
Abstract
N6-methyladenosine (m6A) modification is the most common internal modification of eukaryotic mRNA and is widely involved in many cellular processes, such as RNA transcription, splicing, nuclear transport, degradation, and translation. m6A has been shown to plays important roles in the initiation and progression of various cancers. The altered metabolic programming of cancer cells promotes their cell-autonomous proliferation and survival, leading to an indispensable hallmark of cancers. Accumulating evidence has demonstrated that this epigenetic modification exerts extensive effects on the cancer metabolic network by either directly regulating the expression of metabolic genes or modulating metabolism-associated signaling pathways. In this review, we summarized the regulatory mechanisms and biological functions of m6A and its role in cancer metabolic reprogramming.
Similar content being viewed by others

Introduction
N6-methyladenosine (m6A) is the most prevalent type of RNA modification of eukaryotic mRNAs [1, 2] and plays an important role in many biological functions including tissue development [3], naive pluripotency and stem cell differentiation [4], the heat shock response [5] and DNA damage [6]. m6A has been increasingly implicated in various human diseases such as obesity [7], diabetes [8], infertility [9], metabolic syndrome, and cancers [10,11,12]. In various cancers, m6A functions as a promoter or suppressor in cancers by regulating the expression of cancer-related genes, which may affect the initiation [13], proliferation [14], differentiation [15], metastasis [16] and metabolic reprogramming of cancer cells [17]. In 2011, He et al. discovered that fat mass and obesity-associated protein (FTO) exhibited efficient demethylation of m6A residues in RNA in vitro [18]. Based on the finding that the internal amino acid sequence of FTO was similar to the active domains of DNA demethylases, a second m6A demethylase, Alk B homolog 5 (ALKBH5), was identified and confirmed [19]. Since then, major insights into the biological functions and regulatory mechanisms of m6A have been reported. Although FTO was found to be more active on N6,2′-O-dimethyladenosine (m6Am; at the cap + 1 position) than on m6A in internal mRNA in experiments [20], the FTO-mediated demethylation events that act on internal m6A are more important. This finding is because the fact that total cap m6Am level is less than 1/20 of that of the internal m6A, and approximately 95% of the observed m6A increases occurred on internal sites when FTO was knocked down in AML cells. Moreover, Darnel and Ke et al. concluded that the methylation and demethylation of m6A were constrained in the nucleus and that no specific sites or demethylation of specific m6A residues were added to the mRNA in the cytoplasm [21]. Several groups have argued over the reversibility of m6A methylation in vivo [21, 22]. Currently, little is known about when the m6A actually occurs in the formation of cellular mRNA; hence, additional investigation is required. The field is still in its infancy. Thus, the possible function of the nuclear methylations and apparent demethylations requires further investigation.
Given that altered metabolism is a core hallmark of cancer, one of the recent research hotspots in the cancer field is metabolic reprogramming. This article summarizes the current understanding of m6A and cancer metabolic alterations, together with the crosstalk between m6A and nutrition, metabolism, and tumorigenesis. We further discuss whether m6A could be used in a therapeutic strategy targeting cancer metabolic reprogramming. These findings can be used to develop clinical guidelines and a novel therapeutic approach for cancer treatments involving early diagnosis, long-term follow-up, and prognosis.
The regulatory mechanisms of m6A
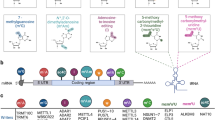
m6A mainly occurs at the consensus motif DRACH (D corresponds to A, G or U; R corresponds to G or A; H corresponds to A, C or U) [23, 24], and it is enriched in the 5′-untranslated region (5′-UTR), 3′-untranslated region (3′-UTR) and coding DNA sequence (CDS) proximal to the stop codon of mRNAs [25, 26]. The effectors of m6A include ‘writers’, ‘readers’ and ‘erasers’ (Fig. 1). The writer methyltransferase adds m6A methylation on target RNAs via the methyl groups of S-adenosylmethionine (SAM) transferase [27]. The methyltransferase complex comprises the catalytic subunit methyltransferase like 3 (METTL3) and the catalytically inactive but structurally stabilizing subunit METTL14. The methyltransferase domains of METTL3 (MTD3, residues 357–580) and METTL14 (MTD14, residues 111–456) engage in extensive contact with each other to form a stable heterodimer, which, with the inclusion of the two Cys-Cys-Cys-His (CCCH)-type zinc-binding (ZFD) motifs of METTL3, can catalyze the addition of m6A [28, 29]. The ZFD serves as the target recognition domain for its special binding to the GGACH consensus sequence in the RNA [30, 31] and thus is responsible for the methyltransferase activity of the METTL3-METTL14 complex. For normal m6A modification to occur in cells, the METTL3-METTL14 complex also needs to associate with additional factors, such as tumor 1-associated protein (WTAP) [29, 32], KIAA1429 (also called Virilizer), RNA binding motif protein 15 (RBM15), the E3 ubiquitin ligase HAKAI, zinc finger CCCH domain-containing protein 13 (ZC3H13), and etc.[33,34,35].

