doi.org/10.1093/nar/gky1012
König J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B et al. Iclip reveals the function of hnrnp particles in splicing at individual nucleotide resolution.Nat Struct Mol Biol. 2010 2010Jul;17(7):909–15. doi: https://doi.org/10.1038/nsmb.1838
Alarcón CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. Hnrnpa2b1 is a mediator of m(6)a-dependent nuclear rna processing events.Cell. 2015 2015Sep 10;162(6):1299–308. doi: https://doi.org/10.1016/j.cell.2015.08.011
Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-methyladenosine-dependent rna structural switches regulate rna-protein interactions.Nature. 2015 2015 Feb26;518(7540):560–64. doi: https://doi.org/10.1038/nature14234
Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O et al. 5’ utr m(6)a promotes cap-independent translation.Cell. 2015 2015 Nov5;163(4):999–1010. doi: https://doi.org/10.1016/j.cell.2015.10.012
Choe J, Lin S, Zhang W, Liu Q, Wang L, Ramirez-Moya J et al. Mrna circularization by mettl3-eif3h enhances translation and promotes oncogenesis.Nature. 2018 2018Sep;561(7724):556–60. doi: https://doi.org/10.1038/s41586-018-0538-8
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011 2011 Mar 4;144(5):646–74. doi: https://doi.org/10.1016/j.cell.2011.02.013
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000 2000 Jan 7;100(1):57–70. doi: https://doi.org/10.1016/s0092-8674(00)81683-9
Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022 2022 Jan;12(1):31–46. doi: https://doi.org/10.1158/2159-8290.CD-21-1059
Cao Y. Adipocyte and lipid metabolism in cancer drug resistance. J Clin Invest. 2019 2019 Jul 2;129(8):3006-17. doi: https://doi.org/10.1172/JCI127201
Guerra F, Arbini AA, Moro L. Mitochondria and cancer chemoresistance. Biochim Biophys Acta Bioenerg. 2017 2017 Aug;1858(8):686–99. doi: https://doi.org/10.1016/j.bbabio.2017.01.012
Porporato PE, Filigheddu N, Pedro J, Kroemer G, Galluzzi L. Mitochondrial metabolism and cancer.Cell Res. 2018 2018Mar;28(3):265–80. doi: https://doi.org/10.1038/cr.2017.155
DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation.Cell Metab. 2008 2008Jan;7(1):11–20. doi: https://doi.org/10.1016/j.cmet.2007.10.002
Yang D, Chang S, Li F, Ma M, Yang J, Lv X et al. M(6) a transferase kiaa1429-stabilized linc00958 accelerates gastric cancer aerobic glycolysis through targeting glut1. Iubmb Life. 2021 2021 Nov;73(11):1325-33. doi: https://doi.org/10.1002/iub.2545
Chen H, Gao S, Liu W, Wong CC, Wu J, Wu J et al. Rna n(6)-methyladenosine methyltransferase mettl3 facilitates colorectal cancer by activating the m(6)a-glut1-mtorc1 axis and is a therapeutic target.Gastroenterology. 2021 2021Mar;160(4):1284–300. doi: https://doi.org/10.1053/j.gastro.2020.11.013
Liu H, Lyu H, Jiang G, Chen D, Ruan S, Liu S et al. Alkbh5-mediated m6a demethylation of glut4 mrna promotes glycolysis and resistance to her2-targeted therapy in breast cancer.Cancer Res. 2022 2022 Nov2;82(21):3974–86. doi: https://doi.org/10.1158/0008-5472.CAN-22-0800
Wang Q, Guo X, Li L, Gao Z, Su X, Ji M et al. N(6)-methyladenosine mettl3 promotes cervical cancer tumorigenesis and warburg effect through ythdf1/hk2 modification.Cell Death Dis. 2020 2020 Oct24;11(10):911. doi: https://doi.org/10.1038/s41419-020-03071-y
Li F, He C, Yao H, Zhao Y, Ye X, Zhou S et al. Glutamate from nerve cells promotes perineural invasion in pancreatic cancer by regulating tumor glycolysis through hk2 mrna-m6a modification.Pharmacol Res. 2023 2023Jan;187:106555. doi: https://doi.org/10.1016/j.phrs.2022.106555
Yu H, Zhao K, Zeng H, Li Z, Chen K, Zhang Z et al. N(6)-methyladenosine (m(6)a) methyltransferase wtap accelerates the warburg effect of gastric cancer through regulating hk2 stability.Biomed Pharmacother. 2021 2021Jan;133:111075. doi: https://doi.org/10.1016/j.biopha.2020.111075
Sun X, Li Q, Yang L. Sevoflurane inhibits lncrna hotair-modulated stability of hk2 mrna in a m6a-dependent manner to dampen aerobic glycolysis and proliferation in lung cancer. Biomed Res Int 2022. 2022;2022:4668774. https://doi.org/10.1155/2022/4668774.
