
Abstract
Deubiquitinases (DUBs) are enzymes that control the stability, interactions or localization of most cellular proteins by removing their ubiquitin modification. In recent years, some DUBs, such as USP7, USP9X and USP10, have been identified as promising therapeutic targets in hematological malignancies. Importantly, some potent inhibitors targeting the oncogenic DUBs have been developed, showing promising inhibitory efficacy in preclinical models, and some have even undergone clinical trials. Different DUBs perform distinct function in diverse hematological malignancies, such as oncogenic, tumor suppressor or context-dependent effects. Therefore, exploring the biological roles of DUBs and their downstream effectors will provide new insights and therapeutic targets for the occurrence and development of hematological malignancies. We summarize the DUBs involved in different categories of hematological malignancies including leukemia, multiple myeloma and lymphoma. We also present the recent development of DUB inhibitors and their applications in hematological malignancies. Together, we demonstrate DUBs as potential therapeutic drug targets in hematological malignancies.
Similar content being viewed by others

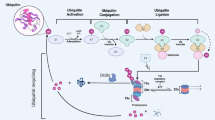
Background
The ubiquitin-proteasome system (UPS) regulates many cellular functions, including proteasomal degradation, selective autophagy, cell signaling, receptor trafficking and endocytosis, DNA damage response, cell cycle, cell survival, cell proliferation and cell death et al. [1,2,3]. The dysfunction or dysregulation of UPS is related to a series of pathologies and diseases, including hematological malignancies [4]. Inhibiting the activity of components of the UPS by bortezomib or carfilzomib has been shown as a therapeutic strategy for the treatment of multiple myeloma (MM) and mantle-cell lymphoma (MCL) [5]. Indeed, the essential components of the UPS consist of ubiquitinases, deubiquitinases and 26S proteasome [6]. Until now, many E3 ubiquitin ligases have been implicated in the pathogenesis of hematological malignancies [7,8,9,10]. Reversely, deubiquitinases (DUBs), contains ~ 100 family proteins, can remove the ubiquitin chains from the substrates and thereby control the stability, interactions or localization of the substrates [11, 12]. DUBs are classified in to six families according to their domain structure: the ubiquitin-specific proteases (USPs, 58 members), the ubiquitin carboxy-terminal hydrolases (UCHs, 4 members), the ovarian tumor-related proteases (OTUs, 16 members), the Machado-Joseph disease protein domain proteases (MJDs, 4 members), the JAB1/PAB1/MPN-domain containing metallo-enzyme (JAMMs, 12 members), and the motif interacting with Ub-containing novel DUB family (MINDYs, 4 members) (Fig. 1) [12]. Therefore, more and more evidences indicate that dysregulation of DUBs plays an important role in the pathogenesis of hematological malignancies [13, 14]. Also, some DUBs are important in the process of differentiation from hematopoietic stem cells (HSCs) to all blood-cell lineages, and the related disorders are associated with hematological malignancies [15,16,17,18]. However, most of DUBs and their roles in the progression of hematological malignancies have not been explored broadly. In this review, we will focus on the function of DUBs in different types of hematological malignancies and the development of targeted small molecule inhibitors.

Human DUBs. Human DUBs are composed of cysteine proteases and metalo proteases. According to their domain structure, DUBs are classified in to six families: the ubiquitin-specific proteases (USPs), the ubiquitin carboxy-terminal hydrolases (UCHs), the ovarian tumor-related proteases (OTUs), the Machado-Joseph disease protein domain proteases (MJDs), the JAB1/PAB1/MPN-domain containing metallo-enzyme (JAMMs), and the motif interacting with Ub-containing novel DUB family (MINDYs)
DUBs in chronic myeloid leukemia
Chronic myeloid leukemia (CML) is a myeloproliferative neoplasm that accounts for approximately 15% of newly diagnosed cases of leukemia in adults [19]. BCR-ABL is responsible for 95% of all diagnosed CML cases, which acts as a constitutively active protein tyrosine kinase [20]. Delightfully, most of the CML patients can achieve complete remission after receiving tyrosine kinase inhibitors (TKIs) treatment, and some patients even discontinue the TKIs treatment to achieve treatment-free remission (TFR) [21, 22]. However, drug resistance and relapse restrict the application of TKIs in treating CML through BCR-ABL-dependent and -independent mechanism [23]. The BCR-ABL-dependent mechanism is mediated through BCR-ABL gene amplification, mutation of the ABL kinase domain and MDR1 upregulation [24]. The BCR-ABL-independent mechanism has still not been well understood until now. Leukemia stem cells (LSCs) in CML are not dependent on BCR-ABL kinase activity for their survival and are insensitive to TKIs in a BCR-ABL-independent manner, therefore leading to relapse [25]. In addition, the aberrant activation of RAS/MAPK or PI3K signaling pathways also contributes to BCR-ABL-independent TKI resistance [26, 27]. Some DUBs have been identified to be associated with BCR-ABL signaling pathway and others could overcome BCR-ABL-dependent or -independent TKI resistance. The DUBs related to CML are shown in Fig. 2.

DUBs in chronic myeloid leukemia. The red arrows represent oncogenes and the green arrows represent tumor suppressors in CML. The surrounding text describes the substrates or signaling pathways or effects of DUBs
USP7 is involved in DNA damage response, DNA replication, epigenetics and viral infections [28]. More importantly, USP7 has been identified as a potential therapeutic target for a variety of cancers due to its role in tumorigenesis [29]. Several small molecule specific inhibitors of USP7 have been developed [30, 31]. In CML, USP7 physically interacts with BCR-ABL and is phosphorylated (Tyr243) by BCR-ABL. Phosphorylated USP7 gains increased deubiquitinating activity, thus promoting the nuclear exclusion of phosphatase and tensin homolog (PTEN), which disrupts PTEN’s tumor suppressive function in CML cells [32, 33]. It is also demonstrated that USP7 interacts with BCR-ABL and blocks its polyubiquitination and degradation, thereby inhibiting its downstream signaling transduction [34]. Furthermore, USP7 is expressed mostly in the nucleus of normal CD34+ cells, but in primary CML CD34+ cells, it is expressed both in the nuclear bodies and cytoplasm [35]. Together, USP7 binds to BCR-ABL in the cytosol and regulates PTEN de-ubiquitination via a PML network in the nuclear bodies.
USP9X acts as an oncogene or tumor suppressor in human cancers, and it also plays a role in the context of normal development and the biological consequences of the disease [36]. USP9X inhibition by the small molecule WP1130 induces BCR-ABL ubiquitination and trafficking, leading to apoptosis in both imatinib-sensitive and -resistant CML cells. Although WP1130 inhibits USP9X activity, USP9X isn’t involved in the increased ubiquitination of BCR-ABL. USP9X silencing in CML cells decreases anti-apoptotic protein MCL-1, which increases sensitivity to imatinib and other apoptotic stimuli [37, 38].
USP10 is a deubiquitinase of S-phase kinase-associated protein 2 (SKP2), which interacts with BCR-ABL and is required for the activation of BCR-ABL in CML. SKP2, an E3 ligase, enhances the activity of BCR-ABL by promoting the K63-linked ubiquitination of BCR-ABL [39]. Although USP10 deubiquitinates and stabilizes SKP2, it cannot directly reduce the ubiquitination of BCR-ABL. Together, USP10 inhibition suppresses the proliferation of imatinib-sensitive and -resistant CML cells.
USP15 is involved in various cellular pathways associated with cancer and other related diseases [40]. USP15 is significantly down-regulated in CML, and its inhibition decreases de-ubiquitination of caspase-6 and promotes the degradation of caspase-6, which attenuates CML cell apoptosis and contributes to imatinib resistance [41].
USP18 (UBP43) is an ISG15-specific isopeptidase, the expression of which is activated by interferon (IFN) [42]. Usp18-deficient bone marrow cells show significant delay of CML development in BCR-ABL retroviral transduction/transplantation assay compared with wild-type ones. However, Usp18 and IFN receptor R1 (Ifnar1) double deficient bone marrow cells with p210 BCR-ABL transduction reverse the original resistance to CML disease development, which indicates the important role of type 1 IFN signaling in the resistance to CML development in Usp18-deficient bone marrow cells [43].
USP25 plays an important role in the regulation of innate immune response, autoimmunity and tumorigenesis by interacting with different tumor necrosis factor (TNF) receptor-related factor (TRAF) proteins [44]. BCR-ABL degradation has been considered as a strategy to overcome TKIs resistance, so inhibiting the deubiquitinases of BCR-ABL may be effective. USP25 deubiquitinates and suppresses the degradation of BCR-ABL, and its depletion inhibits BCR-ABL-mediated signaling and cell proliferation [45].
USP47 is involved in cell survival [46], cell proliferation [47, 48], DNA damage repair [23, 49, 50], epithelial-mesenchymal transition [51], and inflammation [52, 53]. It is demonstrated that Usp47 knockout significantly prolongs the survival of BCR-ABL and BCR-ABLT315I-induced CML mice by reducing leukemia stem/progenitor cells. Mechanism studies have shown that USP47 deubiquitinates and stabilizes Y-box binding protein 1(YB-1) and participates in DNA damage repair in CML cells [23]. It is indicated that USP47 is required for BCR-ABL-induced CML and can overcome BCR-ABL-independent drug resistance.
A20 expression is significantly downregulated in CML CD34+ cells compared to normal bone marrow CD34+ cells. Overexpression of A20 inhibits cell proliferation, cell cycle, and promotes apoptosis in CML CD34+ cells. Furthermore, A20 overexpression significantly reduces the NF-κB signal pathways (P65 and IκBα phosphorylation) in K562 and CML CD34+ cells [54].
BAP1, down-regulated in CML at the transcriptional level, is a deubiquitinase interacting with the DNA repair regulator BRCA1 [55]. It is demonstrated that BAP1 is a major link with the BCR-ABL-induced downregulation of BRCA1 in CML.
Taken together, targeting some deubiquitinases, such as USP7, USP9X, USP10, USP15, USP25 and USP47, can effectively overcome TKIs resistance in CML, and some deubiquitinases play an important role in maintaining the self-renewal ability of CML stem/progenitor cells. For patients with TKIs intolerance or relapse, the use of deubiquitinase inhibitors alone or in combination with TKIs may be effective in solving clinical problems.
DUBs in acute myeloid leukemia
Acute myeloid leukemia (AML) is a disease characterized by the clonal proliferation of primitive hematopoietic stem or progenitor cells. The abnormal differentiation of bone marrow cells leads to an increase in the number of immature malignant cells and a decrease in the number of differentiated red blood cells, platelets and white blood cells [56]. Although some new drugs are applied in the treatment of AML patients, the standard therapy for AML patients is still chemotherapy. However, some patients are intolerable for such treatment, especially elderly patients, while the effect seems limited [57,58,59]. Indeed, most patients receiving chemotherapy experience relapse, which is caused by chemo-resistant leukemia cells (RLCs) regrowth [60]. In addition, leukemia stem cells (LSCs) and drug resistance (FLT3 inhibitors, BCL2 inhibitors etc.) are still difficult problems for the treatment of AML [61,62,63]. Further research on the molecular mechanism of pathogenesis will contribute to the therapeutic effect in AML. The DUBs related to AML are shown in Fig. 3.

DUBs in acute myeloid leukemia. The red arrows represent oncogenes and the green arrows represent tumor suppressors in AML. USP22 acts as an oncogene or tumor suppressor in AML, depending on the context. The surrounding text describes the substrates or related proteins of DUBs in AML
USP1 is frequently overexpressed in several cancer tissue types [64]. In AML cells, USP1 deubiquitinates inhibitor of DNA binding 1 (ID1) and rescues it from proteasome degradation, which plays a role in cellular transformation. USP1 inhibitor, SJB2–043, promotes ID1 degradation and inhibits primary AML cell growth by disrupting the homologous recombination [65].
USP2 is identified as a fusion partner of MLL (also known as KMT2A) in infant acute myeloid leukemia by using a whole transcriptome sequencing analysis [66]. Another study shows that acute leukemia patients are recurrently associated with MLL-USP2 fusion alleles as well as MLL fusion partnerships with USP8, AF4, and AF9 [67, 68]. MLL-USP2 fusion protein can potentially contribute to MLL-r leukemogenesis through three mechanisms: (a) USP2 stabilizes MLL to protect it from ubiquitin-proteasome degradation, (b) dominant negative inhibition of wild-type MLL transcription, and (c) regulate cell cycle, cell proliferation or other cell function by USP2.
USP7, which is known as a PTEN deubiquitinating enzyme, directly interacts with nucleophosmin (NPM1) /mutated nucleophosmin (NPMc+) in AML. NPM1 prevents USP7-mediated deubiquitination of PTEN in the nucleus, which promotes the shuttle of PTEN to the cytoplasm. PTEN is kept in the cytoplasm by USP7 deubiquitination activity, which is regulated by NPMc+ [69]. Furthermore, USP7 inhibition sensitizes AML to chemotherapy by interacting with and modulating CHK1 protein [70]. Also, USP7 deubiquitinases the non-canonical PRC1.1 polycomb complex to target loci and function in gene regulation in AML cells [71]. Together, USP7 is a promising therapeutic target in AML.
USP9X inhibits the K63-linked poly-ubiquitination of FLT3-ITD, while FLT3-ITD reversely phosphorylates USP9X to enhance ubiquitination and proteasomal degradation. Inhibition of USP9X by its inhibitor WP1130 or G9 shows potent anti-leukemia effects in FLT3-ITD-driven cells by blocking downstream signaling events of FLT3 [72].