The process and molecular functions of m6A methylation. The effectors in m6A include ‘writers’, ‘readers’ and ‘erasers’. Writer methyltransferase installs m6A methylation on target RNAs via the methyl groups of S-adenosylmethionine (SAM) transferase. FTO and ALKBH5 were two major RNA demethylases that catalyze the removal of m6A on RNA in a Fe(II)/α-KG (α-ketoglutarate)-dependent manner. Methyltransferases and demethylases cooperate in modulating the distribution and abundance of m6A in RNAs, meanwhile the ‘readers’ specifically recognize and bind m6A-RNAs to control their fate and regulate downstream functions
However, FTO and ALKBH5 are two major RNA demethylases that catalyze the removal of m6A on RNA in an Fe(II)/α-KG (α-ketoglutarate)-dependent manner [36]. Structurally, ALKBH5 preferably demethylates m6A single-stranded RNA (ssRNA) over double-stranded DNA (dsDNA) because the loop (amino acids 229–243) causes a steric clash with the complementary strand of dsDNA. The m6A base is predicted to pack against His-204 and is in a pocket composed of Arg-130 and Tyr-139 in ALKBH5, which contributes to the m6A recognition [37]. In addition, the in vivo substrates of FTO include m6A and cap m6Am in mRNA, m6A and m6Am in snRNA, and m1A in tRNA. FTO exhibits a preference for the nucleobase m6Am over internal m6A in ssRNA, and the key residues (such as E234) in the catalytic pocket of FTO, rather than the structural differences of the ribose ring, function in nucleobase selection and recognition. The sequence and the tertiary structure of RNA affect the catalytic activity of FTO [38]. Methyltransferases and demethylases cooperate in modulating the distribution and abundance of m6A in RNAs, while the ‘readers’ specifically recognize and bind m6A-RNAs to control their fate and regulate downstream functions [39]. One class of direct and robust m6A readers are proteins containing the YT521-B homology (YTH) domain, including YTH domain family 1 − 3 (YTHDF1 − 3) [40] and YTH domain containing 1 − 2 (YTHDC1 − 2) in humans [41]. Other class of direct m6A binding proteins include the heterogeneous nuclear ribonucleoprotein A2B1( HNRNPA2B1) [42]; insulin-like growth factor 2 (IGF2) mRNA-binding proteins 1, 2 and 3 (IGF2BP1/2/3) [43], eukaryotic initiation factor 3 (eIF3) [44], and proline rich coiled-coil 2A (PRRC2A) [45]. In addition, there are other m6A readers such as HNRNPC [46] and HNRNPG [47, 48], which, rather than directly recognize the m6A group, accessibly bind the RNA-binding motifs upon m6A methylation of the RNA [48].
Overall, the installation, demethylation and recognition of m6A involved many effectors and pathways. Further studies need to be conducted to uncover new available factors in m6A modification with the goal of attaining a thorough understanding of this epigenetic modification.
The biological functions of m6A
m6A is involved in many cellular RNA processes, including transcription, splicing, nuclear transport, degradation, and translation (Fig. 1 and Table 1).
m6A modulates mRNA maturation
Chemical inhibition of m6A formation caused changes in the ratio between precursor and mature mRNAs, and m6A was observed to be significantly enriched in both multi-isoform genes and alternatively spliced exons [49]. Immunofluorescence analysis showed that METTL3, METTL14, WTAP, FTO, and ALKBH5 colocalized well in nuclear speckles, the site where splicing factors accumulate [50]. These results suggest a potential role of m6A in mRNA splicing. A combination of transcriptome analyses and m6A-seq revealed that m6A is enriched in exonic regions flanking 5′- and 3′-splice sites, spatially overlapping with mRNA splicing regulatory serine/arginine-rich (SR) protein binding motifs. Modulating the expression of METTL3, ALKBH5 and FTO induces large-scale alterations in splicing patterns [23, 34, 49,50,51]. When ALKBH5 was depleted and the demethylation activity was diminished, serine-arginine protein kinase 1 (SRPK1) translocated from nuclear locations to dot-like cytoplasmic sites. Because SRPK1 is one of the main kinases responsible for the phosphorylation of alternative splicing factor/splicing factor 2 (ASF/SF2) [52], the depletion of ALKBH5 induced the ASF/SF2 switch from splicing factors to export adaptor proteins to promote mRNA export [50]. YTHDC1 promotes the exon inclusion of targeted mRNAs by facilitating serine and arginine rich splicing factor 3 (SRSF3) while blocking SRSF10 mRNA binding, demonstrating how YTHDC1 directly regulates mRNA splicing by bridging interactions of trans- and cis-regulatory elements [53]. In addition, HNRNPA2B1 binds to ‘RGm6AC’-containing sites on nuclear pri-miRNAs and interacts with the DGCR8 protein, a component of the pri-miRNA microprocessor complex, to facilitate the processing and maturation of pri-miRNAs [42].
However, m6A indirectly regulates mRNA splicing by modulating the structure of mRNA. m6A residues within RNA stems can destabilize the thermostability of model RNA duplexes without precluding Watson–Crick base pairing and make them more single-stranded or accessible [54,55,56], thus enhancing their interactions with HNRNPC. Consequently, m6A functions as an mRNA structure remodeler to affect mRNA maturation through interference with post-transcriptional regulator binding activities [57]. Taken together, these observations demonstrate that m6A modulates RNA maturation in direct and indirect ways.
m6A modulates RNA degradation and stability
Degradation plays a fundamental role in maintaining cellular homeostasis, as both a surveillance mechanism eliminating aberrant mRNAs or during RNA processing generating mature transcripts [58]. The deadenylation-dependent decay pathway is used by most mRNAs in eukaryotes [59]. Deadenylation is triggered by deadenylases, including among others, the CCR4–NOT complex in mammals [60]. Additionally, there are three predominant forms of cotranslational mRNA surveillance: nonsense-mediated decay (NMD), no-go decay (NGD), and non-stop decay (NSD) [61]. m6A ‘readers’ proteins were widely reported to bind m6A-methylated mRNA and control RNA decay in an m6A methylation-dependent manner. The carboxyl terminal domain of YTHDF2 selectively binds to m6A-containing mRNA, while the amino-terminal domain recruits the CCR4-NOT complex through the SH domain of CNOT1, the scaffolding subunit of the CCR4-NOT complex. This recruitment is essential for the deadenylation of m6A-containing mRNAs by two deadenylase subunits, CAF1 and CCR [62]. Additionally, the P/Q/N-rich amino-terminus of YTHDF2 localizes the YTHDF2-m6A-mRNA complex to more specialized mRNA decay machinery (P bodies, etc.) for committed degradation [63]. Subsequent studies proved that CCR4-NOT deadenylation complex proteins were notable binding partners of all three DF proteins, indicating the idea that all DF paralogs had a common role in mRNA degradation [64]. In addition, m6A modification around the start codon of SRSFs is involved in degradation through METTL3 and YTHDC1 mediation of NMD, regulating the expression of SRSFs [65]. Mechanistically, in METTL3-KD cells, premature termination (i.e., stop) codons (PTCs) in the mRNAs of SRSFs occur by exon inclusion or skipping upon METTL3 depletion. These mRNAs with PTCs are subjected to NMD. Reduced expression of SRSFs induces alternative splicing isoform switches of related oncogenes, such as BCL-XS and NCOR2, promoting glioblastoma multiforme (GBM) growth and progression.