Shen C, Xuan B, Yan T, Ma Y, Xu P, Tian X et al. M(6)a-dependent glycolysis enhances colorectal cancer progression.Mol Cancer. 2020 2020 Apr3;19(1):72. doi: https://doi.org/10.1186/s12943-020-01190-w
Li Y, He L, Wang Y, Tan Y, Zhang F. N(6)-methyladenosine methyltransferase kiaa1429 elevates colorectal cancer aerobic glycolysis via hk2-dependent manner.Bioengineered. 2022 2022May;13(5):11923–32. doi: https://doi.org/10.1080/21655979.2022.2065952
Wang Y, Yu Z, Shi W, Shen J, Guan Y, Ni F. Hla complex p5 upregulation is correlated with poor prognosis and tumor progression in esophageal squamous cell carcinoma.Bioengineered. 2022 2022Apr;13(4):9301–11. doi: https://doi.org/10.1080/21655979.2022.2051854
Ou B, Liu Y, Yang X, Xu X, Yan Y, Zhang J. C5ar1-positive neutrophils promote breast cancer glycolysis through wtap-dependent m6a methylation of eno1.Cell Death Dis. 2021 2021 Jul26;12(8):737. doi: https://doi.org/10.1038/s41419-021-04028-5
Ma L, Xue X, Zhang X, Yu K, Xu X, Tian X et al. The essential roles of m(6)a rna modification to stimulate eno1-dependent glycolysis and tumorigenesis in lung adenocarcinoma.J Exp Clin Cancer Res. 2022 2022 Jan25;41(1):36. doi: https://doi.org/10.1186/s13046-021-02200-5
Li J, Zhu L, Shi Y, Liu J, Lin L, Chen X. M6a demethylase fto promotes hepatocellular carcinoma tumorigenesis via mediating pkm2 demethylation. Am J Transl Res 2019. 2019;11(9):6084–92.
Yao X, Li W, Li L, Li M, Zhao Y, Fang D et al. Ythdf1 upregulation mediates hypoxia-dependent breast cancer growth and metastasis through regulating pkm2 to affect glycolysis. Cell Death Dis. 2022 2022 Mar 23;13(3):258. doi: https://doi.org/10.1038/s41419-022-04711-1
Urbańska K, Orzechowski A. Unappreciated role of ldha and ldhb to control apoptosis and autophagy in tumor cells.Int J Mol Sci. 2019 2019 Apr27;20(9). doi: https://doi.org/10.3390/ijms20092085
Zhang K, Zhang T, Yang Y, Tu W, Huang H, Wang Y, et al. N(6)-methyladenosine-mediated ldha induction potentiates chemoresistance of colorectal cancer cells through metabolic reprogramming. Theranostics 2022. 2022;12(10):4802–17. https://doi.org/10.7150/thno.73746.
Yuan B, Zhou J. N(6)-methyladenosine (m(6)a) reader igf2bp1 facilitates clear-cell renal cell carcinoma aerobic glycolysis. Peerj 2023. 2023;11:e14591. https://doi.org/10.7717/peerj.14591.