USP10 is identified as a critical DUB to deubiquinate and stabilize FLT3-ITD more than wild-type FLT3 in AML. USP10 inhibition by HBX19818 shows anti-leukemia effect in FLT3-ITD positive AML cells and mouse models. It works synergistically with FLT3 inhibitors and overcomes FLT3 inhibitor resistance [73]. Another USP10 inhibitor, Wu-5, also shows anti-AML effect and overcomes FLT3 inhibitor resistance and synergistically enhances the anti-leukemia effect of crenolanib through targeting both FLT3 and AMPKα pathway [74]. Spleen tyrosine kinase (SYK), stabilized by USP10, is critical for AML transformation and maintenance of the leukemia clone in AML patients. Highly activated SYK is found in FLT3-ITD positive AML, which facilitates Myc transcriptional programs and is critical for TKI resistance [75, 76]. MLL-USP10 fusion has been identified in an adolescent case of relapsed AML-M5a with t (11,16) (q23;q24) [77]. Until now, USP2, USP8, and USP10 have been found to be fused with MLL, which indicates that DUBs may be potential targets of MLL-r leukemia.
The inhibitor of USP14 and UCHL5, b-AP15, inhibits organ infiltration in an AML mouse model [78]. Another compound NiPT, which potently inhibits USP14 and UCHL5 activity, induces cytotoxicity and proteasome inhibition in AML cells [79]. Aurora kinase B (Aurora B), a mitotic checkpoint kinase, has been found to be overexpressed in several types of leukemia [80]. USP14 inhibits chemotherapeutic drugs-induced apoptosis in leukemia cells by deubiquitinating Aurora B [81].
USP15 is highly expressed in human leukemia cells, which interacts with and stabilizes FUS in AML cells [15]. FUS, an RNA/DNA-binding protein, has been reported to promote HSC self-renewal [82]. During normal hematopoiesis, USP15 depletion impairs hematopoietic stem and progenitor cells (HSPCs) proliferation in vitro and reconstitution potential in vivo [15]. In MLL-AF9 leukemia, TIFAB deubiquitination regulated by USP15, decreases p53 signaling and correspondingly promotes leukemia cell function and development of leukemia [83].
USP18 was cloned from leukemia fusion protein AML1-ETO-expressing mice, which blocks cytokine-induced terminal differentiation of monocytic cells [84]. It is demonstrated that USP18 stabilizes PML/RARα protein and inhibits cell apoptosis in all-trans-retinoic acid (RA)-sensitive and RA-resistant acute promyelocytic leukemia (APL) cells [85]. However, other researchers found that USP18 could oppose UBE1L-dependent PML/RARα degradation by targeting the PML domain, rather than RA-dependent PML/RARα degradation [86].
USP22 mRNA expression is significantly induced in FLT3-ITD compared to FLT3-WT AML CD34+ cells by c-Myc, which leads to reduced ubiquitination and enhanced stability of SIRT1 [87]. In contrast, USP22 deficiency blocks myeloid differentiation, therefore promoting AML in Ras-driven myeloproliferative neoplasm. Mechanistically, USP22 deubiquitinates PU.1 and promotes its target genes expression [88]. Together, USP22 shows different functions in the process of leukemia transformation or pathology.
USP28 overexpression not only inhibits AML cell proliferation but also sensitizes AML cells to 5′-azacytidine (5′-AZA)-induced apoptosis. USP28 binds to uridine-cytidine kinase 1 (UCK1) via KLHL2 (an ubiquitin E3 ligase) and antagonizes KLHL2-mediated poly-ubiquitination of UCK1, which has an established role in activating 5′-AZA [89].
USP37 interacts with the promyelocytic leukemia zinc finger (PLZF)/the retinoic acid receptor alpha (RARA) fusion proteins and stabilizes its protein levels. Acute promyelocytic leukemia (APL) patients with PLZF/RARA fusion protein are largely resistant to all-trans retinoic acid (ATRA) treatment and with poor prognosis [90].
USP42 (intro 1) was found to be fused with RUNX1 (intron 7) (t (7,21)(p22;q22)/RUNX1-USP42) in AML, but the function of USP42 was unknown [91]. However, later studies showed that exon 6 or exon 7 of RUNX1 was fused to exon 3 of USP42 with aberrant expression of CD56 and CD7 [92, 93]. AML patients with RUNX1-USP42 show poor prognosis [94]. It is suggested that USP42 may play an important role in AML-ETO positive AML patients.
USP48 is involved in ATRA-induced APL cell differentiation. ATRA treatment in APL cell lines causes nucleus translocation in 24 h and deregulation of USP48. USP48 overexpression inhibits APL cells proliferation and promotes ATRA-mediated differentiation [95].
BRCA1/BRCA2-containing complex 3 (BRCC3) is a member of the JAMMs family capable of cleaving Lys-63 linked poly-ubiquitin chain [96]. BRCC3 mutations result in improved proliferation in AML1-ETO positive AML cell lines and unlimited self-renewal in mouse hematopoietic progenitor cells in vitro. However, BRCC3-mutated AML1-ETO positive AML patients show favorable outcome, since BRCC3 inactivation may lead to an impaired capability of the BRCA1-A complex to repair DNA damage and subsequently higher sensitivity to DNA damaging chemotherapy [97]. There may be significant differences in the pathogenesis of AML between mice and humans.
A20 expression was up-regulated in AML derived DCs (AML-DCs), which are differentiated from AML leukemia cells [98]. Also, A20 expression was increased during monocyte-macrophage differentiation (THP-1 cell) [99]. However, the expression of A20 was lower in T cells from patients with AML than those in the healthy controls [100]. Together, A20 acts as a tumor suppressor in AML.
BAP1 depletion inhibits the growth of myeloid leukemia cells with ASXL1 mutations or MLL-fusions. ASXL1 mutations or MLL-fusions are associated with poor prognosis in a variety of myeloid neoplasms. Mechanistically, the C-terminally truncated mutant ASXL1 (ASXL1-MT) forms a complex with BAP1 and induces the up-regulation of HOXA gene and IRF8 by removing H2AK119 ubiquitination [101, 102].
Taken together, some deubiquitinases, such as USP9X, USP10, USP18 and USP37, are directly associated with leukemic fusion proteins. Also, other deubiquitinases, such as USP2, USP8, USP10 and USP42, fuse with leukemia-associated genes, indicating that deubiquitinases play an important role in the pathogenesis of AML.
DUBs in acute lymphoblastic leukemia
Acute lymphoblastic leukemia (ALL) is caused by malignant proliferation of lymphoid cells (including T and B cells) blocked at an early stage of differentiation that can invade bone marrow, blood and extramedullary sites [103]. The first-line treatment for ALL is chemotherapy and typically includes three phases: induction, consolidation, and maintenance [104]. Radiation therapy can be used for patients with evidence of central nervous system (CNS) or testicular leukemia, while allogeneic hematopoietic cell transplantation and growth factor therapy are complementary to all treatments [105, 106]. New immunotherapeutic strategies, such as monoclonal antibodies and chimeric antigen receptor (CAR) T cells, are also being developed [107, 108]. However, the management of ALL in adults and elderly ALL patients (> 60 years old) is still challenging despite the great progress made in the past decades. The DUBs related to ALL are shown in Fig. 4.

DUBs in acute lymphoblastic leukemia. The red arrows represent oncogenes and the green arrows represent tumor suppressors in ALL. The surrounding text describes the substrates or signaling pathways involved in DUBs of different types of ALL
USP2/USP8-MLL rearrangement is identified in infant patients with B-ALL [66, 109]. The majority of infant ALL is characterized by MLL rearrangements (~ 70 to 80%, with a poor prognosis) in acute lymphoblastic leukemia. Targeting USP2/USP8 may be important to improve the outcomes of MLL-rearranged leukemia.
USP7 is identified as a susceptible locus for T-ALL in a genome-wide association study (GWAS) [110] and is significantly up-regulated in T-ALL cells [111]. Mechanistically, USP7 deubiquitinates and stabilizes NOTCH1, which leads to the decrease of the transcriptional levels of NOTCH1 targets and blocks T-ALL cell growth [111, 112]. However, USP7 is frequently mutated in pediatric T-ALL, with somatic heterozygous loss-of-function mutations (haploinsufficiency) predominantly affecting the subgroup that has aberrant TAL1 oncogene activation. Haploinsufficiency of USP7 promotes cell growth and transcriptionally down-regulates E-proteins targets by interacting with TAL1 [113]. Specifically, USP7 interacts with p190 BCR-ABL in Philadelphia chromosome-positive (Ph+) ALL, and decreased USP7 activity is associated with p53 protein stability [114].
USP9X inhibition reduces leukemia cell growth via repressing mTORC1 activity, enhances spontaneous apoptosis and overcomes glucocorticoid resistance in B-ALL [115]. However, USP9X level is positively associated with survival in T-ALL patients and knockdown of USP9X does not induce apoptosis and growth inhibition in T-ALL cells [116]. It is reported that pharmacological or genetic inhibition of USP9X, as well as treatment with low-dose ruxolitinib, may promote the survival of CRLF2-positive B-ALL with down syndrome (DS-ALL) cells, potentially by restricting JAK signaling [117]. These findings suggest that USP9X may play different roles in different types of ALL.
USP24 inhibition significantly induces growth inhibition and apoptosis in T-ALL cells. WP1130 blocks USP24 activity by directly interacting with the activity site pocket of USP24 in T-ALL cells. Mechanistically, USP24 inhibition regulates Mcl-1 stability to accelerate the collapse of mitochondrial transmembrane potential [116].
USP44 expression is elevated in T-ALL. USP44 overexpression leads to whole chromosomal instability, as well as increased cyclin B prior to mitosis, via regulating Cdc20-APC/C activity [118].
UCHL1 is decreased in the differentiation process of the lymphoblastic leukemia cell line, and its expression is independent of the apoptotic pathway [119]. However, the mechanism by which UCHL1 is involved in differentiation has not been elucidated.
A20 is expressed at high levels in ALL patients and cell lines, which promotes proliferation, regulates cell cycle progression and induces chemotherapy resistance [120]. A20 is also overexpressed in adult B-ALL, which may be related to the pathogenesis of B-ALL [121].
CYLD is significantly down-regulated in primary T-ALL [122]. Overexpression of CYLD could abolish the ability of constitutively active Notch1 in T-ALL [123]. Interestingly, cleaved CYLD overexpression in normal human peripheral blood mononuclear cells has the inherent capacity to program the genome of these cells resulting in T-cell lineage ALL [124]. Together, CYLD acts as a tumor suppressor in ALL.
Taken together, USP7, USP9X, USP24 and USP44 have been shown to promote the development of ALL as oncogenic genes. However, USP7 and USP9X also act as tumor suppressor genes in ALL. Therefore, targeting different deubiquitinases may have a role in certain types of ALL.
DUBs in chronic lymphocytic leukemia
Chronic lymphocytic leukemia (CLL) is the most frequent adult leukemia, which is initiated by specific genomic alterations that impair apoptosis of clonal B-cells [125]. With the development of kinase inhibitors that target B-cell receptor signaling, CLL therapy has transformed during the past decade. For example, Bruton’s tyrosine kinase (BTK) and isoform-selective phosphatidylinositol 3-kinase (PI3K) inhibitors disrupt B-cell receptor signaling. Besides, BCL2 (B-cell lymphoma 2) has emerged as another important therapeutic target [126]. New molecularly targeted drugs have shown good advantages over the previous chemotherapy regimens or monoclonal antibody regimens for elderly and high-risk CLL patients. However, drug resistance, side effects and high costs are currently existing problems.
USP7 is overexpressed in CLL compared with normal donors. Inhibition of USP7 partially impairs DNA repair by homologous recombination, which disrupts the stability of E3 ligase RAD18 and leads to the accumulation of DNA damage, therefore killing CLL cells independently of ataxia telangiectasia mutated (ATM) and p53 [127, 128]. USP7 is also shown to promote PTEN delocalization from the nucleus with consequent loss of part of its tumor suppressive function in a p53 dispensable manner [129]. Together, targeting USP7 is an effective strategy to kill CLL cells.
CYLD is considerably down-regulated in CLL cells compared to normal B cells, which correlates with lower overall survival (OS) in CLL patients [130, 131]. Lymphoid enhancer-binding factor 1(LEF1), a downstream effector of the Wnt/β-catenin pathway, can repress the transcription of CYLD and down-regulated CYLD inhibits TNF-α-induced necroptosis [130, 132]. Moreover, alternative splicing of CYLD is detected in many B-cell CLL patients’ samples, which can lead to CD5+ B-cell expansion through sustained NF-κB signaling [133].
A20 negatively regulates NF-κB activity in several types of B-cell malignancies. However, neither mutations nor aberrant DNA methylation is found at the A20 locus during gene analysis in CLL cells, while the expression of A20 is normal [134, 135]. These results suggest that A20 may not play a significant role in the pathogenesis of CLL.
DUBs in multiple myeloma
Multiple myeloma (MM) is the second most common hematological malignancy characterized by the accumulation of monoclonal plasma cells that produce M protein in the bone marrow [136]. The use of novel agents (such as proteasome inhibitors, immunomodulatory drugs and antibodies targeting cell surface molecules), as well as high-dose therapy and autologous stem cell transplantation (ASCT) in younger patients, has significantly improved the prognosis of patients with multiple myeloma [137]. However, most patients experience multiple relapses and eventually die of the disease itself or treatment-related complications, especially the elderly patients [138]. The discovery of new pathogenesis will help to develop new therapeutic drugs to overcome the problems of drug resistance and recurrence. Reports on the mechanisms by which deubiquitinating enzymes regulate the pathogenesis of MM have revealed the great potential for targeting deubiquitinates. The DUBs related to MM are shown in Fig. 5.

DUBs in multiple myeloma. The red arrows represent oncogenes and the green arrows represent tumor suppressors in MM. The surrounding text describes the substrates or signaling pathways of DUBs in MM
USP1, which participates in DNA damage response and cell differentiation, is overexpressed in some MM cases and is associated with poor prognosis. SJB3-019A is a selective inhibitor of USP1, which can decrease cell viability, trigger apoptosis, overcome bortezomib resistance in MM cells, and down-regulate renewal/survival related proteins and even promote MM stem cells differentiation [139].