Nevertheless, other relevant proteins, such as insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs) and ELAVL1 (also known as HUR), promote the stability and storage of target mRNAs under various physiological conditions and thus affect gene expression output. The mRNA decay of C-MYC, Fascin Actin-Bundling Protein 1(FSCN1), Thymidine Kinase 1(TK1), and Myristoylated alanine-rich protein kinase C substrate Like 1 (MARCKSL1) was accelerated upon knockdown of IGF2BPs in HepG2 cells. Moreover, the mRNA stabilizing function of IGF2BPs was supported by its cofactors HuR and matrin 3 (MATR3) [43]. HuR is a well-established RNA stabilizer protein that binds to the U-rich regions at the 3′-UTR of thousands of transcripts and blocks miRNA targeting. Visvanathan et al. demonstrated that HuR interacted with SOX2 mRNA containing m6A to block the miRNA-dependent mRNA degradation and therefore to increase the mRNA stability [66]. Another RNA immunoprecipitation (RIP) analysis indicated increased HuR binding at the IGFBP3 3′-UTR in METTL3 or METTL14 knockdown cells with decreased m6A levels, suggesting that demethylation accompanies HuR binding. Because the predicted motifs of m6A and HuR binding sites differ substantially and the endogenous m6A and HuR sites do not always colocalize, spatial constraints may control m6A and HuR binding [67]. Thus, m6A methylation has complex and sophisticated functions involving both stabilization and destabilization of RNAs. More investigations are needed to identify how to equilibrate and coordinate these two processes in mammalian cells.
m6A modulates RNA translation
In addition to the control of mRNA degradation, regulation of translation is critical for managing the quantity and duration of gene expression in eukaryotic cells. mRNA nuclear export decreased when METTL3 was silenced [68] but accelerated upon ALKBH5 knockdown [50]. Furthermore, the fragile X mental retardation protein (FMRP), an m6A reader, promoted the nuclear export of methylated mRNAs in a CRM1-dependent manner during neural differentiation [69]. These results proved that m6A played an important role in RNA nuclear export, thus modulating mRNA translocation in humans. Wang et al. showed that YTHDF1 promoted the ribosome loading of mRNA and directly accelerated the translation initiation rate of target mRNAs in cells, possibly via the association of YTHDF1 with the translation initiation complex [70]. However, when Jaffrey and his team reanalyzed the previously published data from Wang et al. together with their own independent ribosome profiling datasets and performed new polysome fraction analysis, they claimed that none of the DF proteins (including YTHDF1, YTHDF2 and YTHDF3) directly promoted translation of m6A-mRNAs in Hela cells. Instead, their major function was to mediate mRNA degradation [64]. The inconsistent results may be due to bioinformatic and technical issues. m6A, however, affects mRNA translation through DF-independent mechanisms. m6A could impact 3′-UTR length, indirectly affecting translation [21]. Moreover, the m6A in the 5′-UTR recruits eIF3 to promote translation [44]. Although the presence of an m6A within a codon does not perturb canonical base-pairing in the final step of tRNA accumulation, the steric effects caused by decreased thermodynamic stability of modified A-U pairs can block the tRNA accommodation and translation elongation [71].rRNA with m6A in the mature ribosome has been implicated in the regulation and activity tuning of protein synthesis because it tends to localize in functionally important regions [72]. ZCCHC4, a new m6A methyltransferase, catalyzed m6A4220 methylation in human 28S rRNA to affect global translation activity, which is required for cell proliferation and tumor growth. It also interacts with a subset of mRNAs [73, 74]. Consequently, m6A affects nuclear export and translation by regulating the biological behaviors of mRNA, tRNA, and rRNA.
Methods for detection of m6A methylation
To date, most methods for global m6A detection have relied on immunoprecipitation of methylated RNAs using m6A-recognizing antibodies in a technique called methylated RNA immunoprecipitation sequencing (MeRIP-Seq/m6A-Seq) [75]. Although these methods have yielded unprecedented insights into the location and regulation of m6A in cellular RNAs, they have several limitations. Novel methods combined with multifield technologies have emerged (Fig. 2a–d), benefiting from the development of the high throughput sequencing and the liquid chromatography-tandem mass spectrometry (LC-MS/MS).

Methods for m6A methylation detection. a MeRIP-seq/m6A-seq; b Mapping m6A at individual-nucleotide resolution using crosslinking and immunoprecipitation: miCLIP and m6ACE-seq; c Site-specific cleavage and radioactive labeling followed by ligation-assisted extraction and thin-layer chromatography (SCARLET); d Antibody-independent m6A identification methods: (m6A-REF-seq) or MAZTER-seq
MeRIP-seq/m6A-seq
MeRIP-seq/m6A-seq is by now the most extensively used molecular tool in m6A research. This technique uses anti-m6A antibodies to capture and enrich the m6A-containing RNA fragments (Fig. 2a), followed by high-throughput sequencing to profile m6A distributions in mammalian transcriptomes [76]. MeRIP-seq cannot identify the strict locations of the m6A in global transcriptomes because the resolution is 100–200 nt. Moreover, in the immunoprecipitation, large amounts of input RNA prevent global m6A detection in rare biological materials such as pathological tissues or early embryos [66]. Thus, novel methods are needed to unambiguously determine the m6A status at single-nucleotide resolution to further understand the biological function of this highly abundant modification.