Qing Y, Dong L, Gao L, Li C, Li Y, Han L et al. R-2-hydroxyglutarate attenuates aerobic glycolysis in leukemia by targeting the fto/m(6)a/pfkp/ldhb axis.Mol Cell. 2021 2021 Mar4;81(5):922–39. doi: https://doi.org/10.1016/j.molcel.2020.12.026
Yu H, Yang X, Tang J, Si S, Zhou Z, Lu J et al. Alkbh5 inhibited cell proliferation and sensitized bladder cancer cells to cisplatin by m6a-ck2α-mediated glycolysis.Mol Ther Nucleic Acids. 2021 2021 Mar5;23:27–41. doi: https://doi.org/10.1016/j.omtn.2020.10.031
Li XD, Wang MJ, Zheng JL, Wu YH, Wang X, Jiang XB. Long noncoding rna just proximal to x-inactive specific transcript facilitates aerobic glycolysis and temozolomide chemoresistance by promoting stability of pdk1 mrna in an m6a-dependent manner in glioblastoma multiforme cells.Cancer Sci. 2021 2021Nov;112(11):4543–52. doi: https://doi.org/10.1111/cas.15072
Li Z, Peng Y, Li J, Chen Z, Chen F, Tu J et al. N(6)-methyladenosine regulates glycolysis of cancer cells through pdk4.Nat Commun. 2020 2020May 22;11(1):2578. doi: https://doi.org/10.1038/s41467-020-16306-5
Zhang C, Chen L, Liu Y, Huang J, Liu A, Xu Y, et al. Downregulated mettl14 accumulates bptf that reinforces super-enhancers and distal lung metastasis via glycolytic reprogramming in renal cell carcinoma. Theranostics 2021. 2021;11(8):3676–93. https://doi.org/10.7150/thno.55424.
Hu C, Liu T, Han C, Xuan Y, Jiang D, Sun Y, et al. Hpv e6/e7 promotes aerobic glycolysis in cervical cancer by regulating igf2bp2 to stabilize m(6)a-myc expression. Int J Biol Sci 2022. 2022;18(2):507–21. https://doi.org/10.7150/ijbs.67770.
Wang Y, Lu JH, Wu QN, Jin Y, Wang DS, Chen YX et al. Lncrna linris stabilizes igf2bp2 and promotes the aerobic glycolysis in colorectal cancer.Mol Cancer. 2019 2019 Dec2;18(1):174. doi: https://doi.org/10.1186/s12943-019-1105-0
Zhang Q, Zhang Y, Chen H, Sun LN, Zhang B, Yue DS et al. Mettl3-induced dlgap1-as2 promotes non-small cell lung cancer tumorigenesis through m(6)a/c-myc-dependent aerobic glycolysis.Cell Cycle. 2022 2022Dec;21(24):2602–14. doi: https://doi.org/10.1080/15384101.2022.2105885
Luo F, Lin K. N(6)-methyladenosine (m(6)a) reader igf2bp1 accelerates gastric cancer aerobic glycolysis in c-myc-dependent manner. Exp Cell Res. 2022 2022 Aug 1;417(1):113176. doi: https://doi.org/10.1016/j.yexcr.2022.113176
Yang N, Wang T, Li Q, Han F, Wang Z, Zhu R et al. Hbxip drives metabolic reprogramming in hepatocellular carcinoma cells via mettl3-mediated m6a modification of hif-1α.J Cell Physiol. 2021 2021May;236(5):3863–80. doi: https://doi.org/10.1002/jcp.30128
Green NH, Galvan DL, Badal SS, Chang BH, LeBleu VS, Long J et al. Mthfd2 links rna methylation to metabolic reprogramming in renal cell carcinoma.Oncogene. 2019 2019Aug;38(34):6211–25. doi: https://doi.org/10.1038/s41388-019-0869-4
Shmakova A, Frost M, Batie M, Kenneth NS, Rocha S. Pbrm1 cooperates with ythdf2 to control hif-1α protein translation.Cells. 2021 2021 Jun8;10(6). doi: https://doi.org/10.3390/cells10061425