USP5 and OTUB1 stabilize c-Maf and promote myeloma cell proliferation and survival [140,141,142,143], which indicates that USP5/c-Maf or OTUB1/c-Maf axis could be a potential target for myeloma therapy. Mebendazole down-regulates the expression of USP5 and disrupts the interaction between USP5 and c-Maf, which results in an increase in the level of c-Maf ubiquitination and subsequent c-Maf degradation [140]. MiR-125a inhibits the expression of USP5, thus reducing the proliferation and survival of MM cells [144]. Bortezomib-induced peripheral neuropathy (BIPN) is one of the most serious side effects associated with bortezomib treatment [145]. Interestingly, USP5 is found to be upregulated during this pathological process to stabilize Cav3.2, a T-type calcium channel essential for BIPN, in the mouse model [146]. Nanchangmycin (Nam), a polyketide antibiotic, is identified to inhibit c-Maf activity in the presence of OTUB1, indicating that Nam has the potential to treat MM by targeting the OTUB1/c-Maf axis [147].
USP7 overexpression predicts poor prognosis and USP7 inhibition overcomes bortezomib resistance in MM [148, 149]. USP7 is involved in bortezomib resistance by stabilizing NEK2 or repressing IκBα, and thus activating NF-κB signaling pathway [149, 150]. USP7 also promotes MM cell growth and inhibits cell apoptosis through other mechanisms. For example, USP7 stabilizes transcription factor Maf family members (c-Maf and MafA), which are highly expressed in MM and contribute to the invasion, adhesion and migration of MM cells [151]. USP7 stimulates DNA methyltransferase 1 (DNMT1) activity and blockade of USP7 enhances anti-MM activity of oxidative epigenetic agent RRx-001 [152]. Similarly, P5091, another USP7 inhibitor, inhibits MM cell growth and increases cell apoptosis both in vitro and in vivo [148, 151]. Interestingly, other USP7 inhibitors upregulate the transcription of genes that are normally silenced by the epigenetic suppressor complex polycomb inhibitor 2 (PRC2) and enhance the activity of PIM and PI3K inhibitors as well as DNA-damaging agents [153].
USP9X can remove the Lys48-linked poly-ubiquitin chains on Mcl-1 and thus prevent its degradation. It also promotes cell survival and its overexpression in MM is associated with poor clinical outcomes [154, 155]. USP24 can also stabilize Mcl-1 and promote MM cell survival independent of USP9X. WP1130, a small molecule inhibitor targeting USP9X, induces MM cell apoptosis both in vitro and in vivo [156]. Other leading compounds T5165804 and CP2005 inhibit USP9X activity at higher nanomolar potency against MM cell lines, compared with WP1130 [157].
USP14 and UCHL5, targeting the 19S regulatory subunit of the proteasome and affecting the proteasomal uptake of protein substrate for degradation, are highly expressed in MM and their inhibition by b-AP15 overcomes bortezomib resistance [78, 158, 159]. USP14 is involved in cell adhesion-mediated drug resistance (CAM-DR) of multiple myeloma cells by acting as a bridge between Bcl-xl apoptotic pathway and Wnt-signaling pathway [160].
USP15 expression is upregulated in MM patients compared with normal health ones. USP15 silencing induces MM cell proliferation inhibition and apoptosis via inhibiting NF-κB pathway. In turn, NF-κB p65 can thus promote USP15 expression, which forms a positive feedback loop [161].
UCHL1 is highly expressed in many MM cases and acts as a poor prognostic factor [162, 163]. UCHL1 impairs mTORC1 activity and increases mTORC2-mediated phosphorylation of the proliferative kinase Akt, and thus promotes the survival of MM cell [164].
PMSD14/Rpn11 is a component of the 26S proteasome, a multi-protein complex that catalyzes the degradation of ubiquitinated intracellular proteins [165]. Inhibition of PMSD14 activates the caspase cascade and endoplasmic stress response signals to trigger MM cell apoptosis and overcome bortezomib resistance [166].
Aberrant activation of the NF-κB pathway has been observed in MM [167], while CYLD is a negative regulator of NF-κB pathway, and the status of CYLD at 16q12 is highly correlated with the clinical outcome of MM patients [168]. In addition to affecting NF-κB signaling, CYLD loss or low expression also sensitizes MM cells to Wnt ligands, indicating another possible tumor-suppressive mechanism of CYLD in MM [169]. Moreover, it’s worth noting that the inhibitory effects of proteasome inhibitors on NF-κB signaling, which could be observed during MM treatment, might be mediated, at least in part, by the cellular accumulation of CYLD [170].
A20, a suppressor of the NF-κB pathway, is frequently down-regulated in MM, as a result of gene copy number reduction [171]. Berbamine treatment results in an increased expression of A20, which inhibits the proliferation of MM cells [172].
Taken together, deubiquitinase inhibitors have been shown to be effective in overcoming bortezomib resistance, so the combination of deubiquitinase inhibitors with existing therapies will become a new therapeutic strategy.
DUBs in Hodgkin lymphoma
Hodgkin Lymphoma (HL) is a type of lymphoid malignancy derived from B cells of the germinal center or postgerminal center, which is characterized by a low number of malignant cells and a high number of immune effector cells in the tumor microenvironment [173]. Primary HL can be cured with radiation therapy and multiagent chemotherapy, and even recurrent or refractory HL can be effectively treated or cured with high-dose chemotherapy and autologous hematopoietic stem cell transplantation. In addition, immunotherapy with antibody-drug conjugates and immune checkpoint inhibitors have been shown to be effective against HL [174]. Due to the satisfying therapeutic effects of current therapies against HL, there are few studies on deubiquitination enzyme in HL.
Classical Hodgkin lymphoma (cHL), a subtype of HL accounting for 95% cases, is characterized by the presence of less than 1% malignant mononucleated Hodgkin and multinucleated Reed-Sternberg cells (HRS cells) mixed with nonneoplastic cells [175]. NF-κB signaling pathway, which is regulated by A20 and CYLD, is very important for the survival and proliferation of HRS cells [176]. Mutation or deletion of A20 gene exists in nearly 40% of HL cases, contributing to constitutive NF-κB activity in cHL cells, while A20 reconstitution confers cytotoxicity to A20-deficient cHL cells [177,178,179]. Deletion or DNA copy number loss of CYLD are detected in one of four commonly used cHL cell lines and biallelic gene mutations of CYLD are detected at low frequency [180, 181], suggesting that impaired CYLD function may contribute to some cHL cases.
DUBs in non-Hodgkin lymphoma
Non-Hodgkin Lymphoma (NHL) accounts for about 90% of all lymphomas, which can be subdivided into B-cell lymphomas and T-cell/ NK-cell lymphomas. The classification of NHL is complex and constantly evolving, with more than 80 different subtypes listed in the latest classification of the World Health Organization [182, 183]. Each type of NHL has its unique histological and biological characteristics, as well as different treatment strategies and clinical results, making NHL a complex disease [184]. From the perspective of treatment, NHL is divided into high grade and low grade lymphoma. Low grade NHL patients can be cured by surgical excision or radiotherapy, but most patients appear in advanced stages or transform to high grade disease [185]. For high grade NHL patients, chemotherapy is still the main treatment method, and some patients will also receive radiotherapy. For B cell-derived lymphomas, CD20 monoclonal antibody immunotherapy will also be used in combination [186, 187]. However, drug resistance and relapse during or after chemotherapy are major problems need to be solved, especially for T-cell/ NK-cell lymphomas. The DUBs related to NHL are shown in Fig. 6.

DUBs in non-hodgkin lymphoma. The red arrows represent oncogenes and the green arrows represent tumor suppressors in NHL. The surrounding text describes the substrates or signaling pathways involved in DUBs of different types of lymphoma. CRLs: cullin-RING E3 ubiquitin ligases
USP2 inhibition by ML364 induces an increase in cellular cyclin D1 degradation and causes cell cycle arrest in mantle cell lymphoma (MCL) cell line (Mino cell) [188]. In addition, USP2 stabilizes Mdm2, which antagonizes the pro-apoptotic activity of p53 and possibly contributes to therapeutic resistance in cutaneous T-cell lymphoma (CTCL) [189].
USP9X was found to be highly expressed in samples obtained from patients with aggressive B-cell lymphoma. Knockdown of USP9X leads to suppressed lymphoma growth and increased sensitivity to chemotherapy by destabilizing X-linked inhibitor of apoptosis protein (XIAP) in B-cell lymphoma, independently of Mcl-1 [190]. In diffuse large B cell lymphoma (DLBCL) and follicular lymphoma (FL), USP9X deubiquitinates Mcl-1 and protects it from degrading, while high-level Mcl-1 is associated with malignant B-cell proliferation and poor outcomes in B-cell lymphoma patients [154].
USP14 and UCHL5 are aberrantly expressed in the cytoplasm of DLBCL cells [191]. USP14 and UCHL5 inhibition by b-AP15 could induce cell apoptosis and suppress cell migration in both ABC- and GBC-subtypes of DLBCL [192]. In addition, USP14 inhibitor IU1 leads to tumor regression in malignant lymphomas of thymus (MLT) mouse model [193].
USP21 is overexpressed in DLBCL and promotes cell proliferation by maintaining the stability of enhancer of zeste homolog2 (EZH2), an oncogenic molecule in NHL [194, 195]. Targeted inhibition of USP21 activity resulted in a reduction in EZH2 and thus inhibited cell growth, independent of EZH2 mutations. Another DUB targeting EZH2, USP36, was found in nasal natural killer /T cell lymphoma (NKTL). USP36 stabilizes EZH2 by removing the K222-linked poly-ubiquitin chain from EZH2 [196].
USP34 expression is significantly higher in DLBCL than that in reactive lymphatic hyperplasia. USP34 overexpression is associated with age, germinal center B-cell-like (GCB) subtypes, multiple extranodal involvements, and higher International Prognostic Index (IPI) scores in DLBCL [197]. Also, USP34 gene amplification was detected in transformed DLBCL (tDLBCL) [198].
UCHL1 was detected to be highly expressed in both Burkitt’s lymphoma (BL) and DLBCL, which is correlated with a poor clinical outcome [199]. Knockdown of UCHL1 using specific shRNA in BL cells leads to reduced cell proliferation, as well as strong LFA-1-dependent homotypic adhesion [200]. Mechanistically, aberrantly expressed UCHL1 could activate the Akt pathway via down-regulating the antagonistic phosphatase PHLPP1 and Akt signaling [201], as well as promoting mTORC2 assembly [202]. At the same time, UCHL1 can bypass mTORC1 to promote the assembly of eIF4F, a translational regulator downstream to Akt and mTOR, which is required for MYC-driven B-cell malignancy in mice [202].
CSN5 is identified the protease as a positive regulator of oncogenes and negative regulator of tumor suppressors [203,204,205]. CSN5i-3, a CSN5 inhibitor, inhibits the growth of human-derived cell line SU-DHL-1 (anaplastic large-cell lymphoma, ALCL) in a xenograft model [206]. Also, CSN5 knockdown inhibits the growth of DLBCL cells partially through the CSN5-HSP90 pathway [207].
A20 mutations and/or deletions are linked with various subtypes of B-cell or T-cell lymphomas, including mucosal-associated lymphoid tissue (MALT) lymphoma, DLBCL, MCL, FL and NKTL [208,209,210]. In NKTL, monoallelic deletion of A20 occurs in 18% cases, while no biallelic deletion was detected [175]. A20 mutations occur in more than 50% of ABC-DLBCL and a small fraction of GCB-DLBCL [211]. Up-regulating A20 with phorbol myristate acetate (PMA) and lonomycin (IONO) could effectively induce cell cycle arrest at G0/G1 phase, suppress cell proliferation and induce apoptosis in OCI-LY1, a DLBCL cell line [212]. Re-expression of A20 in DLBCL cell lines carrying biallelic inactivation of the A20 gene leads to cell apoptosis and impairs NF-κB signaling [211]. After A20 knockdown, NF-κB signaling in T cells can be activated by CD3 alone, rather than CD3/CD28 co-stimulation [213]. However, A20 deficiency is not a prognostic factor in all of our NKTL cohorts. It is associated with shorter PFS in the high IPI subgroup [175].
The cleavage of CYLD by MALT1 is blocked by the selective inhibitor of MALT, which inhibits the growth of ABC-DLBCL cells both in vitro and in vivo [214]. CYLD anomalies are also detected in adult T-cell leukemia/lymphoma (ATLL) cells, as hyper-phosphorylated CYLD losses its function of removing the ubiquitin chains on RIP1, leading to continuous activation of the NF-κB pathway [215]. Furthermore, CYLD phosphorylation down-regulation by BTK inhibitors induces cell apoptosis and tumor growth inhibition in non-GCB-DLBCL, especially in the rituximab-resistant relapsed/refractory cases [216].
Taken together, there are many types of NHL, and some deubiquitinases have shown great significance in certain types of NHL. However, the pathogenesis role of deubiquitinases in NHL and relative combination therapy need further investigation.
DUB inhibitors in hematological malignancies
Unlike E3 ligases, most of DUBs can be drugged according to their cysteine proteases or metalo proteases [12, 217]. To date, several DUBs, particularly those in the USP family, have been successfully targeted as small molecule inhibitors showing cytotoxic effects in vitro and in vivo. In addition to their potential therapeutic implications, the use of inhibitors has contributed greatly to understanding the role DUBs plays in various processes. In Table 1, we list the inhibitors of DUBs that have been reported so far and whether they are used in hematological malignancies.
Although DUBs are promising drug targets for the treatment of hematological malignancies, the development of DUB specific inhibitors has not been an easy task. This is because: 1) DUBs show complex structural characteristics in their catalytic domain and similarities among family members, which makes it difficult to target; 2) Some DUBs have high molecular weight, which leads to difficulties in crystal formation and obtaining complete crystal structure; 3) DUBs may undergo conformational changes after ubiquitin binding, suggesting the flexibility of their active sites, which poses challenges to the prediction and computer simulation of small molecules and leads to inefficiency; 4) The mechanism of DUBs regulation is complex, involving allosteric regulation of catalytic activity and/or substrate-mediated catalysis.