Mapping m6A at individual-nucleotide resolution using crosslinking and immunoprecipitation
In m6A individual-nucleotide resolution crosslinking and immunoprecipitation (miCLIP), extracted cellular RNA is cropped and crosslinked to an anti-m6A antibody using ultraviolet light (Fig. 2b). Antibody-crosslinked RNA fragments are then purified and converted into a cDNA library according to the PAR-CLIP protocol [77]. Then, crosslink-induced truncations or mutations after reverse transcription are investigated to identify exact m6A sites in the transcriptome. miCLIP detects m6A with high specificity and sensitivity [24]. Concurrently, using crosslinking and immunoprecipitation, m6A-CLIP [78] and m6A-crosslinking-exonucleasesequencing (m6ACE-seq) [79], different teams adopted techniques similar to miCLIP to map m6A at quantitative single-base resolution. m6A sites identified by m6ACE-seq exhibited significant overlap with sites identified by previous single-base resolution m6A-sequencing methods [79]. The sensitivity and specificity of m6ACE-seq were further validated by comparison with an orthogonal sequencing-independent single-base-resolution m6A mapping technique, site-specific cleavage and radioactive labeling followed by ligation-assisted extraction and thin-layer chromatography, known as SCARLET [80].
Site-specific cleavage and radioactive labeling followed by ligation-assisted extraction and thin-layer chromatography
The sequencing methods only offer the distribution of m6A at a transcriptome-wide scale, but they cannot quantitatively detect the m6A fraction in the specific location, which are becoming increasingly important. SCARLET can accurately determine the m6A signature at single nucleotide resolution in any mRNA or lncRNA (Fig. 2c). Purification of the RNA of interest is not needed. Under the guidance of a complementary 2′-OMe/2′-H chimeric oligonucleotide, RNase H is applied to cut RNA, achieving site-specific cleavage 5′ to the candidate site. Before splint-ligating to a 116-nucleotide single-stranded DNA oligonucleotide using DNA ligase, the RNA fragment is radiolabeled using 32P. All RNA samples are treated with RNase T1/A for complete digestion except for the 32P-labeled candidate site. The 117/118-mers band on the denaturing electrophoresis gel is harvested and eluted and then digested by nuclease P1 into mononucleotides containing 5′ phosphate. Finally, thin-layer chromatography is performed to determine the m6A signature. SCARLET requires only common and readily available lab equipment and material, which makes it ready and available for researchers to investigate the dynamics and biology of RNA modification [80, 81].
Antibody-independent m6A identification methods
Currently, most of the commonly used high-sensitivity LC-MS/MS and blotting methods are m6A antibody dependent, suffering from poor reproducibility and complicated processes. Additionally, it is difficult to quantify the level of methylation because of the affinity variation and batch effects of antibodies. Therefore, novel methods are still needed for whole transcriptome m6A identification and quantification to elucidate the dynamics and cellular functions of m6A in post-transcriptomic regulation. m6A-sensitive RNA-endoribonuclease-facilitated sequencing (m6A-REF-seq) [82] or MAZTER-seq [83] relies on the fact that MazF (Fig. 2d), an Escherichia coli toxin and RNA endoribonuclease, is sensitive to m6A modification in the ACA motif. This enzyme specifically identifies and cleaves the unmethylated ACA motifs while leaving methylated (m6A)CA motifs intact [84]. With MAZTER-MINE (https://github.com/SchwartzLab/mazter_mine), a computational pipeline, data from paired-end sequencing are analyzed to identify and quantify methylation sites following end repair, ligation, reverse transcription, and cDNA amplification [83]. Rapid and simplified experimental design without antibody-enrichment substantially reduces the starting RNA amount and sample preparation time, which addresses the limitations of current antibody-dependent methods. In addition, this method can capture the subtle changes of m6A during metabolic processes, highly advancing the dynamic studies of RNA m6A modification in different life stages.
m6A methylation in cancer metabolic reprogramming
Reprogramming energy metabolism is one of the hallmarks of cancer, in addition to mutants, proliferative signaling, invasion and metastasis, and angiogenesis [85]. By taking advantage of the existing metabolic networks, cancer cells selectively activate or inhibit the metabolic pathways (e.g., aerobic glycolysis [86], disordered lipid metabolism [87], glutamine-dependent anaplerosis [88], and so on) (Table 2) to accelerate the proliferative capabilities. Thus, metabolite influx is altered and metabolites shunt into pathways that support biosynthesis to meet bioenergetic needs [89]. As the most abundant internal RNA modification, m6A plays an indispensable role in cancer metabolic reprogramming through either direct regulation of nutrient transporters and metabolic enzymes or indirect control of metabolic oncogenes and key components of metabolic pathways.
m6A in glucose uptake and glycolysis
Aerobic glycolysis in cancer is activated under hypoxic conditions. Hypoxia could broadly increase the m6A of polyA + mRNA of certain genes that are closely associated with glycolysis (e.g., GLUT1 and MYC) [90, 91]. MYC induce the expression of glucose transporters and most glycolytic enzymes and can prominently drive aerobic glycolysis [92, 93]. The methyltransferase METTL3 upregulates MYC expression at multiple levels; for instance, METTL3 upregulates MYC mRNA stability by addition of methylation mainly around the stop codon and 3′-UTR [94], MYC mRNA elongation through AFF4 [95], and MYC transcription. Chen et al. found that m6A modification was directly correlated with activating the glycolytic pathway in colorectal cancer. Mechanistically, METTL3 increased the stability of HK2 and GLUT1 mRNA transcripts due to the m6A recognition of IGF2BP2/3. The METTL3-HK2/GLUT1-IGF2BP axis plays a critical role in the pathogenesis of colorectal cancer [17]. Moreover, FTO was shown to trigger the m6A demethylation of PKM2 mRNA and accelerated translation, leading to tumorigenesis of hepatocellular carcinoma [96].