Chen B, Hong Y, Gui R, Zheng H, Tian S, Zhai X et al. N6-methyladenosine modification of circ_0003215 suppresses the pentose phosphate pathway and malignancy of colorectal cancer through the mir-663b/dlg4/g6pd axis.Cell Death Dis. 2022 2022 Sep20;13(9):804. doi: https://doi.org/10.1038/s41419-022-05245-2
Zhang Y, Xu L, Ren Z, Liu X, Song J, Zhang P et al. Linc01615 maintains cell survival in adaptation to nutrient starvation through the pentose phosphate pathway and modulates chemosensitivity in colorectal cancer.Cell Mol Life Sci. 2022 2022 Dec28;80(1):20. doi: https://doi.org/10.1007/s00018-022-04675-7
Liu Z, Chen Y, Wang L, Ji S. Alkbh5 promotes the proliferation of glioma cells via enhancing the mrna stability of g6pd.Neurochem Res. 2021 2021Nov;46(11):3003–11. doi: https://doi.org/10.1007/s11064-021-03408-9
Sheng H, Li Z, Su S, Sun W, Zhang X, Li L et al. Yth domain family 2 promotes lung cancer cell growth by facilitating 6-phosphogluconate dehydrogenase mrna translation.Carcinogenesis. 2020 2020 Jul10;41(5):541–50. doi: https://doi.org/10.1093/carcin/bgz152
Zhao Y, Feng F, Guo QH, Wang YP, Zhao R. Role of succinate dehydrogenase deficiency and oncometabolites in gastrointestinal stromal tumors. World J Gastroenterol. 2020 2020 Sep 14;26(34):5074-89. doi: https://doi.org/10.3748/wjg.v26.i34.5074
Santos CR, Schulze A. Lipid metabolism in cancer. Febs J 2012. 2012 Aug;279(15):2610–23. https://doi.org/10.1111/j.1742-4658.2012.08644.x.
Abramson HN. The lipogenesis pathway as a cancer target.J Med Chem. 2011 2011 Aug25;54(16):5615–38. doi: https://doi.org/10.1021/jm2005805
Röhrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer 2016. 2016 Nov;16(11):732–49. https://doi.org/10.1038/nrc.2016.89.
Nath A, Li I, Roberts LR, Chan C. Elevated free fatty acid uptake via cd36 promotes epithelial-mesenchymal transition in hepatocellular carcinoma. Sci Rep. 2015 2015 Oct 1;5:14752. doi: https://doi.org/10.1038/srep14752
Zhao J, Zhi Z, Wang C, Xing H, Song G, Yu X et al. Exogenous lipids promote the growth of breast cancer cells via cd36. Oncol Rep. 2017 2017 Oct;38(4):2105-15. doi: https://doi.org/10.3892/or.2017.5864
Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis.Nat Rev Cancer. 2007 2007Oct;7(10):763–77. doi: https://doi.org/10.1038/nrc2222
Koizume S, Miyagi Y. Lipid droplets: a key cellular organelle associated with cancer cell survival under normoxia and hypoxia.Int J Mol Sci. 2016 2016 Aug31;17(9). doi: https://doi.org/10.3390/ijms17091430
Guo H, Wang B, Xu K, Nie L, Fu Y, Wang Z et al. M(6)a reader hnrnpa2b1 promotes esophageal cancer progression via up-regulation of acly and acc1. Front Oncol. 2020 2020;10:553045. doi: https://doi.org/10.3389/fonc.2020.553045
Yang Y, Cai J, Yang X, Wang K, Sun K, Yang Z et al. Dysregulated m6a modification promotes lipogenesis and development of non-alcoholic fatty liver disease and hepatocellular carcinoma. Mol Ther. 2022 2022 Jun 1;30(6):2342-53. doi: https://doi.org/10.1016/j.ymthe.2022.02.021