Conclusions and perspectives
In this review, we have shown that DUBs are attractive drug targets in hematological malignancies, and some DUB inhibitors have shown favorable anti-hematologic effects both in vitro and in vivo. However, some DUBs, such as USP9X and USP22, display both oncogenic and tumor suppressor properties in hematological malignancies. To further clarify the role of DUBs in hematological malignancies, more studies should be focused on the location and substrate specificity of DUBs, as well as their interaction with other oncoproteins. Moreover, further analysis of the ubiquitin chain cleavage mode, the chain linkage specificity of DUBs, and the ubiquitin chain architecture on substrate proteins will help to understand the different biological functions of DUBs. In addition, understanding the role of DUBs in hematopoietic stem cells (HSCs) maintenance and differentiation would also contribute to elucidation of the pathogenesis of hematological malignancies.
DUBs are involved in a variety of cellular life activities, including DNA methylation, DNA damage repair, survival, differentiation, and apoptosis. This functional diversity indicates: 1) DUBs inhibition may overcome drug resistance and relapse, which are the major problems in the clinical treatment of hematological malignancies; 2) Targeted inhibition of some DUBs can eliminate leukemia stem cells and minimal residual disease; 3) Therapeutic DUB inhibitors can be used in combination with other anticancer therapies.
To improve the specificity of DUB inhibitors, allosteric inhibitors might be an attractive alternative, or by blocking the interactions between DUBs and specific substrate.
Overall, advances in the field of deubiquitinases further pave the way for targeted therapies, and we believe that the development of DUB inhibitors could bring clinical benefits to patients with hematological malignancies.
Availability of data and materials
Not applicable.
Abbreviations
- DUB:
-
Deubiquitinase
- UPS:
-
Ubiquitin-proteasome system
- MM:
-
Multiple myeloma
- MCL:
-
Mantle-cell lymphoma
- USP:
-
Ubiquitin-specific protease
- UCH:
-
Ubiquitin carboxy-terminal hydrolase
- OTU:
-
Ovarian tumor-related protease
- MJD:
-
Machado-Joseph disease protein domain protease
- JAMM:
-
JAB1/PAB1/MPN-domain containing metallo-enzyme
- MINDY:
-
Motif interacting with Ub-containing novel DUB family
- HSC:
-
Hematopoietic stem cell
- CML:
-
Chronic myeloid leukemia
- TKI:
-
Tyrosine kinase inhibitor
- TFR:
-
Treatment-free remission
- PTEN:
-
Phosphatase and tensin homolog
- SKP2:
-
S-phase kinase-associated protein 2
- AML:
-
Acute myeloid leukemia
- NPM:
-
Nucleophosmin
- HSPC:
-
Hematopoietic stem and progenitor cell
- RA:
-
All-trans-retinoic acid
- APL:
-
Acute promyelocytic leukemia
- CNS:
-
Central nervous system
- CAR:
-
Chimeric antigen receptor
- ALL:
-
Acute lymphoblastic leukemia
- CLL:
-
Chronic lymphocytic leukemia
- ATM:
-
Ataxia telangiectasia mutated
- OS:
-
Overall survival
- ASCT:
-
Autologous stem cell transplantation
- HL:
-
Hodgkin lymphoma
- cHL:
-
Classical Hodgkin lymphoma
- NHL:
-
Non-hodgkin lymphoma
- DLBCL:
-
Diffuse large B cell lymphoma
- FL:
-
Follicular lymphoma
- MLT:
-
Malignant lymphomas of thymus
- NKTL:
-
Natural killer /T cell lymphoma
- GCB:
-
Germinal center B-cell-like
- IPI:
-
International prognostic index
- BL:
-
Burkitt’s lymphoma
- ALCL:
-
Anaplastic large-cell lymphoma
- ATLL:
-
Adult T-cell leukemia/lymphoma
References
Damgaard RB. The ubiquitin system: from cell signalling to disease biology and new therapeutic opportunities. Cell Death Differ. 2021;28(2):423–6. https://doi.org/10.1038/s41418-020-00703-w.
Ciechanover A. The unravelling of the ubiquitin system. Nat Rev Mol Cell Biol. 2015;16(5):322–4. https://doi.org/10.1038/nrm3982.
Merbl Y, Refour P, Patel H, Springer M, Kirschner MW. Profiling of ubiquitin-like modifications reveals features of mitotic control. Cell. 2013;152(5):1160–72. https://doi.org/10.1016/j.cell.2013.02.007.
Sahasrabuddhe AA, Elenitoba-Johnson KS. Role of the ubiquitin proteasome system in hematologic malignancies. Immunol Rev. 2015;263(1):224–39. https://doi.org/10.1111/imr.12236.
Di Costanzo A, Del Gaudio N, Conte L, Altucci L. The Ubiquitin Proteasome System in Hematological Malignancies: New Insight into Its Functional Role and Therapeutic Options. Cancers (Basel). 2020;12(7). https://doi.org/10.3390/cancers12071898.
Park J, Cho J, Song EJ. Ubiquitin-proteasome system (UPS) as a target for anticancer treatment. Arch Pharm Res. 2020;43(11):1144–61. https://doi.org/10.1007/s12272-020-01281-8.
Chen BB, Glasser JR, Coon TA, Zou C, Miller HL, Fenton M, et al. F-box protein FBXL2 targets cyclin D2 for ubiquitination and degradation to inhibit leukemic cell proliferation. Blood. 2012;119(13):3132–41. https://doi.org/10.1182/blood-2011-06-358911.
Duan S, Cermak L, Pagan JK, Rossi M, Martinengo C, di Celle PF, et al. FBXO11 targets BCL6 for degradation and is inactivated in diffuse large B-cell lymphomas. Nature. 2012;481(7379):90–3. https://doi.org/10.1038/nature10688.
Meissner B, Kridel R, Lim RS, Rogic S, Tse K, Scott DW, et al. The E3 ubiquitin ligase UBR5 is recurrently mutated in mantle cell lymphoma. Blood. 2013;121(16):3161–4. https://doi.org/10.1182/blood-2013-01-478834.
Ramkumar C, Cui H, Kong Y, Jones SN, Gerstein RM, Zhang H. Smurf2 suppresses B-cell proliferation and lymphomagenesis by mediating ubiquitination and degradation of YY1. Nat Commun. 2013;4:2598. https://doi.org/10.1038/ncomms3598.
Clague MJ, Urbe S, Komander D. Breaking the chains: deubiquitylating enzyme specificity begets function. Nat Rev Mol Cell Biol. 2019;20(6):338–52. https://doi.org/10.1038/s41580-019-0099-1.
Harrigan JA, Jacq X, Martin NM, Jackson SP. Deubiquitylating enzymes and drug discovery: emerging opportunities. Nat Rev Drug Discov. 2018;17(1):57–78. https://doi.org/10.1038/nrd.2017.152.
Gutierrez-Diaz BT, Gu W, Ntziachristos P. Deubiquitinases: pro-oncogenic activity and therapeutic targeting in blood malignancies. Trends Immunol. 2020;41(4):327–40. https://doi.org/10.1016/j.it.2020.02.004.
Sarodaya N, Karapurkar J, Kim KS, Hong SH, Ramakrishna S. The Role of Deubiquitinating Enzymes in Hematopoiesis and Hematological Malignancies. Cancers (Basel). 2020;12(5). https://doi.org/10.3390/cancers12051103.
van den Berk P, Lancini C, Company C, Serresi M, Sanchez-Bailon MP, Hulsman D, et al. USP15 Deubiquitinase safeguards hematopoiesis and genome integrity in hematopoietic stem cells and leukemia cells. Cell Rep. 2020;33(13):108533. https://doi.org/10.1016/j.celrep.2020.108533.
Xiong Z, Xia P, Zhu X, Geng J, Wang S, Ye B, et al. Glutamylation of deubiquitinase BAP1 controls self-renewal of hematopoietic stem cells and hematopoiesis. J Exp Med. 2020;217(2). https://doi.org/10.1084/jem.20190974.
Donaghy R, Han X, Rozenova K, Lv K, Jiang Q, Doepner M, et al. The BRISC deubiquitinating enzyme complex limits hematopoietic stem cell expansion by regulating JAK2 K63-ubiquitination. Blood. 2019;133(14):1560–71. https://doi.org/10.1182/blood-2018-10-877563.
Higuchi M, Kawamura H, Matsuki H, Hara T, Takahashi M, Saito S, et al. USP10 is an essential Deubiquitinase for hematopoiesis and inhibits apoptosis of Long-term hematopoietic stem cells. Stem Cell Rep. 2016;7(6):1116–29. https://doi.org/10.1016/j.stemcr.2016.11.003.
Apperley JF. Chronic myeloid leukaemia. Lancet. 2015;385(9976):1447–59. https://doi.org/10.1016/S0140-6736(13)62120-0.
Braun TP, Eide CA, Druker BJ. Response and resistance to BCR-ABL1-targeted therapies. Cancer Cell. 2020;37(4):530–42. https://doi.org/10.1016/j.ccell.2020.03.006.
Ross DM, Hughes TP. Treatment-free remission in patients with chronic myeloid leukaemia. Nat Rev Clin Oncol. 2020;17(8):493–503. https://doi.org/10.1038/s41571-020-0367-1.
Shih YT, Cortes JE, Kantarjian HM. Treatment value of second-generation BCR-ABL1 tyrosine kinase inhibitors compared with imatinib to achieve treatment-free remission in patients with chronic myeloid leukaemia: a modelling study. Lancet Haematol. 2019;6(8):e398–408. https://doi.org/10.1016/S2352-3026(19)30087-0.
Lei H, Xu HZ, Shan HZ, Liu M, Lu Y, Fang ZX, et al. Targeting USP47 overcomes tyrosine kinase inhibitor resistance and eradicates leukemia stem/progenitor cells in chronic myelogenous leukemia. Nat Commun. 2021;12(1):51. https://doi.org/10.1038/s41467-020-20259-0.
O'Hare T, Eide CA, Deininger MW. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood. 2007;110(7):2242–9. https://doi.org/10.1182/blood-2007-03-066936.
Hamilton A, Helgason GV, Schemionek M, Zhang B, Myssina S, Allan EK, et al. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood. 2012;119(6):1501–10. https://doi.org/10.1182/blood-2010-12-326843.
Quentmeier H, Eberth S, Romani J, Zaborski M, Drexler HG. BCR-ABL1-independent PI3Kinase activation causing imatinib-resistance. J Hematol Oncol. 2011;4:6. https://doi.org/10.1186/1756-8722-4-6.
Bigenzahn JW, Collu GM, Kartnig F, Pieraks M, Vladimer GI, Heinz LX, et al. LZTR1 is a regulator of RAS ubiquitination and signaling. Science. 2018;362(6419):1171–7. https://doi.org/10.1126/science.aap8210.
Pozhidaeva A, Bezsonova I. USP7: structure, substrate specificity, and inhibition. DNA Repair (Amst). 2019;76:30–9. https://doi.org/10.1016/j.dnarep.2019.02.005.
Nininahazwe L, Liu B, He C, Zhang H, Chen ZS. The emerging nature of ubiquitin-specific protease 7 (USP7): a new target in cancer therapy. Drug Discov Today. 2021;26(2):490–502. https://doi.org/10.1016/j.drudis.2020.10.028.
Kategaya L, Di Lello P, Rouge L, Pastor R, Clark KR, Drummond J, et al. USP7 small-molecule inhibitors interfere with ubiquitin binding. Nature. 2017;550(7677):534–8. https://doi.org/10.1038/nature24006.
Gavory G, O'Dowd CR, Helm MD, Flasz J, Arkoudis E, Dossang A, et al. Discovery and characterization of highly potent and selective allosteric USP7 inhibitors. Nat Chem Biol. 2018;14(2):118–25. https://doi.org/10.1038/nchembio.2528.
Morotti A, Panuzzo C, Crivellaro S, Pergolizzi B, Familiari U, Berger AH, et al. BCR-ABL disrupts PTEN nuclear-cytoplasmic shuttling through phosphorylation-dependent activation of HAUSP. Leukemia. 2014;28(6):1326–33. https://doi.org/10.1038/leu.2013.370.
Song MS, Salmena L, Carracedo A, Egia A, Lo-Coco F, Teruya-Feldstein J, et al. The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature. 2008;455(7214):813–7. https://doi.org/10.1038/nature07290.
Jiang S, Wang X, He Y, Huang H, Cao B, Zhang Z, et al. Suppression of USP7 induces BCR-ABL degradation and chronic myelogenous leukemia cell apoptosis. Cell Death Dis. 2021;12(5):456. https://doi.org/10.1038/s41419-021-03732-6.
Morotti A, Panuzzo C, Crivellaro S, Carra G, Guerrasio A, Saglio G. HAUSP compartmentalization in chronic myeloid leukemia. Eur J Haematol. 2015;94(4):318–21. https://doi.org/10.1111/ejh.12422.
Murtaza M, Jolly LA, Gecz J, Wood SA. La FAM fatale: USP9X in development and disease. Cell Mol Life Sci. 2015;72(11):2075–89. https://doi.org/10.1007/s00018-015-1851-0.
Sun H, Kapuria V, Peterson LF, Fang D, Bornmann WG, Bartholomeusz G, et al. Bcr-Abl ubiquitination and Usp9x inhibition block kinase signaling and promote CML cell apoptosis. Blood. 2011;117(11):3151–62. https://doi.org/10.1182/blood-2010-03-276477.
Bartholomeusz GA, Talpaz M, Kapuria V, Kong LY, Wang S, Estrov Z, et al. Activation of a novel Bcr/Abl destruction pathway by WP1130 induces apoptosis of chronic myelogenous leukemia cells. Blood. 2007;109(8):3470–8. https://doi.org/10.1182/blood-2006-02-005579.