In addition, m6A has broad effects on glycolysis-associated signaling pathways. A RIP assay with an antibody against m6A showed that the overexpression of PIK3CB containing the rs142933486-T allele (PIK3CB[T]) decreased the m6A level of PIK3CB compared with that of PIK3CB[G], indicating that the missense variant rs142933486 G > T in PIK3CB reduced the m6A level. Mechanistically, the variant is located 3 bp from a predicted m6A site. m6A is enriched at the consensus motif of DRACH; herein, D corresponds to A, G and U. Therefore, the G > T base change may disrupt the recognition by the m6A ‘writers’ complex (METTL3-METTL14-WTAP) and ‘erasers’, reducing m6A levels. However, m6A-methylated PIK3CB is recognized by YTHDF2, substantially decreasing the mRNA and protein expression by influencing its mRNA stability. The rs142933486 G > T in PIK3CB in turn enhances PIK3CB expression [97]. PIK3CB further activates the AKT pathway, whose downstream transcription factors can mediate glycolytic enzymes [98, 99]. Moreover, m6A methylation normally attenuates AKT activity in the endometrium by promoting the m6A-dependent translation of PHLPP2 and m6A-dependent degradation of transcripts encoding subunits of mTORC2, increasing the proliferation and tumorigenicity of endometrial cancer cells [14]. Other reported studies in multiple tumor types have also confirmed the association between m6A and AKT signaling, including that in leukemia cells [100] and clear cell renal cell carcinoma [101]. Notably, YTHDF2 functioned as a tumor suppressor in HCC by negatively modulating EGFR mRNA stability via its binding to the m6A site in the 3′-UTR of EGFR mRNA, in turn impairing the MEK/ERK pathway and consequently impeding the cell proliferation and growth [102]. METTL3 elimination inhibited the proteasome-mediated IκBα degradation and p65 phosphorylation, thereby restraining NF-κB nuclear translocation and leading to its transcriptional repression. Mechanistically, METTL3 installed m6A methylation on MYD88 mRNA to positively regulate MYD88 expression, allowing in the activation of NF-κB signaling [103]. METTL3 also activated NF-κB signaling by promoting the expression of IKBKB and RELA by regulating translational efficiency [94]. As we described above, the “writers”, “erasers” or “readers” induce m6A fluctuation of various mRNAs, indicating critical roles in glucose metabolism via glycolytic enzymes or associated signaling pathways.
m6A in lipid metabolism
Increased de novo fatty acid synthesis and alternation of fatty acid uptake and catabolism elevate the rate of lipogenesis, allowing tumor cells to maintain their high proliferative rate. Lipid metabolic reprogramming allows cancers to adjust the metabolic demands toward the synthesis of macromolecules, the main lipids for the biogenesis of membranes and various signaling incentives to support tumorigenesis [104, 105]. ACC1, ACLY, DGAT2, EHHADH, FASN, FOXO, PGC1A, and SIRT1 are critical for the regulation of fatty acid synthesis and oxidation; however, they were dramatically decreased in the livers of mice with hepatocyte-specific METTL3 knockout. Additionally, the levels of two important regulators of cholesterol metabolism, CD36 and LDLR, were also downregulated in these mice due to the improvement in expression of FASN [106]. One recent study suggested that METTL3-mediated m6A modification led to LINC00958 upregulation by stabilizing its RNA transcript, which subsequently activated the miR-3619-5p/HDGF axis to facilitate lipogenesis in HCC. Key enzymes in lipogenesis, including SREBP1, FASN, SCD1, and ACC1, were also affected by LINC00958. These results delineated the m6A-involved regulatory mechanisms in lipogenesis of HCC [107]. Chen et al. showed that FTO increased lipid accumulation by a novel FTO/SREBP1c/CIDEC signaling pathway in an m6A-dependent manner in HepG2 cells and provided insight into the molecular mechanism of FTO in hepatic lipogenesis [108]. YTHDF2 could also bind to the mRNA of lipogenic genes, including SREBP1c, FASN, SCD1, and ACC1, to decrease their mRNA stability and inhibit gene expression [109].
In addition, m6A is closely associated with several signaling pathways to regulate lipid metabolism. m6A modification promoted the translation of protein phosphatase 1A, magnesium-dependent, alpha isoform (PPM1A), a negative AMPK regulator, but decreased the expression of calcium/calmodulin-dependent protein kinase kinase 2 (CAMKK2), a positive AMPK regulator, by reducing its RNA stability [110]. Thus, m6A modification resulted in reduced AMPK activity. AMPK affected PARK2 mRNA stability in a YTHDF2-dependent manner through FTO-dependent demethylation of m6A. Furthermore, m6A was highly enriched in FAM225A and enhanced its RNA stability [111]. In summary, m6A plays an important role in the lipid metabolic reprogramming of cancer.
m6A in glutamine metabolism
Glutamine, as a source of carbon and nitrogen for biomass accumulation, participates in biosynthesis, energetics, and cellular homeostasis, reinforcing tumor growth and vitality [112]. Glutamine can be converted into α-KG to replenish the TCA cycle through two mechanisms: glutamate dehydrogenase (GLUD1) or transaminases [113]. FTO and ALKBH5 are α-KG-dependent dioxygenases and competitively inhibited by the structurally related metabolite D-2-hydorxyglutarate (D2-HG), which aberrantly accumulated in isocitrate dehydrogenase 1 or 2 (IDH1/2)-mutant tumors [114, 115]. Therefore, the effects of m6A on cancer pathogenesis need to be interpreted in the context of glutamine metabolism, but related studies are limited. Interactions between m6A and glutamine catalytic enzymes and signaling pathways remain to be explored.