Sun D, Zhao T, Zhang Q, Wu M, Zhang Z. Fat mass and obesity-associated protein regulates lipogenesis via m(6) a modification in fatty acid synthase mrna.Cell Biol Int. 2021 2021Feb;45(2):334–44. doi: https://doi.org/10.1002/cbin.11490
Yang Z, Yu GL, Zhu X, Peng TH, Lv YC. Critical roles of fto-mediated mrna m6a demethylation in regulating adipogenesis and lipid metabolism: implications in lipid metabolic disorders.Genes Dis. 2022 2022Jan;9(1):51–61. doi: https://doi.org/10.1016/j.gendis.2021.01.005
Conte G, Mele M, Chessa S, Castiglioni B, Serra A, Pagnacco G et al. Diacylglycerol acyltransferase 1, stearoyl-coa desaturase 1, and sterol regulatory element binding protein 1 gene polymorphisms and milk fatty acid composition in italian brown cattle.J Dairy Sci. 2010 2010Feb;93(2):753–63. doi: https://doi.org/10.3168/jds.2009-2581
Bravard A, Lefai E, Meugnier E, Pesenti S, Disse E, Vouillarmet J, et al. Fto is increased in muscle during type 2 diabetes, and its overexpression in myotubes alters insulin signaling, enhances lipogenesis and ros production, and induces mitochondrial dysfunction. Diabetes 2011. 2011 Jan;60(1):258–68. https://doi.org/10.2337/db10-0281.
Yabe D, Komuro R, Liang G, Goldstein JL, Brown MS. Liver-specific mrna for insig-2 down-regulated by insulin: implications for fatty acid synthesis.Proc Natl Acad Sci U S a. 2003 2003 Mar18;100(6):3155–60. doi: https://doi.org/10.1073/pnas.0130116100
Chen Z, Wu L, Zhou J, Lin X, Peng Y, Ge L, et al. N6-methyladenosine-induced errγ triggers chemoresistance of cancer cells through upregulation of abcb1 and metabolic reprogramming. Theranostics 2020. 2020;10(8):3382–96. https://doi.org/10.7150/thno.40144.
Peng H, Chen B, Wei W, Guo S, Han H, Yang C et al. N(6)-methyladenosine (m(6)a) in 18s rrna promotes fatty acid metabolism and oncogenic transformation.Nat Metab. 2022 2022Aug;4(8):1041–54. doi: https://doi.org/10.1038/s42255-022-00622-9
Zhen L, Pan W. Alkbh5 inhibits the sirt3/acc1 axis to regulate fatty acid metabolism via an m6a-igf2bp1-dependent manner in cervical squamous cell carcinoma. Clin Exp Pharmacol Physiol. 2023 2023 Jan 27. doi: https://doi.org/10.1111/1440-1681.13754
Zhu S, Wang JZ, Chen D, He YT, Meng N, Chen M et al. An oncopeptide regulates m(6)a recognition by the m(6)a reader igf2bp1 and tumorigenesis.Nat Commun. 2020 2020 Apr3;11(1):1685. doi: https://doi.org/10.1038/s41467-020-15403-9
Hassanein M, Hoeksema MD, Shiota M, Qian J, Harris BK, Chen H et al. Slc1a5 mediates glutamine transport required for lung cancer cell growth and survival. Clin Cancer Res. 2013 2013 Feb 1;19(3):560–70. doi: https://doi.org/10.1158/1078-0432.CCR-12-2334
Matés JM, Segura JA, Martín-Rufián M, Campos-Sandoval JA, Alonso FJ, Márquez J. Glutaminase isoenzymes as key regulators in metabolic and oxidative stress against cancer. Curr Mol Med. 2013 2013 May;13(4):514–34. doi: https://doi.org/10.2174/1566524011313040005
Xiao Y, Thakkar KN, Zhao H, Broughton J, Li Y, Seoane JA et al. The m(6)a rna demethylase fto is a hif-independent synthetic lethal partner with the vhl tumor suppressor. Proc Natl Acad Sci U S a. 2020 2020 Sep 1;117(35):21441-49. doi: https://doi.org/10.1073/pnas.2000516117