Liao Y, Liu N, Xia X, Guo Z, Li Y, Jiang L, et al. USP10 modulates the SKP2/Bcr-Abl axis via stabilizing SKP2 in chronic myeloid leukemia. Cell Discov. 2019;5:24. https://doi.org/10.1038/s41421-019-0092-z.
Das T, Song EJ, Kim EE. The Multifaceted Roles of USP15 in Signal Transduction. Int J Mol Sci. 2021;22(9). https://doi.org/10.3390/ijms22094728.
Nie ZY, Yao M, Yang Z, Yang L, Liu XJ, Yu J, et al. De-regulated STAT5A/miR-202-5p/USP15/Caspase-6 regulatory axis suppresses CML cell apoptosis and contributes to Imatinib resistance. J Exp Clin Cancer Res. 2020;39(1):17. https://doi.org/10.1186/s13046-019-1502-7.
Mustachio LM, Lu Y, Kawakami M, Roszik J, Freemantle SJ, Liu X, et al. Evidence for the ISG15-specific Deubiquitinase USP18 as an antineoplastic target. Cancer Res. 2018;78(3):587–92. https://doi.org/10.1158/0008-5472.CAN-17-1752.
Yan M, Luo JK, Ritchie KJ, Sakai I, Takeuchi K, Ren R, et al. Ubp43 regulates BCR-ABL leukemogenesis via the type 1 interferon receptor signaling. Blood. 2007;110(1):305–12. https://doi.org/10.1182/blood-2006-07-033209.
Lin D, Zhang M, Zhang MX, Ren Y, Jin J, Zhao Q, et al. Induction of USP25 by viral infection promotes innate antiviral responses by mediating the stabilization of TRAF3 and TRAF6. Proc Natl Acad Sci U S A. 2015;112(36):11324–9. https://doi.org/10.1073/pnas.1509968112.
Shibata N, Ohoka N, Tsuji G, Demizu Y, Miyawaza K, Ui-Tei K, et al. Deubiquitylase USP25 prevents degradation of BCR-ABL protein and ensures proliferation of Ph-positive leukemia cells. Oncogene. 2020;39(19):3867–78. https://doi.org/10.1038/s41388-020-1253-0.
Peschiaroli A, Skaar JR, Pagano M, Melino G. The ubiquitin-specific protease USP47 is a novel beta-TRCP interactor regulating cell survival. Oncogene. 2010;29(9):1384–93. https://doi.org/10.1038/onc.2009.430.
Zhang B, Yin Y, Hu Y, Zhang J, Bian Z, Song M, et al. MicroRNA-204-5p inhibits gastric cancer cell proliferation by downregulating USP47 and RAB22A. Med Oncol. 2015;32(1):331. https://doi.org/10.1007/s12032-014-0331-y.
Yu L, Dong L, Wang Y, Liu L, Long H, Li H, et al. Reversible regulation of SATB1 ubiquitination by USP47 and SMURF2 mediates colon cancer cell proliferation and tumor progression. Cancer Lett. 2019;448:40–51. https://doi.org/10.1016/j.canlet.2019.01.039.
Parsons JL, Dianova II, Khoronenkova SV, Edelmann MJ, Kessler BM, Dianov GL. USP47 is a deubiquitylating enzyme that regulates base excision repair by controlling steady-state levels of DNA polymerase beta. Mol Cell. 2011;41(5):609–15. https://doi.org/10.1016/j.molcel.2011.02.016.
Ka HI, Lee S, Han S, Jeong AL, Park JY, Joo HJ, et al. Deubiquitinase USP47-stabilized splicing factor IK regulates the splicing of ATM pre-mRNA. Cell Death Discov. 2020;6:34. https://doi.org/10.1038/s41420-020-0268-1.
Silvestrini VC, Thome CH, Albuquerque D, de Souza PC, Ferreira GA, Lanfredi GP, et al. Proteomics analysis reveals the role of ubiquitin specific protease (USP47) in epithelial to Mesenchymal transition (EMT) induced by TGFbeta2 in breast cells. J Proteome. 2020;219:103734. https://doi.org/10.1016/j.jprot.2020.103734.
Palazon-Riquelme P, Worboys JD, Green J, Valera A, Martin-Sanchez F, Pellegrini C, et al. USP7 and USP47 deubiquitinases regulate NLRP3 inflammasome activation. EMBO Rep. 2018;19(10). https://doi.org/10.15252/embr.201744766.
Ashton-Beaucage D, Lemieux C, Udell CM, Sahmi M, Rochette S, Therrien M. The Deubiquitinase USP47 stabilizes MAPK by counteracting the function of the N-end rule ligase POE/UBR4 in Drosophila. PLoS Biol. 2016;14(8):e1002539. https://doi.org/10.1371/journal.pbio.1002539.
Jia Q, Sun H, Xiao F, Sai Y, Li Q, Zhang X, et al. miR-17-92 promotes leukemogenesis in chronic myeloid leukemia via targeting A20 and activation of NF-kappaB signaling. Biochem Biophys Res Commun. 2017;487(4):868–74. https://doi.org/10.1016/j.bbrc.2017.04.144.
Dkhissi F, Aggoune D, Pontis J, Sorel N, Piccirilli N, LeCorf A, et al. The downregulation of BAP1 expression by BCR-ABL reduces the stability of BRCA1 in chronic myeloid leukemia. Exp Hematol. 2015;43(9):775–80. https://doi.org/10.1016/j.exphem.2015.04.013.
Khwaja A, Bjorkholm M, Gale RE, Levine RL, Jordan CT, Ehninger G, et al. Acute myeloid leukaemia. Nat Rev Dis Primers. 2016;2:16010. https://doi.org/10.1038/nrdp.2016.10.
Dohner H, Estey EH, Amadori S, Appelbaum FR, Buchner T, Burnett AK, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115(3):453–74. https://doi.org/10.1182/blood-2009-07-235358.
De Kouchkovsky I, Abdul-Hay M. 'Acute myeloid leukemia: a comprehensive review and 2016 update. Blood Cancer J. 2016;6(7):e441. https://doi.org/10.1038/bcj.2016.50.
Vadakekolathu J, Minden MD, Hood T, Church SE, Reeder S, Altmann H, et al. Immune landscapes predict chemotherapy resistance and immunotherapy response in acute myeloid leukemia. Sci Transl Med. 2020;12(546). https://doi.org/10.1126/scitranslmed.aaz0463.
Farge T, Saland E, de Toni F, Aroua N, Hosseini M, Perry R, et al. Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. 2017;7(7):716–35. https://doi.org/10.1158/2159-8290.CD-16-0441.
Gebru MT, Wang HG. Therapeutic targeting of FLT3 and associated drug resistance in acute myeloid leukemia. J Hematol Oncol. 2020;13(1):155. https://doi.org/10.1186/s13045-020-00992-1.
Nair R, Salinas-Illarena A, Baldauf HM. New strategies to treat AML: novel insights into AML survival pathways and combination therapies. Leukemia. 2021;35(2):299–311. https://doi.org/10.1038/s41375-020-01069-1.
Liu H. Emerging agents and regimens for AML. J Hematol Oncol. 2021;14(1):49. https://doi.org/10.1186/s13045-021-01062-w.
Garcia-Santisteban I, Peters GJ, Giovannetti E, Rodriguez JA. USP1 deubiquitinase: cellular functions, regulatory mechanisms and emerging potential as target in cancer therapy. Mol Cancer. 2013;12:91. https://doi.org/10.1186/1476-4598-12-91.
Mistry H, Hsieh G, Buhrlage SJ, Huang M, Park E, Cuny GD, et al. Small-molecule inhibitors of USP1 target ID1 degradation in leukemic cells. Mol Cancer Ther. 2013;12(12):2651–62. https://doi.org/10.1158/1535-7163.MCT-13-0103-T.
Ikeda J, Shiba N, Tsujimoto SI, Yoshida M, Nakabayashi K, Ogata-Kawata H, et al. Whole transcriptome sequencing reveals a KMT2A-USP2 fusion in infant acute myeloid leukemia. Genes Chromosomes Cancer. 2019;58(9):669–72. https://doi.org/10.1002/gcc.22751.
Meyer C, Lopes BA, Caye-Eude A, Cave H, Arfeuille C, Cuccuini W, et al. Human MLL/KMT2A gene exhibits a second breakpoint cluster region for recurrent MLL-USP2 fusions. Leukemia. 2019;33(9):2306–40. https://doi.org/10.1038/s41375-019-0451-7.
Meyer C, Lopes B, Caye-Eude A, Cave H, Arfeuille C, Sutton R, et al. MLL-USP2: an underestimated new entity of MLL-rearranged leukemia identified by NGS analysis. Blood. 2018;132. https://doi.org/10.1182/blood-2018-99-112594.
Noguera NI, Song MS, Divona M, Catalano G, Calvo KL, Garcia F, et al. Nucleophosmin/B26 regulates PTEN through interaction with HAUSP in acute myeloid leukemia. Leukemia. 2013;27(5):1037–43. https://doi.org/10.1038/leu.2012.314.
Cartel M, Mouchel PL, Gotanegre M, David L, Bertoli S, Mansat-De Mas V, et al. Inhibition of ubiquitin-specific protease 7 sensitizes acute myeloid leukemia to chemotherapy. Leukemia. 2021;35(2):417–32. https://doi.org/10.1038/s41375-020-0878-x.
Maat H, Atsma TJ, Hogeling SM, Rodriguez Lopez A, Jaques J, Olthuis M, et al. The USP7-TRIM27 axis mediates non-canonical PRC1.1 function and is a druggable target in leukemia. iScience. 2021;24(5):102435. https://doi.org/10.1016/j.isci.2021.102435.
Akiyama H, Umezawa Y, Ishida S, Okada K, Nogami A, Miura O. Inhibition of USP9X induces apoptosis in FLT3-ITD-positive AML cells cooperatively by inhibiting the mutant kinase through aggresomal translocation and inducing oxidative stress. Cancer Lett. 2019;453:84–94. https://doi.org/10.1016/j.canlet.2019.03.046.
Weisberg EL, Schauer NJ, Yang J, Lamberto I, Doherty L, Bhatt S, et al. Inhibition of USP10 induces degradation of oncogenic FLT3. Nat Chem Biol. 2017;13(12):1207–15. https://doi.org/10.1038/nchembio.2486.
Yu M, Fang ZX, Wang WW, Zhang Y, Bu ZL, Liu M, et al. Wu-5, a novel USP10 inhibitor, enhances crenolanib-induced FLT3-ITD-positive AML cell death via inhibiting FLT3 and AMPK pathways. Acta Pharmacol Sin. 2021;42(4):604–12. https://doi.org/10.1038/s41401-020-0455-x.
Puissant A, Fenouille N, Alexe G, Pikman Y, Bassil CF, Mehta S, et al. SYK is a critical regulator of FLT3 in acute myeloid leukemia. Cancer Cell. 2014;25(2):226–42. https://doi.org/10.1016/j.ccr.2014.01.022.
Yang J, Meng C, Weisberg E, Case A, Lamberto I, Magin RS, et al. Inhibition of the deubiquitinase USP10 induces degradation of SYK. Br J Cancer. 2020;122(8):1175–84. https://doi.org/10.1038/s41416-020-0731-z.
Zerkalenkova E, Lebedeva S, Kazakova A, Baryshev P, Meyer C, Marschalek R, et al. A case of pediatric acute myeloid leukemia with t (11;16)(q23;q24) leading to a novel KMT2A-USP10 fusion gene. Genes Chromosomes Cancer. 2018;57(10):522–4. https://doi.org/10.1002/gcc.22646.
D'Arcy P, Brnjic S, Olofsson MH, Fryknas M, Lindsten K, De Cesare M, et al. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat Med. 2011;17(12):1636–40. https://doi.org/10.1038/nm.2536.
Zhao C, Chen X, Zang D, Lan X, Liao S, Yang C, et al. A novel nickel complex works as a proteasomal deubiquitinase inhibitor for cancer therapy. Oncogene. 2016;35(45):5916–27. https://doi.org/10.1038/onc.2016.114.
Goldenson B, Crispino JD. The aurora kinases in cell cycle and leukemia. Oncogene. 2015;34(5):537–45. https://doi.org/10.1038/onc.2014.14.
Song C, Ma R, Yang X, Pang S. The Deubiquitinating enzyme USP14 regulates leukemic chemotherapy drugs-induced cell apoptosis by suppressing Ubiquitination of Aurora kinase B. Cell Physiol Biochem. 2017;42(3):965–73. https://doi.org/10.1159/000478679.
Sugawara T, Oguro H, Negishi M, Morita Y, Ichikawa H, Iseki T, et al. FET family proto-oncogene Fus contributes to self-renewal of hematopoietic stem cells. Exp Hematol. 2010;38(8):696–706. https://doi.org/10.1016/j.exphem.2010.04.006.
Niederkorn M, Hueneman K, Choi K, Varney ME, Romano L, Pujato MA, et al. TIFAB regulates USP15-mediated p53 signaling during stressed and malignant hematopoiesis. Cell Rep. 2020;30(8):2776–90 e6. https://doi.org/10.1016/j.celrep.2020.01.093.
Liu LQ, Ilaria R Jr, Kingsley PD, Iwama A, van Etten RA, Palis J, et al. A novel ubiquitin-specific protease, UBP43, cloned from leukemia fusion protein AML1-ETO-expressing mice, functions in hematopoietic cell differentiation. Mol Cell Biol. 1999;19(4):3029–38. https://doi.org/10.1128/mcb.19.4.3029.
Guo Y, Dolinko AV, Chinyengetere F, Stanton B, Bomberger JM, Demidenko E, et al. Blockade of the ubiquitin protease UBP43 destabilizes transcription factor PML/RARalpha and inhibits the growth of acute promyelocytic leukemia. Cancer Res. 2010;70(23):9875–85. https://doi.org/10.1158/0008-5472.CAN-10-1100.