Conclusion and perspectives
Studies in the past few decades have shown that aberrant distribution and abundances of m6A drive tumorigenesis, at least in part through the control of cell metabolism [17, 96, 97]. Future studies aimed at refining our molecular map of the crucial regulatory nodes connecting m6A to the metabolic networks in different cancers will help reveal metabolic dependencies and novel therapeutic strategies. Due to the rapid development of technology methods for m6A, researchers have productively discovered many mechanisms of how m6A in cancer metabolism, including aerobic glycolysis, disordered lipid metabolism and glutamine-dependent anaplerosis. However, many challenges remain. The function of m6A in various cancers are still controversial. These functions are characterized by fluctuating distribution of m6A on different regions of mRNAs and subcellular readers responding to target genes that participate in different cellular processes. Moreover, with regard to the same target, the acceleration of m6A in the target could result in altered RNA splicing and increased translational capability, leading to upregulated mRNA. However, methylation at other loci may decrease the mRNA level because of enhanced m6A-dependent degradation. Given the extensive crosstalk among metabolic networks, it is particularly important to maintain homeostasis among various metabolic processes. Although there have been breakthroughs in the studies of glutamine metabolism in tumorigenesis and progression, the role of m6A may be less substantial.
In conclusion, elucidation of the molecular mechanisms underlying m6A in RNAs and its effects on cancer metabolic reprogramming could provide a better understanding of the epigenetics and abnormal metabolic characteristics of cancers. Additionally, these results may help predict cancer risk, achieve early diagnosis, track the prognosis of tumors fate, and ultimately provide novel therapeutic approaches.
Availability of data and materials
Not applicable.
References
Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci USA. 1974;71(10):3971–5.
Perry RP, Kelley DE. Existence of methylated messenger RNA in mouse L cells. Cell. 1974;1(1):37–42.
Wang Y, et al. N6-methyladenosine RNA modification regulates embryonic neural stem cell self-renewal through histone modifications. Nat Neurosci. 2018;21(2):195–206.
Shay G, et al. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science. 2015;347(6225):1002–6.
Jun Z, et al. Dynamic m6A mRNA methylation directs translational control of heat shock response. Nature. 2015;526(7574):591–4.
Xiang Y, et al. RNA m6A methylation regulates the ultraviolet-induced DNA damage response. Nature. 2017;543(7646):573–6.
Church C, et al. Overexpression of Fto leads to increased food intake and results in obesity. Nat Genet. 2010;42(12):1086–92.
Yang Y, et al. Glucose is involved in the dynamic regulation of m6A in patients with type 2 diabetes. J Clin Endocrinol Metab. 2019;104(3):665–73.
Yang Y, et al. Increased N6-methyladenosine in Human Sperm RNA as a Risk Factor for Asthenozoospermia. Scientific reports. 2016;6:24345–24345.
Lin X, et al. RNA m(6)A methylation regulates the epithelial mesenchymal transition of cancer cells and translation of Snail. Nature communications. 2019;10(1):2065–2065.
Li J, et al. N6-methyladenosine regulates the expression and secretion of TGFβ1 to affect the epithelial-mesenchymal transition of cancer cells. Cells. 2020;9(2):296.
Yue B, et al. METTL3-mediated N6-methyladenosine modification is critical for epithelial-mesenchymal transition and metastasis of gastric cancer. Molecular cancer. 2019;18(1):142–142.
Lin S, et al. The m6A methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell. 2016;62(3):335–45.
Liu J, et al. m6A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat Cell Biol. 2018;20(9):1074–83.
Chen J, et al. m6A regulates neurogenesis and neuronal development by modulating histone methyltransferase Ezh2. Genomics, Proteomics Bioinformatics. 2019;17(2):154–68.
Ma J-Z, et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N6-methyladenosine-dependent primary MicroRNA processing. Hepatology. 2017;65(2):529–43.
Shen C, et al. m6A-dependent glycolysis enhances colorectal cancer progression. Mol Cancer. 2020;19(1):72–72.
Jia G, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885–7.
Fu Y, et al. The AlkB domain of mammalian ABH8 catalyzes hydroxylation of 5-methoxycarbonylmethyluridine at the wobble position of tRNA. Angew Chem Int Ed Engl. 2010;49(47):8885–8.
Mauer J, et al. Reversible methylation of m(6)A(m) in the 5' cap controls mRNA stability. Nature. 2017;541(7637):371–5.
Ke S, et al. m(6)A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 2017;31(10):990–1006.
Darnell RB, Ke S, Darnell JE Jr. Pre-mRNA processing includes N(6) methylation of adenosine residues that are retained in mRNA exons and the fallacy of "RNA epigenetics". RNA. 2018;24(3):262–7.
Dominissini D, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–6.
Linder B, et al. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 2015;12(8):767–72.
Chen S, et al. WTAP promotes osteosarcoma tumorigenesis by repressing HMBOX1 expression in an m(6)A-dependent manner. Cell Death Dis. 2020;11(8):659.
Li Z, et al. N(6)-methyladenosine regulates glycolysis of cancer cells through PDK4. Nat Commun. 2020;11(1):2578.
Bedi RK, et al. Small-molecule inhibitors of METTL3, the major human epitranscriptomic writer. ChemMedChem. 2020;15(9):744–8.
Huang J, et al. Solution structure of the RNA recognition domain of METTL3-METTL14 N(6)-methyladenosine methyltransferase. Protein Cell. 2019;10(4):272–84.
Wang P, Doxtader KA, Nam Y. Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases. Mol Cell. 2016;63(2):306–17.
Fu M, Blackshear PJ. RNA-binding proteins in immune regulation: a focus on CCCH zinc finger proteins. Nat Rev Immunol. 2017;17(2):130–43.
Park S, et al. Structural basis for interaction of the tandem zinc finger domains of human muscleblind with cognate RNA from human cardiac troponin T. Biochemistry. 2017;56(32):4154–68.
Chen Y, et al. WTAP facilitates progression of hepatocellular carcinoma via m6A-HuR-dependent epigenetic silencing of ETS1. Molecular cancer. 2019;18(1):127–127.
Schwartz S, et al. Perturbation of m6A writers reveals two distinct classes of mRNA Methylation at internal and 5′sites. Cell Rep. 2014;8(1):284–96.
Ping XL, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24(2):177–89.
Bokar JA, et al. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA. 1997;3(11):1233–47.
Niu Y, et al. RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol Cancer. 2019;18(1):46.
Xu C, et al. Structures of human ALKBH5 demethylase reveal a unique binding mode for specific single-stranded N6-methyladenosine RNA demethylation. J Biol Chem. 2014;289(25):17299–311.