Chen P, Liu XQ, Lin X, Gao LY, Zhang S, Huang X. Targeting ythdf1 effectively re-sensitizes cisplatin-resistant colon cancer cells by modulating gls-mediated glutamine metabolism.Mol Ther Oncolytics. 2021 2021 Mar26;20:228–39. doi: https://doi.org/10.1016/j.omto.2021.01.001
Weng H, Huang F, Yu Z, Chen Z, Prince E, Kang Y et al. The m(6)a reader igf2bp2 regulates glutamine metabolism and represents a therapeutic target in acute myeloid leukemia. Cancer Cell. 2022 2022 Dec 12;40(12):1566-82. doi: https://doi.org/10.1016/j.ccell.2022.10.004
Han L, Dong L, Leung K, Zhao Z, Li Y, Gao L et al. Mettl16 drives leukemogenesis and leukemia stem cell self-renewal by reprogramming bcaa metabolism.Cell Stem Cell. 2023 2023 Jan5;30(1):52–68. doi: https://doi.org/10.1016/j.stem.2022.12.006
Zhuang C, Zhuang C, Luo X, Huang X, Yao L, Li J et al. N6-methyladenosine demethylase fto suppresses clear cell renal cell carcinoma through a novel fto-pgc-1α signalling axis. J Cell Mol Med. 2019 2019 Mar;23(3):2163-73. doi: https://doi.org/10.1111/jcmm.14128
Panayiotou C, Solaroli N, Karlsson A. The many isoforms of human adenylate kinases.Int J Biochem Cell Biol. 2014 2014Apr;49:75–83. doi: https://doi.org/10.1016/j.biocel.2014.01.014
Liu X, Gonzalez G, Dai X, Miao W, Yuan J, Huang M et al. Adenylate kinase 4 modulates the resistance of breast cancer cells to tamoxifen through an m(6)a-based epitranscriptomic mechanism.Mol Ther. 2020 2020 Dec2;28(12):2593–604. doi: https://doi.org/10.1016/j.ymthe.2020.09.007
Jia C, Guo Y, Chen Y, Wang X, Xu Q, Zhang Y et al. Hnrnpa2b1-mediated m6a modification of tlr4 mrna promotes progression of multiple myeloma.J Transl Med. 2022 2022 Nov18;20(1):537. doi: https://doi.org/10.1186/s12967-022-03750-8
Huang J, Sun W, Wang Z, Lv C, Zhang T, Zhang D et al. Fto suppresses glycolysis and growth of papillary thyroid cancer via decreasing stability of apoe mrna in an n6-methyladenosine-dependent manner.J Exp Clin Cancer Res. 2022 2022Jan 28;41(1):42. doi: https://doi.org/10.1186/s13046-022-02254-z
You Q, Wang F, Du R, Pi J, Wang H, Huo Y et al. M(6) a reader ythdf1-targeting engineered small extracellular vesicles for gastric cancer therapy via epigenetic and immune regulation.Adv Mater. 2023 2023Feb;35(8):e2204910. doi: https://doi.org/10.1002/adma.202204910
Chen B, Ye F, Yu L, Jia G, Huang X, Zhang X et al. Development of cell-active n6-methyladenosine rna demethylase fto inhibitor.J Am Chem Soc. 2012 2012 Oct31;134(43):17963–71. doi: https://doi.org/10.1021/ja3064149
Huang Y, Yan J, Li Q, Li J, Gong S, Zhou H et al. Meclofenamic acid selectively inhibits fto demethylation of m6a over alkbh5. Nucleic Acids Res. 2015 2015 Jan;43(1):373–84. doi: https://doi.org/10.1093/nar/gku1276
Wang T, Hong T, Huang Y, Su H, Wu F, Chen Y et al. Fluorescein derivatives as bifunctional molecules for the simultaneous inhibiting and labeling of fto protein.J Am Chem Soc. 2015 2015 Nov4;137(43):13736–39. doi: https://doi.org/10.1021/jacs.5b06690
Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu H et al. Small-molecule targeting of oncogenic fto demethylase in acute myeloid leukemia.Cancer Cell. 2019 2019 Apr15;35(4):677–91. doi: https://doi.org/10.1016/j.ccell.2019.03.006
Su R, Dong L, Li Y, Gao M, Han L, Wunderlich M et al. Targeting fto suppresses cancer stem cell maintenance and immune evasion.Cancer Cell. 2020 2020 Jul13;38(1):79–96. doi: https://doi.org/10.1016/j.ccell.2020.04.017
Huff S, Kummetha IR, Zhang L, Wang L, Bray W, Yin J et al. Rational design and optimization of m(6)a-rna demethylase fto inhibitors as anticancer agents.J Med Chem. 2022 2022 Aug25;65(16):10920–37. doi: https://doi.org/10.1021/acs.jmedchem.1c02075
Bedi RK, Huang D, Eberle SA, Wiedmer L, Śledź P, Caflisch A. Small-molecule inhibitors of mettl3, the major human epitranscriptomic writer. Chemmedchem. 2020 2020 May 6;15(9):744–48. doi: https://doi.org/10.1002/cmdc.202000011
Moroz-Omori EV, Huang D, Kumar BR, Cheriyamkunnel SJ, Bochenkova E, Dolbois A et al. Mettl3 inhibitors for epitranscriptomic modulation of cellular processes.Chemmedchem. 2021 2021 Oct6;16(19):3035–43. doi: https://doi.org/10.1002/cmdc.202100291
Dolbois A, Bedi RK, Bochenkova E, Müller A, Moroz-Omori EV, Huang D et al. 1,4,9-triazaspiro[5.5]undecan-2-one derivatives as potent and selective mettl3 inhibitors.J Med Chem. 2021 2021 Sep9;64(17):12738–60. doi: https://doi.org/10.1021/acs.jmedchem.1c00773
Yankova E, Blackaby W, Albertella M, Rak J, De Braekeleer E, Tsagkogeorga G et al. Small-molecule inhibition of mettl3 as a strategy against myeloid leukaemia.Nature. 2021 2021May;593(7860):597–601. doi: https://doi.org/10.1038/s41586-021-03536-w
Lee JH, Choi N, Kim S, Jin MS, Shen H, Kim YC. Eltrombopag as an allosteric inhibitor of the mettl3-14 complex affecting the m(6)a methylation of rna in acute myeloid leukemia cells. Pharmaceuticals (Basel). 2022 2022 Apr 1;15(4). doi: https://doi.org/10.3390/ph15040440
Roth M, Will B, Simkin G, Narayanagari S, Barreyro L, Bartholdy B et al. Eltrombopag inhibits the proliferation of leukemia cells via reduction of intracellular iron and induction of differentiation. Blood. 2012 2012 Jul 12;120(2):386–94. doi: https://doi.org/10.1182/blood-2011-12-399667
Lee JH, Kim S, Jin MS, Kim YC. Discovery of substituted indole derivatives as allosteric inhibitors of m(6) a-rna methyltransferase, mettl3-14 complex.Drug Dev Res. 2022 2022May;83(3):783–99. doi: https://doi.org/10.1002/ddr.21910
Xiang M, Liu W, Tian W, You A, Deng D. Rna n-6-methyladenosine enzymes and resistance of cancer cells to chemotherapy and radiotherapy.Epigenomics. 2020 2020May;12(9):801–09. doi: https://doi.org/10.2217/epi-2019-0358
Yan F, Al-Kali A, Zhang Z, Liu J, Pang J, Zhao N, et al. A dynamic n(6)-methyladenosine methylome regulates intrinsic and acquired resistance to tyrosine kinase inhibitors. Cell Res 2018. 2018 Nov;28(11):1062–76. https://doi.org/10.1038/s41422-018-0097-4.
Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y et al. R-2hg exhibits anti-tumor activity by targeting fto/m(6)a/myc/cebpa signaling. Cell. 2018 2018 Jan 11;172(1–2):90–105. doi: https://doi.org/10.1016/j.cell.2017.11.031
Wang J, Qiao Y, Sun M, Sun H, Xie F, Chang H et al. Fto promotes colorectal cancer progression and chemotherapy resistance via demethylating g6pd/parp1. Clin Transl Med. 2022 2022 Mar;12(3):e772. doi: https://doi.org/10.1002/ctm2.772
Liu Y, Liang G, Xu H, Dong W, Dong Z, Qiu Z et al. Tumors exploit fto-mediated regulation of glycolytic metabolism to evade immune surveillance. Cell Metab. 2021 2021 Jun 1;33(6):1221-33. doi: https://doi.org/10.1016/j.cmet.2021.04.001
Pan ZP, Wang B, Hou DY, You RL, Wang XT, Xie WH et al. Mettl3 mediates bone marrow mesenchymal stem cell adipogenesis to promote chemoresistance in acute myeloid leukaemia.Febs Open Bio. 2021 2021Jun;11(6):1659–72. doi: https://doi.org/10.1002/2211-5463.13165
Yang H, Hu Y, Weng M, Liu X, Wan P, Hu Y et al. Hypoxia inducible lncrna-cbslr modulates ferroptosis through m6a-ythdf2-dependent modulation of cbs in gastric cancer.J Adv Res. 2022 2022Mar;37:91–106. doi: https://doi.org/10.1016/j.jare.2021.10.001
Liu Z, Chen Y, Shen T. Evidence based on an integrative analysis of multi-omics data on mettl7a as a molecular marker in pan-cancer.Biomolecules. 2023 2023 Jan18;13(2). doi: https://doi.org/10.3390/biom13020195
Pei H, Dai Y, Yu Y, Tang J, Cao Z, Zhang Y et al. The tumorigenic effect of lncrna afap1-as1 is mediated by translated peptide atmlp under the control of m(6) a methylation. Adv Sci (Weinh). 2023 2023 Mar 4:e2300314. doi: https://doi.org/10.1002/advs.202300314
Duan JL, Chen W, Xie JJ, Zhang ML, Nie RC, Liang H et al. A novel peptide encoded by n6-methyladenosine modified circmap3k4 prevents apoptosis in hepatocellular carcinoma. Mol Cancer. 2022 2022 Apr 2;21(1):93. doi: https://doi.org/10.1186/s12943-022-01537-5
Chen Y, Ling Z, Cai X, Xu Y, Lv Z, Man D et al. Activation of yap1 by n6-methyladenosine-modified circcpsf6 drives malignancy in hepatocellular carcinoma.Cancer Res. 2022 2022 Feb15;82(4):599–614. doi: https://doi.org/10.1158/0008-5472.CAN-21-1628
Wang L, Yi X, Xiao X, Zheng Q, Ma L, Li B. Exosomal mir-628-5p from m1 polarized macrophages hinders m6a modification of circfut8 to suppress hepatocellular carcinoma progression.Cell Mol Biol Lett. 2022 2022 Dec6;27(1):106. doi: https://doi.org/10.1186/s11658-022-00406-9
Acknowledgements
Not applicable.
Funding
This study was funded by National Natural Science Foundation of China (82172976).
Author information
Authors and Affiliations
Contributions
Z.Z. and F.L. conceived and revised the paper. J.H. drafted the manuscript and prepared the figures. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
He, J., Liu, F. & Zhang, Z. Functions of N6-methyladenosine in cancer metabolism: from mechanism to targeted therapy. Biomark Res 11, 40 (2023). https://doi.org/10.1186/s40364-023-00483-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-023-00483-8