Shah SJ, Blumen S, Pitha-Rowe I, Kitareewan S, Freemantle SJ, Feng Q, et al. UBE1L represses PML/RAR {alpha} by targeting the PML domain for ISG15ylation. Mol Cancer Ther. 2008;7(4):905–14. https://doi.org/10.1158/1535-7163.MCT-07-0515.
Li L, Osdal T, Ho Y, Chun S, McDonald T, Agarwal P, et al. SIRT1 activation by a c-MYC oncogenic network promotes the maintenance and drug resistance of human FLT3-ITD acute myeloid leukemia stem cells. Cell Stem Cell. 2014;15(4):431–46. https://doi.org/10.1016/j.stem.2014.08.001.
Melo-Cardenas J, Xu Y, Wei J, Tan C, Kong S, Gao B, et al. USP22 deficiency leads to myeloid leukemia upon oncogenic Kras activation through a PU.1-dependent mechanism. Blood. 2018;132(4):423–34. https://doi.org/10.1182/blood-2017-10-811760.
Zhang H, Huang H, Feng X, Song H, Zhang Z, Shen A, et al. Deubiquitinase USP28 inhibits ubiquitin ligase KLHL2-mediated uridine-cytidine kinase 1 degradation and confers sensitivity to 5′-azacytidine-resistant human leukemia cells. Theranostics. 2020;10(3):1046–59. https://doi.org/10.7150/thno.36503.
Yang WC, Shih HM. The deubiquitinating enzyme USP37 regulates the oncogenic fusion protein PLZF/RARA stability. Oncogene. 2013;32(43):5167–75. https://doi.org/10.1038/onc.2012.537.
Paulsson K, Bekassy AN, Olofsson T, Mitelman F, Johansson B, Panagopoulos I. A novel and cytogenetically cryptic t (7;21)(p22;q22) in acute myeloid leukemia results in fusion of RUNX1 with the ubiquitin-specific protease gene USP42. Leukemia. 2006;20(2):224–9. https://doi.org/10.1038/sj.leu.2404076.
Panagopoulos I, Gorunova L, Brandal P, Garnes M, Tierens A, Heim S. Myeloid leukemia with t(7;21)(p22;q22) and 5q deletion. Oncol Rep. 2013;30(4):1549–52. https://doi.org/10.3892/or.2013.2623.
Paulraj P, Diamond S, Razzaqi F, Ozeran JD, Longhurst M, Andersen EF, et al. Pediatric acute myeloid leukemia with t(7,21)(p22;q22). Genes Chromosomes Cancer. 2019;58(8):551–7. https://doi.org/10.1002/gcc.22740.
Flach J, Shumilov E, Joncourt R, Porret N, Tchinda J, Legros M, et al. Detection of rare reciprocal RUNX1 rearrangements by next-generation sequencing in acute myeloid leukemia. Genes Chromosomes Cancer. 2020;59(4):268–74. https://doi.org/10.1002/gcc.22829.
Li L, Wang Y, Zhang X, Song G, Guo Q, Zhang Z, et al. Deubiquitinase USP48 promotes ATRA-induced granulocytic differentiation of acute promyelocytic leukemia cells. Int J Oncol. 2018;53(2):895–903. https://doi.org/10.3892/ijo.2018.4440.
Huang D, Nagata Y, Grossmann V, Radivoyevitch T, Okuno Y, Nagae G, et al. BRCC3 mutations in myeloid neoplasms. Haematologica. 2015;100(8):1051–7. https://doi.org/10.3324/haematol.2014.111989.
Meyer T, Jahn N, Lindner S, Rohner L, Dolnik A, Weber D, et al. Functional characterization of BRCC3 mutations in acute myeloid leukemia with t(8;21)(q22;q22.1). Leukemia. 2020;34(2):404–15. https://doi.org/10.1038/s41375-019-0578-6.
Zhang X, Su Y, Song H, Yu Z, Zhang B, Chen H. Attenuated A20 expression of acute myeloid leukemia-derived dendritic cells increased the anti-leukemia immune response of autologous cytolytic T cells. Leuk Res. 2014;38(6):673–81. https://doi.org/10.1016/j.leukres.2014.03.011.
Osako M, Itsumi M, Yamaguchi H, Takeuchi H, Yamaoka S. A20 restores phorbol ester-induced differentiation of THP-1 cells in the absence of nuclear factor-kappaB activation. J Cell Biochem. 2018;119(2):1475–87. https://doi.org/10.1002/jcb.26308.
Shi L, Chen S, Lu Y, Wang X, Xu L, Zhang F, et al. Changes in the MALT1-A20-NF-kappaB expression pattern may be related to T cell dysfunction in AML. Cancer Cell Int. 2013;13(1):37. https://doi.org/10.1186/1475-2867-13-37.
Asada S, Goyama S, Inoue D, Shikata S, Takeda R, Fukushima T, et al. Mutant ASXL1 cooperates with BAP1 to promote myeloid leukaemogenesis. Nat Commun. 2018;9(1):2733. https://doi.org/10.1038/s41467-018-05085-9.
Wang L, Birch NW, Zhao ZB, Nestler CM, Kazmer A, Shilati A, et al. Epigenetic targeted therapy of stabilized BAP1 in ASXL1 gain-of-function mutated leukemia. Nat Cancer. 2021;2(5):515. https://doi.org/10.1038/s43018-021-00199-4.
Malard F, Mohty M. Acute lymphoblastic leukaemia. Lancet. 2020;395(10230):1146–62. https://doi.org/10.1016/S0140-6736(19)33018-1.
Brown PA, Wieduwilt M, Logan A, DeAngelo DJ, Wang ES, Fathi A, et al. Guidelines insights: acute lymphoblastic leukemia, version 1.2019. J Natl Compr Cancer Netw. 2019;17(5):414–23. https://doi.org/10.6004/jnccn.2019.0024.
Bassan R, Hoelzer D. Modern therapy of acute lymphoblastic leukemia. J Clin Oncol. 2011;29(5):532–43. https://doi.org/10.1200/JCO.2010.30.1382.
Teachey DT, Hunger SP, Loh ML. Optimizing therapy in the modern age: differences in length of maintenance therapy in acute lymphoblastic leukemia. Blood. 2021;137(2):168–77. https://doi.org/10.1182/blood.2020007702.
Wang Z, Wu Z, Liu Y, Han W. New development in CAR-T cell therapy. J Hematol Oncol. 2017;10(1):53. https://doi.org/10.1186/s13045-017-0423-1.
Rafei H, Kantarjian HM, Jabbour EJ. Recent advances in the treatment of acute lymphoblastic leukemia. Leuk Lymphoma. 2019;60(11):2606–21. https://doi.org/10.1080/10428194.2019.1605071.
Blackburn PR, Smadbeck JB, Znoyko I, Webley MR, Pitel BA, Vasmatzis G, et al. Cryptic and atypical KMT2A-USP2 and KMT2A-USP8 rearrangements identified by mate pair sequencing in infant and childhood leukemia. Genes Chromosomes Cancer. 2020;59(7):422–7. https://doi.org/10.1002/gcc.22842.
Qian M, Zhao X, Devidas M, Yang W, Gocho Y, Smith C, et al. Genome-wide association study of susceptibility loci for T-cell acute lymphoblastic leukemia in children. J Natl Cancer Inst. 2019;111(12):1350–7. https://doi.org/10.1093/jnci/djz043.
Shan H, Li X, Xiao X, Dai Y, Huang J, Song J, et al. USP7 deubiquitinates and stabilizes NOTCH1 in T-cell acute lymphoblastic leukemia. Signal Transduct Target Ther. 2018;3:29. https://doi.org/10.1038/s41392-018-0028-3.
Jin Q, Martinez CA, Arcipowski KM, Zhu Y, Gutierrez-Diaz BT, Wang KK, et al. USP7 cooperates with NOTCH1 to drive the oncogenic transcriptional program in T-cell leukemia. Clin Cancer Res. 2019;25(1):222–39. https://doi.org/10.1158/1078-0432.CCR-18-1740.
Shaw TI, Dong L, Tian L, Qian C, Liu Y, Ju B, et al. Integrative network analysis reveals USP7 haploinsufficiency inhibits E-protein activity in pediatric T-lineage acute lymphoblastic leukemia (T-ALL). Sci Rep. 2021;11(1):5154. https://doi.org/10.1038/s41598-021-84647-2.
Carra G, Panuzzo C, Crivellaro S, Morena D, Taulli R, Guerrasio A, et al. The targetable role of herpes virus-associated ubiquitin-specific protease (HAUSP) in p190 BCR-ABL leukemia. Oncol Lett. 2016;12(5):3123–6. https://doi.org/10.3892/ol.2016.5073.
Zhou M, Wang T, Lai H, Zhao X, Yu Q, Zhou J, et al. Targeting of the deubiquitinase USP9X attenuates B-cell acute lymphoblastic leukemia cell survival and overcomes glucocorticoid resistance. Biochem Biophys Res Commun. 2015;459(2):333–9. https://doi.org/10.1016/j.bbrc.2015.02.115.
Luo H, Jing B, Xia Y, Zhang Y, Hu M, Cai H, et al. WP1130 reveals USP24 as a novel target in T-cell acute lymphoblastic leukemia. Cancer Cell Int. 2019;19:56. https://doi.org/10.1186/s12935-019-0773-6.
Schwartzman O, Savino AM, Gombert M, Palmi C, Cario G, Schrappe M, et al. Suppressors and activators of JAK-STAT signaling at diagnosis and relapse of acute lymphoblastic leukemia in Down syndrome. Proc Natl Acad Sci U S A. 2017;114(20):E4030–E9. https://doi.org/10.1073/pnas.1702489114.
Zhang Y, van Deursen J, Galardy PJ. Overexpression of ubiquitin specific protease 44 (USP44) induces chromosomal instability and is frequently observed in human T-cell leukemia. PLoS One. 2011;6(8):e23389. https://doi.org/10.1371/journal.pone.0023389.
Maki A, Mohammad RM, Smith M, Al-Katib A. Role of ubiquitin carboxyl terminal hydrolase in the differentiation of human acute lymphoblastic leukemia cell line, Reh. Differentiation. 1996;60(1):59–66. https://doi.org/10.1046/j.1432-0436.1996.6010059.x.
Chen S, Xing H, Li S, Yu J, Li H, Liu S, et al. Up-regulated A20 promotes proliferation, regulates cell cycle progression and induces chemotherapy resistance of acute lymphoblastic leukemia cells. Leuk Res. 2015;39(9):976–83. https://doi.org/10.1016/j.leukres.2015.06.004.
Xu Y, Hu J, Wang X, Xuan L, Lai J, Xu L, et al. Overexpression of MALT1-A20-NF-kappaB in adult B-cell acute lymphoblastic leukemia. Cancer Cell Int. 2015;15:73. https://doi.org/10.1186/s12935-015-0222-0.
Espinosa L, Cathelin S, D'Altri T, Trimarchi T, Statnikov A, Guiu J, et al. The notch/Hes1 pathway sustains NF-kappaB activation through CYLD repression in T cell leukemia. Cancer Cell. 2010;18(3):268–81. https://doi.org/10.1016/j.ccr.2010.08.006.
D'Altri T, Gonzalez J, Aifantis I, Espinosa L, Bigas A. Hes1 expression and CYLD repression are essential events downstream of Notch1 in T-cell leukemia. Cell Cycle. 2011;10(7):1031–6. https://doi.org/10.4161/cc.10.7.15067.
Arora M, Kaul D, Varma N, Marwaha RK. Cellular proteolytic modification of tumor-suppressor CYLD is critical for the initiation of human T-cell acute lymphoblastic leukemia. Blood Cells Mol Dis. 2015;54(1):132–8. https://doi.org/10.1016/j.bcmd.2014.07.008.
Hallek M. Chronic lymphocytic leukemia: 2020 update on diagnosis, risk stratification and treatment. Am J Hematol. 2019;94(11):1266–87. https://doi.org/10.1002/ajh.25595.
Burger JA. Treatment of chronic lymphocytic leukemia. N Engl J Med. 2020;383(5):460–73. https://doi.org/10.1056/NEJMra1908213.
Agathanggelou A, Smith E, Davies NJ, Kwok M, Zlatanou A, Oldreive CE, et al. USP7 inhibition alters homologous recombination repair and targets CLL cells independently of ATM/p53 functional status. Blood. 2017;130(2):156–66. https://doi.org/10.1182/blood-2016-12-758219.
Sampath D. Targeting deubiquitinases in CLL. Blood. 2017;130(2):100–1. https://doi.org/10.1182/blood-2017-06-786236.
Carra G, Panuzzo C, Torti D, Parvis G, Crivellaro S, Familiari U, et al. Therapeutic inhibition of USP7-PTEN network in chronic lymphocytic leukemia: a strategy to overcome TP53 mutated/deleted clones. Oncotarget. 2017;8(22):35508–22. https://doi.org/10.18632/oncotarget.16348.
Liu P, Xu B, Shen W, Zhu H, Wu W, Fu Y, et al. Dysregulation of TNFalpha-induced necroptotic signaling in chronic lymphocytic leukemia: suppression of CYLD gene by LEF1. Leukemia. 2012;26(6):1293–300. https://doi.org/10.1038/leu.2011.357.
Wu W, Zhu H, Fu Y, Shen W, Xu J, Miao K, et al. Clinical significance of down-regulated cylindromatosis gene in chronic lymphocytic leukemia. Leuk Lymphoma. 2014;55(3):588–94. https://doi.org/10.3109/10428194.2013.809077.
Wu W, Zhu H, Fu Y, Shen W, Miao K, Hong M, et al. High LEF1 expression predicts adverse prognosis in chronic lymphocytic leukemia and may be targeted by ethacrynic acid. Oncotarget. 2016;7(16):21631–43. https://doi.org/10.18632/oncotarget.7795.