Zhang X, et al. Structural insights into FTO's catalytic mechanism for the demethylation of multiple RNA substrates. Proc Natl Acad Sci U S A. 2019;116(8):2919–24.
Gao Y, et al. Multivalent m(6)A motifs promote phase separation of YTHDF proteins. Cell Res. 2019;29(9):767–9.
Shi H, et al. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 2017;27(3):315–28.
Wojtas MN, et al. Regulation of m(6)A transcripts by the 3'→5' RNA helicase YTHDC2 is essential for a successful meiotic program in the mammalian germline. Mol Cell. 2017;68(2):374–387.e12.
Alarcón CR, et al. HNRNPA2B1 Is a mediator of m(6)A-dependent nuclear RNA processing events. Cell. 2015;162(6):1299–308.
Huang H, et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20(3):285–95.
Meyer KD, et al. 5' UTR m6A promotes cap-independent translation. Cell. 2015;163(4):999–1010.
Wu R, et al. A novel m(6)A reader Prrc2a controls oligodendroglial specification and myelination. Cell Res. 2019;29(1):23–41.
Liu N, et al. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518(7540):560–4.
Geissler R, et al. A widespread sequence-specific mRNA decay pathway mediated by hnRNPs A1 and A2/B1. Genes Dev. 2016;30(9):1070–85.
Liu N, et al. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017;45(10):6051–63.
Zhao X, et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014;24(12):1403–19.
Zheng G, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49(1):18–29.
Nilsen TW. Internal mRNA methylation finally finds functions. Science. 2014;343(6176):1207–8.
Nowak DG, et al. Regulation of vascular endothelial growth factor (VEGF) splicing from pro-angiogenic to anti-angiogenic isoforms: a novel therapeutic strategy for angiogenesis. J Biol Chem. 2010;285(8):5532–40.
Xiao W, et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol Cell. 2016;61(4):507–19.
Spitale RC, et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature. 2015;527(7577):486–90.
Yue W, et al. Landscape and variation of RNA secondary structure across the human transcriptome. Nature. 2014;505(7485):706–9.
Schwartz S, et al. High-resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis. Cell. 2013;155(6):1409–21.
Zhou KI, et al. N 6 -methyladenosine modification in a long noncoding RNA hairpin predisposes its conformation to protein binding. J Mol Biol. 2016;428(5):822–33.
Sokhi UK, et al. Chapter five - human polynucleotide phosphorylase (hPNPaseold-35): should I eat you or not—that is the question? In: Tew KD, Fisher PB, editors. Advances in cancer research. Cambridge: Academic Press; 2013. p. 161–190.
Moraes KCM, Wilusz CJ, Wilusz J. CUG-BP binds to RNA substrates and recruits PARN deadenylase. RNA. 2006;12(6):1084–91.
Slobodin B, et al. Transcription dynamics regulate poly(A) tails and expression of the RNA degradation machinery to balance mRNA levels. Mol Cell. 2020;78(3):434–444.e5.
Shoemaker CJ, Green R. Translation drives mRNA quality control. Nat Struct Mol Biol. 2012;19(6):594–601.
Du H, et al. YTHDF2 destabilizes m6A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat Commun. 2016;7:12626–12626.
Xiao W, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505(7481):117–20.
Zaccara S, Jaffrey SR. A unified model for the function of YTHDF proteins in regulating m6A-modified mRNA. Cell. 2020;181(7):1582–1595.e18.
Li F, et al. N6-methyladenosine modulates nonsense-mediated mRNA decay in human glioblastoma. Cancer Res. 2019;79(22):5785–98.
Visvanathan A, et al. Essential role of METTL3-mediated m6A modification in glioma stem-like cells maintenance and radioresistance. Oncogene. 2018;37(4):522–33.
Wang Y, et al. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol. 2014;16(2):191–8.
Jean-Michel F, et al. RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell. 2013;155(4):793–806.
Edens BM, et al. FMRP modulates neural differentiation through m6A-dependent mRNA nuclear export. Cell Rep. 2019;28(4):845–854.e5.
Wang X, et al. N6-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161(6):1388–99.
Choi J, et al. N6-methyladenosine in mRNA disrupts tRNA selection and translation-elongation dynamics. Nat Struct Mol Biol. 2016;23(2):110–5.
Natchiar SK, et al. Visualization of chemical modifications in the human 80S ribosome structure. Nature. 2017;551(7681):472–7.
Ren W, et al. Structure and regulation of ZCCHC4 in m6A-methylation of 28S rRNA. Nat Commun. 2019;10(1):5042–50.
Ma H, et al. N(6-)Methyladenosine methyltransferase ZCCHC4 mediates ribosomal RNA methylation. Nat Chem Biol. 2019;15(1):88–94.
Meyer KD, et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell. 2012;149(7):1635–46.
Chen K, et al. High-resolution N6-methyladenosine (m6A) map using photo-crosslinking-assisted m6A sequencing. Angew Chem. 2015;54(5):1587–90.
Hafner M, et al. Transcriptome-wide identification of RNA-binding protein and MicroRNA target sites by PAR-CLIP. Cell. 2010;141(1):129–41.
Ke S, et al. A majority of m6A residues are in the last exons, allowing the potential for 3' UTR regulation. Genes Dev. 2015;29(19):2037–53.
Koh CWQ, Goh YT, Goh WSS. Atlas of quantitative single-base-resolution N(6)-methyl-adenine methylomes. Nat Commun. 2019;10(1):5636.
Nian L, et al. Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA. 2013;19(12):1848–56.
Liu W, et al. Identification of a selective DNA ligase for accurate recognition and ultrasensitive quantification of N6-methyladenosine in RNA at one-nucleotide resolution. Chem Sci. 2018;9(13):3354–9.
Zhang Z, et al. Single-base mapping of m6A by an antibody-independent method. Sci Adv. 2019;5(7):eaax0250.