Hahn M, Burckert JP, Luttenberger CA, Klebow S, Hess M, Al-Maarri M, et al. Aberrant splicing of the tumor suppressor CYLD promotes the development of chronic lymphocytic leukemia via sustained NF-kappaB signaling. Leukemia. 2018;32(1):72–82. https://doi.org/10.1038/leu.2017.168.
Frenzel LP, Claus R, Plume N, Schwamb J, Konermann C, Pallasch CP, et al. Sustained NF-kappaB activity in chronic lymphocytic leukemia is independent of genetic and epigenetic alterations in the TNFAIP3 (A20) locus. Int J Cancer. 2011;128(10):2495–500. https://doi.org/10.1002/ijc.25579.
Philipp C, Edelmann J, Buhler A, Winkler D, Stilgenbauer S, Kuppers R. Mutation analysis of the TNFAIP3 (A20) tumor suppressor gene in CLL. Int J Cancer. 2011;128(7):1747–50. https://doi.org/10.1002/ijc.25497.
van de Donk N, Pawlyn C, Yong KL. Multiple myeloma. Lancet. 2021;397(10272):410–27. https://doi.org/10.1016/S0140-6736(21)00135-5.
Kumar SK, Dimopoulos MA, Kastritis E, Terpos E, Nahi H, Goldschmidt H, et al. Natural history of relapsed myeloma, refractory to immunomodulatory drugs and proteasome inhibitors: a multicenter IMWG study. Leukemia. 2017;31(11):2443–8. https://doi.org/10.1038/leu.2017.138.
Rajkumar SV. Multiple myeloma: 2020 update on diagnosis, risk-stratification and management. Am J Hematol. 2020;95(5):548–67. https://doi.org/10.1002/ajh.25791.
Das DS, Das A, Ray A, Song Y, Samur MK, Munshi NC, et al. Blockade of Deubiquitylating enzyme USP1 inhibits DNA repair and triggers apoptosis in multiple myeloma cells. Clin Cancer Res. 2017;23(15):4280–9. https://doi.org/10.1158/1078-0432.CCR-16-2692.
Chen XH, Xu YJ, Wang XG, Lin P, Cao BY, Zeng YY, et al. Mebendazole elicits potent antimyeloma activity by inhibiting the USP5/c-Maf axis. Acta Pharmacol Sin. 2019;40(12):1568–77. https://doi.org/10.1038/s41401-019-0249-1.
Wang S, Juan J, Zhang Z, Du Y, Xu Y, Tong J, et al. Inhibition of the deubiquitinase USP5 leads to c-Maf protein degradation and myeloma cell apoptosis. Cell Death Dis. 2017;8(9):e3058. https://doi.org/10.1038/cddis.2017.450.
Zhang Z, Tong J, Tang X, Juan J, Cao B, Hurren R, et al. The ubiquitin ligase HERC4 mediates c-Maf ubiquitination and delays the growth of multiple myeloma xenografts in nude mice. Blood. 2016;127(13):1676–86. https://doi.org/10.1182/blood-2015-07-658203.
Xu Y, Xu M, Tong J, Tang X, Chen J, Chen X, et al. Targeting the Otub1/c-Maf axis for the treatment of multiple myeloma. Blood. 2021;137(11):1478–90. https://doi.org/10.1182/blood.2020005199.
Wu L, Zhang C, Chu M, Fan Y, Wei L, Li Z, et al. miR-125a suppresses malignancy of multiple myeloma by reducing the deubiquitinase USP5. J Cell Biochem. 2020;121(1):642–50. https://doi.org/10.1002/jcb.29309.
Argyriou AA, Cavaletti G, Bruna J, Kyritsis AP, Kalofonos HP. Bortezomib-induced peripheral neurotoxicity: an update. Arch Toxicol. 2014;88(9):1669–79. https://doi.org/10.1007/s00204-014-1316-5.
Tomita S, Sekiguchi F, Deguchi T, Miyazaki T, Ikeda Y, Tsubota M, et al. Critical role of Cav3.2 T-type calcium channels in the peripheral neuropathy induced by bortezomib, a proteasome-inhibiting chemotherapeutic agent, in mice. Toxicology. 2019;413:33–9. https://doi.org/10.1016/j.tox.2018.12.003.
Xu Y, Sun T, Zeng K, Xu M, Chen J, Xu X, et al. Anti-bacterial and anti-viral nanchangmycin displays anti-myeloma activity by targeting Otub1 and c-Maf. Cell Death Dis. 2020;11(9):818. https://doi.org/10.1038/s41419-020-03017-4.
Chauhan D, Tian Z, Nicholson B, Kumar KG, Zhou B, Carrasco R, et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell. 2012;22(3):345–58. https://doi.org/10.1016/j.ccr.2012.08.007.
Yao Y, Zhang Y, Shi M, Sun Y, Chen C, Niu M, et al. Blockade of deubiquitinase USP7 overcomes bortezomib resistance by suppressing NF-kappaB signaling pathway in multiple myeloma. J Leukoc Biol. 2018;104(6):1105–15. https://doi.org/10.1002/JLB.2A1017-420RR.
Franqui-Machin R, Hao M, Bai H, Gu Z, Zhan X, Habelhah H, et al. Destabilizing NEK2 overcomes resistance to proteasome inhibition in multiple myeloma. J Clin Invest. 2018;128(7):2877–93. https://doi.org/10.1172/JCI98765.
He Y, Wang S, Tong J, Jiang S, Yang Y, Zhang Z, et al. The deubiquitinase USP7 stabilizes Maf proteins to promote myeloma cell survival. J Biol Chem. 2020;295(7):2084–96. https://doi.org/10.1074/jbc.RA119.010724.
Das DS, Ray A, Das A, Song Y, Tian Z, Oronsky B, et al. A novel hypoxia-selective epigenetic agent RRx-001 triggers apoptosis and overcomes drug resistance in multiple myeloma cells. Leukemia. 2016;30(11):2187–97. https://doi.org/10.1038/leu.2016.96.
Ohol YM, Sun MT, Cutler G, Leger PR, Hu DX, Biannic B, et al. Novel, selective inhibitors of USP7 uncover multiple mechanisms of antitumor activity in vitro and in vivo. Mol Cancer Ther. 2020;19(10):1970–80. https://doi.org/10.1158/1535-7163.MCT-20-0184.
Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM, et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature. 2010;463(7277):103–7. https://doi.org/10.1038/nature08646.
Wuilleme-Toumi S, Robillard N, Gomez P, Moreau P, Le Gouill S, Avet-Loiseau H, et al. Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia. 2005;19(7):1248–52. https://doi.org/10.1038/sj.leu.2403784.
Peterson LF, Sun H, Liu Y, Potu H, Kandarpa M, Ermann M, et al. Targeting deubiquitinase activity with a novel small-molecule inhibitor as therapy for B-cell malignancies. Blood. 2015;125(23):3588–97. https://doi.org/10.1182/blood-2014-10-605584.
Peng Z, Maxwell DS, Sun D, Bhanu Prasad BA, Schuber PT Jr, Pal A, et al. Degrasyn-like symmetrical compounds: possible therapeutic agents for multiple myeloma (MM-I). Bioorg Med Chem. 2014;22(4):1450–8. https://doi.org/10.1016/j.bmc.2013.12.048.
Shukla N, Somwar R, Smith RS, Ambati S, Munoz S, Merchant M, et al. Proteasome addiction defined in Ewing sarcoma is effectively targeted by a novel class of 19S proteasome inhibitors. Cancer Res. 2016;76(15):4525–34. https://doi.org/10.1158/0008-5472.CAN-16-1040.
Tian Z, D'Arcy P, Wang X, Ray A, Tai YT, Hu Y, et al. A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance. Blood. 2014;123(5):706–16. https://doi.org/10.1182/blood-2013-05-500033.
Xu X, Liu J, Shen C, Ding L, Zhong F, Ouyang Y, et al. The role of ubiquitin-specific protease 14 (USP14) in cell adhesion-mediated drug resistance (CAM-DR) of multiple myeloma cells. Eur J Haematol. 2017;98(1):4–12. https://doi.org/10.1111/ejh.12729.
Zhou L, Jiang H, Du J, Li L, Li R, Lu J, et al. USP15 inhibits multiple myeloma cell apoptosis through activating a feedback loop with the transcription factor NF-kappaBp65. Exp Mol Med. 2018;50(11):1–12. https://doi.org/10.1038/s12276-018-0180-4.
Hussain S, Bedekovics T, Chesi M, Bergsagel PL, Galardy PJ. UCHL1 is a biomarker of aggressive multiple myeloma required for disease progression. Oncotarget. 2015;6(38):40704–18. https://doi.org/10.18632/oncotarget.5727.
Otsuki T, Yata K, Takata-Tomokuni A, Hyodoh F, Miura Y, Sakaguchi H, et al. Expression of protein gene product 9.5 (PGP9.5)/ubiquitin-C-terminal hydrolase 1 (UCHL-1) in human myeloma cells. Br J Haematol. 2004;127(3):292–8. https://doi.org/10.1111/j.1365-2141.2004.05205.x.
Hussain S, Feldman AL, Das C, Ziesmer SC, Ansell SM, Galardy PJ. Ubiquitin hydrolase UCH-L1 destabilizes mTOR complex 1 by antagonizing DDB1-CUL4-mediated ubiquitination of raptor. Mol Cell Biol. 2013;33(6):1188–97. https://doi.org/10.1128/MCB.01389-12.
Spataro V, Toda T, Craig R, Seeger M, Dubiel W, Harris AL, et al. Resistance to diverse drugs and ultraviolet light conferred by overexpression of a novel human 26 S proteasome subunit. J Biol Chem. 1997;272(48):30470–5. https://doi.org/10.1074/jbc.272.48.30470.
Song Y, Li S, Ray A, Das DS, Qi J, Samur MK, et al. Blockade of deubiquitylating enzyme Rpn11 triggers apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Oncogene. 2017;36(40):5631–8. https://doi.org/10.1038/onc.2017.172.
Demchenko YN, Glebov OK, Zingone A, Keats JJ, Bergsagel PL, Kuehl WM. Classical and/or alternative NF-kappaB pathway activation in multiple myeloma. Blood. 2010;115(17):3541–52. https://doi.org/10.1182/blood-2009-09-243535.
Jenner MW, Leone PE, Walker BA, Ross FM, Johnson DC, Gonzalez D, et al. Gene mapping and expression analysis of 16q loss of heterozygosity identifies WWOX and CYLD as being important in determining clinical outcome in multiple myeloma. Blood. 2007;110(9):3291–300. https://doi.org/10.1182/blood-2007-02-075069.
van Andel H, Kocemba KA, de Haan-Kramer A, Mellink CH, Piwowar M, Broijl A, et al. Loss of CYLD expression unleashes Wnt signaling in multiple myeloma and is associated with aggressive disease. Oncogene. 2017;36(15):2105–15. https://doi.org/10.1038/onc.2016.368.
Ang E, Pavlos NJ, Rea SL, Qi M, Chai T, Walsh JP, et al. Proteasome inhibitors impair RANKL-induced NF-kappaB activity in osteoclast-like cells via disruption of p62, TRAF6, CYLD, and IkappaBalpha signaling cascades. J Cell Physiol. 2009;220(2):450–9. https://doi.org/10.1002/jcp.21787.
Troppan K, Hofer S, Wenzl K, Lassnig M, Pursche B, Steinbauer E, et al. Frequent down regulation of the tumor suppressor gene a20 in multiple myeloma. PLoS One. 2015;10(4):e0123922. https://doi.org/10.1371/journal.pone.0123922.
Liang Y, Xu RZ, Zhang L, Zhao XY. Berbamine, a novel nuclear factor kappaB inhibitor, inhibits growth and induces apoptosis in human myeloma cells. Acta Pharmacol Sin. 2009;30(12):1659–65. https://doi.org/10.1038/aps.2009.167.
Connors JM, Cozen W, Steidl C, Carbone A, Hoppe RT, Flechtner HH, et al. Hodgkin lymphoma. Nat Rev Dis Primers. 2020;6(1):61. https://doi.org/10.1038/s41572-020-0189-6.
Shanbhag S, Ambinder RF. Hodgkin lymphoma: a review and update on recent progress. CA Cancer J Clin. 2018;68(2):116–32. https://doi.org/10.3322/caac.21438.
Ahn H, Yang JM, Jeon YK, Paik JH. Clinicopathologic implications of TNFAIP3/A20 deletions in extranodal NK/T-cell lymphoma. Genes Chromosomes Cancer. 2018;57(5):231–9. https://doi.org/10.1002/gcc.22524.
Bargou RC, Leng C, Krappmann D, Emmerich F, Mapara MY, Bommert K, et al. High-level nuclear NF-kappa B and Oct-2 is a common feature of cultured Hodgkin/reed-Sternberg cells. Blood. 1996;87(10):4340–7.
Schmitz R, Hansmann ML, Bohle V, Martin-Subero JI, Hartmann S, Mechtersheimer G, et al. TNFAIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J Exp Med. 2009;206(5):981–9. https://doi.org/10.1084/jem.20090528.
Nomoto J, Hiramoto N, Kato M, Sanada M, Maeshima AM, Taniguchi H, et al. Deletion of the TNFAIP3/A20 gene detected by FICTION analysis in classical Hodgkin lymphoma. BMC Cancer. 2012;12:457. https://doi.org/10.1186/1471-2407-12-457.
Song Z, Wei W, Xiao W, Al-Saleem ED, Nejati R, Chen L, et al. Essential role of the linear ubiquitin chain assembly complex and TAK1 kinase in A20 mutant Hodgkin lymphoma. Proc Natl Acad Sci U S A. 2020;117(46):28980–91. https://doi.org/10.1073/pnas.2014470117.