Garcia-Campos MA, et al. Deciphering the "m(6)A Code" via Antibody-Independent Quantitative Profiling. Cell. 2019;178(3):731–747.e16.
Imanishi M, et al. Detection of N6-methyladenosine based on the methyl-sensitivity of MazF RNA endonuclease. Chem Commun. 2017;53(96):12930–3.
Hanahan D, Robert A. Weinberg, hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Gupta A, et al. PAK2-c-Myc-PKM2 axis plays an essential role in head and neck oncogenesis via regulating Warburg effect. Cell Death Dis. 2018;9(8):825–39.
Liu Q, et al. Targeting lipid metabolism of cancer cells: a promising therapeutic strategy for cancer. Cancer Lett. 2017;401:39–45.
Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16(10):619–34.
DeBerardinis RJ, et al. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7(1):11–20.
Fry NJ, et al. N6-methyladenosine is required for the hypoxic stabilization of specific mRNAs. RNA. 2017;23(9):1444–555.
Priolo C, et al. AKT1 and MYC induce distinctive metabolic fingerprints in human prostate cancer. Can Res. 2014;74(24):7198–204.
Hu S, et al. 13C-pyruvate imaging reveals alterations in glycolysis that precede c-Myc-induced tumor formation and regression. Cell Metab. 2011;14(1):131–42.
David CJ, et al. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010;463(7279):364–8.
Cheng M, et al. The m6A methyltransferase METTL3 promotes bladder cancer progression via AFF4/NF-κB/MYC signaling network. Oncogene. 2019;38(19):3667–800.
Luo Z, et al. The super elongation complex family of RNA polymerase II elongation factors: gene target specificity and transcriptional output. Mol Cell Biol. 2012;32(13):2608–17.
Li J, et al. m6A demethylase FTO promotes hepatocellular carcinoma tumorigenesis via mediating PKM2 demethylation. Am J Transl Res. 2019;11(9):6084–92.
Tian J, et al. N6-methyladenosine mRNA methylation of PIK3CB regulates AKT signalling to promote PTEN-deficient pancreatic cancer progression. Gut. 2020. https://doi.org/10.1136/gutjnl-2019-320179.
Chae YC, et al. Mitochondrial akt regulation of hypoxic tumor reprogramming. Cancer Cell. 2016;30(2):257–72.
Düvel K, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39(2):171–83.
Vu LP, et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. 2017;23(11):1369–76.
Zhou J, et al. Gene signatures and prognostic values of m6A regulators in clear cell renal cell carcinoma—a retrospective study using TCGA database. Aging. 2019;11(6):1633–47.
Zhong L, et al. YTHDF2 suppresses cell proliferation and growth via destabilizing the EGFR mRNA in hepatocellular carcinoma. Cancer Lett. 2019;442:252–61.
Yu J, et al. The m6A methyltransferase METTL3 cooperates with demethylase ALKBH5 to regulate osteogenic differentiation through NF-κB signaling. Mol Cell Biochem. 2020;463(1):203–10.
Ray U, Roy SS. Aberrant lipid metabolism in cancer cells – the role of oncolipid-activated signaling. FEBS J. 2018;285(3):432–43.
Alwarawrah Y, et al. Fasnall, a selective FASN inhibitor, shows potent anti-tumor activity in the MMTV-Neu model of HER2(+) breast cancer. Cell Chem Biol. 2016;23(6):678–88.
Xie W, et al. METTL3 inhibits hepatic insulin sensitivity via N6-methyladenosine modification of Fasn mRNA and promoting fatty acid metabolism. Biochem Biophys Res Commun. 2019;518(1):120–6.
Zuo X, et al. M6A-mediated upregulation of LINC00958 increases lipogenesis and acts as a nanotherapeutic target in hepatocellular carcinoma. J Hemato Oncol. 2020;13(1):5.
Chen A, et al. FTO promotes SREBP1c maturation and enhances CIDEC transcription during lipid accumulation in HepG2 cells. Biochim Biophys Acta. 2018;1863(5):538–48.
Zhou B, et al. N6-methyladenosine reader protein Ythdc2 suppresses liver steatosis via regulation of mRNA stability of lipogenic genes. Hepatology. 2020. https://doi.org/10.1002/hep.31220.
Chen Y, et al. m6A mRNA methylation regulates testosterone synthesis through modulating autophagy in Leydig cells. Autophagy. 2020. https://doi.org/10.1080/15548627.2020.1720431.
Zheng Z-Q, et al. Long noncoding RNA FAM225A promotes nasopharyngeal carcinoma tumorigenesis and metastasis by acting as ceRNA to sponge miR-590-3p/miR-1275 and upregulate ITGB3. Can Res. 2019;79(18):4612–26.
Huang W, et al. A proposed role for glutamine in cancer cell growth through acid resistance. Cell Res. 2013;23(5):724–7.
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–33.
Elkashef SM, et al. IDH mutation, competitive inhibition of FTO, and RNA methylation. Cancer Cell. 2017;31(5):619–20.
Su R, et al. R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell. 2018;172(1–2):90–105.e23.
Liu J, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10(2):93–5.
Vu LP, et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. 2017;23(11):1369–76.
Acknowledgements
We thank the teachers of Center of Clinical Laboratory of the First Affiliated Hospital of Soochow University.
Funding
The study was supported by the grants from National Natural Science Foundation of China (no. 81501425), the Science and technology project of Suzhou (no. SLT201921), and the Science and technology project of Changzhou (no. QN201926), and Project of Changzhou No.2 People's Hospital (no. 2019K007).
Author information
Authors and Affiliations
Contributions
XH designed the research and wrote the paper. XH searched and read the literature. LW and QH provided essential suggestion and revision. QH had primary responsibility for final content. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
No potential conflicts of interest were disclosed.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Han, X., Wang, L. & Han, Q. Advances in the role of m6A RNA modification in cancer metabolic reprogramming. Cell Biosci 10, 117 (2020). https://doi.org/10.1186/s13578-020-00479-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-020-00479-z