Feys T, Poppe B, De Preter K, Van Roy N, Verhasselt B, De Paepe P, et al. A detailed inventory of DNA copy number alterations in four commonly used Hodgkin's lymphoma cell lines. Haematologica. 2007;92(7):913–20. https://doi.org/10.3324/haematol.11073.
Schmidt A, Schmitz R, Giefing M, Martin-Subero JI, Gesk S, Vater I, et al. Rare occurrence of biallelic CYLD gene mutations in classical Hodgkin lymphoma. Genes Chromosomes Cancer. 2010;49(9):803–9. https://doi.org/10.1002/gcc.20789.
Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375–90. https://doi.org/10.1182/blood-2016-01-643569.
de Leval L, Jaffe ES. Lymphoma classification. Cancer J. 2020;26(3):176–85. https://doi.org/10.1097/PPO.0000000000000451.
Shankland KR, Armitage JO, Hancock BW. Non-Hodgkin lymphoma. Lancet. 2012;380(9844):848–57. https://doi.org/10.1016/S0140-6736(12)60605-9.
Al-Tourah AJ, Gill KK, Chhanabhai M, Hoskins PJ, Klasa RJ, Savage KJ, et al. Population-based analysis of incidence and outcome of transformed non-Hodgkin's lymphoma. J Clin Oncol. 2008;26(32):5165–9. https://doi.org/10.1200/JCO.2008.16.0283.
Coiffier B, Lepage E, Briere J, Herbrecht R, Tilly H, Bouabdallah R, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med. 2002;346(4):235–42. https://doi.org/10.1056/NEJMoa011795.
Chiappella A, Martelli M, Angelucci E, Brusamolino E, Evangelista A, Carella AM, et al. Rituximab-dose-dense chemotherapy with or without high-dose chemotherapy plus autologous stem-cell transplantation in high-risk diffuse large B-cell lymphoma (DLCL04): final results of a multicentre, open-label, randomised, controlled, phase 3 study. Lancet Oncol. 2017;18(8):1076–88. https://doi.org/10.1016/S1470-2045(17)30444-8.
Davis MI, Pragani R, Fox JT, Shen M, Parmar K, Gaudiano EF, et al. Small molecule inhibition of the ubiquitin-specific protease USP2 accelerates cyclin D1 degradation and leads to cell cycle arrest in colorectal Cancer and mantle cell lymphoma models. J Biol Chem. 2016;291(47):24628–40. https://doi.org/10.1074/jbc.M116.738567.
Wei T, Biskup E, Gjerdrum LM, Niazi O, Odum N, Gniadecki R. Ubiquitin-specific protease 2 decreases p53-dependent apoptosis in cutaneous T-cell lymphoma. Oncotarget. 2016;7(30):48391–400. https://doi.org/10.18632/oncotarget.10268.
Engel K, Rudelius M, Slawska J, Jacobs L, Ahangarian Abhari B, Altmann B, et al. USP9X stabilizes XIAP to regulate mitotic cell death and chemoresistance in aggressive B-cell lymphoma. EMBO Mol Med. 2016;8(8):851–62. https://doi.org/10.15252/emmm.201506047.
Delforoush M, Berglund M, Edqvist PH, Sundstrom C, Gullbo J, Enblad G. Expression of possible targets for new proteasome inhibitors in diffuse large B-cell lymphoma. Eur J Haematol. 2017;98(1):52–6. https://doi.org/10.1111/ejh.12784.
Jiang L, Sun Y, Wang J, He Q, Chen X, Lan X, et al. Proteasomal cysteine deubiquitinase inhibitor b-AP15 suppresses migration and induces apoptosis in diffuse large B cell lymphoma. J Exp Clin Cancer Res. 2019;38(1):453. https://doi.org/10.1186/s13046-019-1446-y.
Ma YS, Wang XF, Zhang YJ, Luo P, Long HD, Li L, et al. Inhibition of USP14 Deubiquitinating activity as a potential therapy for tumors with p53 deficiency. Mol Ther Oncolytics. 2020;16:147–57. https://doi.org/10.1016/j.omto.2019.12.013.
Ma H, Luo X, Zhou P, He N, Zhou J, Liu M, et al. USP21 promotes cell proliferation by maintaining the EZH2 level in diffuse large B-cell lymphoma. J Clin Lab Anal. 2021;35(3):e23693. https://doi.org/10.1002/jcla.23693.
Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016;22(2):128–34. https://doi.org/10.1038/nm.4036.
Li B, Yan J, Phyu T, Fan S, Chung TH, Mustafa N, et al. MELK mediates the stability of EZH2 through site-specific phosphorylation in extranodal natural killer/T-cell lymphoma. Blood. 2019;134(23):2046–58. https://doi.org/10.1182/blood.2019000381.
Li C, Huang L, Lu H, Wang W, Chen G, Gu Y, et al. Expression and clinical significance of ubiquitinspecificprocessing protease 34 in diffuse large Bcell lymphoma. Mol Med Rep. 2018;18(5):4543–54. https://doi.org/10.3892/mmr.2018.9447.
Kwiecinska A, Ichimura K, Berglund M, Dinets A, Sulaiman L, Collins VP, et al. Amplification of 2p as a genomic marker for transformation in lymphoma. Genes Chromosomes Cancer. 2014;53(9):750–68. https://doi.org/10.1002/gcc.22184.
Bedekovics T, Hussain S, Feldman AL, Galardy PJ. UCH-L1 is induced in germinal center B cells and identifies patients with aggressive germinal center diffuse large B-cell lymphoma. Blood. 2016;127(12):1564–74. https://doi.org/10.1182/blood-2015-07-656678.
Rolen U, Freda E, Xie J, Pfirrmann T, Frisan T, Masucci MG. The ubiquitin C-terminal hydrolase UCH-L1 regulates B-cell proliferation and integrin activation. J Cell Mol Med. 2009;13(8B):1666–78. https://doi.org/10.1111/j.1582-4934.2008.00501.x.
Hussain S, Foreman O, Perkins SL, Witzig TE, Miles RR, van Deursen J, et al. The de-ubiquitinase UCH-L1 is an oncogene that drives the development of lymphoma in vivo by deregulating PHLPP1 and Akt signaling. Leukemia. 2010;24(9):1641–55. https://doi.org/10.1038/leu.2010.138.
Hussain S, Bedekovics T, Liu Q, Hu W, Jeon H, Johnson SH, et al. UCH-L1 bypasses mTOR to promote protein biosynthesis and is required for MYC-driven lymphomagenesis in mice. Blood. 2018;132(24):2564–74. https://doi.org/10.1182/blood-2018-05-848515.
Cope GA, Deshaies RJ. Targeted silencing of Jab1/Csn5 in human cells downregulates SCF activity through reduction of F-box protein levels. BMC Biochem. 2006;7:1. https://doi.org/10.1186/1471-2091-7-1.
Lee YH, Judge AD, Seo D, Kitade M, Gomez-Quiroz LE, Ishikawa T, et al. Molecular targeting of CSN5 in human hepatocellular carcinoma: a mechanism of therapeutic response. Oncogene. 2011;30(40):4175–84. https://doi.org/10.1038/onc.2011.126.
Zhong G, Li H, Shan T, Zhang N. CSN5 silencing inhibits invasion and arrests cell cycle progression in human colorectal cancer SW480 and LS174T cells in vitro. Int J Clin Exp Pathol. 2015;8(3):2809–15.
Schlierf A, Altmann E, Quancard J, Jefferson AB, Assenberg R, Renatus M, et al. Targeted inhibition of the COP9 signalosome for treatment of cancer. Nat Commun. 2016;7:13166. https://doi.org/10.1038/ncomms13166.
Pulvino M, Chen L, Oleksyn D, Li J, Compitello G, Rossi R, et al. Inhibition of COP9-signalosome (CSN) deneddylating activity and tumor growth of diffuse large B-cell lymphomas by doxycycline. Oncotarget. 2015;6(17):14796–813. https://doi.org/10.18632/oncotarget.4193.
Kato M, Sanada M, Kato I, Sato Y, Takita J, Takeuchi K, et al. Frequent inactivation of A20 in B-cell lymphomas. Nature. 2009;459(7247):712–6. https://doi.org/10.1038/nature07969.
Chanudet E, Huang Y, Ichimura K, Dong G, Hamoudi RA, Radford J, et al. A20 is targeted by promoter methylation, deletion and inactivating mutation in MALT lymphoma. Leukemia. 2010;24(2):483–7. https://doi.org/10.1038/leu.2009.234.
Ando M, Sato Y, Takata K, Nomoto J, Nakamura S, Ohshima K, et al. A20 (TNFAIP3) deletion in Epstein-Barr virus-associated lymphoproliferative disorders/lymphomas. PLoS One. 2013;8(2):e56741. https://doi.org/10.1371/journal.pone.0056741.
Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature. 2009;459(7247):717–21. https://doi.org/10.1038/nature07968.
Yang W, Li Y, Li P, Wang L. PMA/IONO affects diffuse large B-cell lymphoma cell growth through upregulation of A20 expression. Oncol Rep. 2016;36(2):1069–75. https://doi.org/10.3892/or.2016.4899.
Duwel M, Welteke V, Oeckinghaus A, Baens M, Kloo B, Ferch U, et al. A20 negatively regulates T cell receptor signaling to NF-kappaB by cleaving Malt1 ubiquitin chains. J Immunol. 2009;182(12):7718–28. https://doi.org/10.4049/jimmunol.0803313.
Lim SM, Jeong Y, Lee S, Im H, Tae HS, Kim BG, et al. Identification of beta-Lapachone analogs as novel MALT1 inhibitors to treat an aggressive subtype of diffuse large B-cell lymphoma. J Med Chem. 2015;58(21):8491–502. https://doi.org/10.1021/acs.jmedchem.5b01415.
Xu X, Kalac M, Markson M, Chan M, Brody JD, Bhagat G, et al. Reversal of CYLD phosphorylation as a novel therapeutic approach for adult T-cell leukemia/lymphoma (ATLL). Cell Death Dis. 2020;11(2):94. https://doi.org/10.1038/s41419-020-2294-6.
Xu X, Wei T, Zhong W, Ang R, Lei Y, Zhang H, et al. Down-regulation of cylindromatosis protein phosphorylation by BTK inhibitor promotes apoptosis of non-GCB-diffuse large B-cell lymphoma. Cancer Cell Int. 2021;21(1):195. https://doi.org/10.1186/s12935-021-01891-2.
Schauer NJ, Magin RS, Liu X, Doherty LM, Buhrlage SJ. Advances in discovering Deubiquitinating enzyme (DUB) inhibitors. J Med Chem. 2020;63(6):2731–50. https://doi.org/10.1021/acs.jmedchem.9b01138.
Altun M, Kramer HB, Willems LI, McDermott JL, Leach CA, Goldenberg SJ, et al. Activity-based chemical proteomics accelerates inhibitor development for deubiquitylating enzymes. Chem Biol. 2011;18(11):1401–12. https://doi.org/10.1016/j.chembiol.2011.08.018.
Kuang X, Xiong J, Lu T, Wang W, Zhang Z, Wang J. Inhibition of USP1 induces apoptosis via ID1/AKT pathway in B-cell acute lymphoblastic leukemia cells. Int J Med Sci. 2021;18(1):245–55. https://doi.org/10.7150/ijms.47597.
Liang Q, Dexheimer TS, Zhang P, Rosenthal AS, Villamil MA, You C, et al. A selective USP1-UAF1 inhibitor links deubiquitination to DNA damage responses. Nat Chem Biol. 2014;10(4):298–304. https://doi.org/10.1038/nchembio.1455.
Antonenko SV, Telegeev GD. Inhibition of USP1, a new partner of Bcr-Abl, results in decrease of Bcr-Abl level in K562 cells. Exp Oncol. 2020;42(2):109–14. https://doi.org/10.32471/exp-oncology.2312-8852.vol-42-no-2.14533.
Ohayon S, Refua M, Hendler A, Aharoni A, Brik A. Harnessing the oxidation susceptibility of deubiquitinases for inhibition with small molecules. Angew Chem Int Ed Engl. 2015;54(2):599–603. https://doi.org/10.1002/anie.201408411.
Magiera K, Tomala M, Kubica K, De Cesare V, Trost M, Zieba BJ, et al. Lithocholic acid Hydroxyamide destabilizes Cyclin D1 and induces G0/G1 arrest by inhibiting Deubiquitinase USP2a. Cell Chem Biol. 2017;24(4):458–70 e18. https://doi.org/10.1016/j.chembiol.2017.03.002.
Tomala MD, Magiera-Mularz K, Kubica K, Krzanik S, Zieba B, Musielak B, et al. Identification of small-molecule inhibitors of USP2a. Eur J Med Chem. 2018;150:261–7. https://doi.org/10.1016/j.ejmech.2018.03.009.
Chuang SJ, Cheng SC, Tang HC, Sun CY, Chou CY. 6-Thioguanine is a noncompetitive and slow binding inhibitor of human deubiquitinating protease USP2. Sci Rep. 2018;8(1):3102. https://doi.org/10.1038/s41598-018-21476-w.
Okada K, Ye YQ, Taniguchi K, Yoshida A, Akiyama T, Yoshioka Y, et al. Vialinin a is a ubiquitin-specific peptidase inhibitor. Bioorg Med Chem Lett. 2013;23(15):4328–31. https://doi.org/10.1016/j.bmcl.2013.05.093.
Weinstock J, Wu J, Cao P, Kingsbury WD, McDermott JL, Kodrasov MP, et al. Selective dual inhibitors of the Cancer-related Deubiquitylating proteases USP7 and USP47. ACS Med Chem Lett. 2012;3(10):789–92. https://doi.org/10.1021/ml200276j.
Deng J, Liang L, Yi H, Su T, Yang Z, Nie L, et al. USP7 inhibition inhibits proliferation and induces megakaryocytic differentiation in MDS cells by upregulating gelsolin. Br J Haematol. 2020;190(3):418–29. https://doi.org/10.1111/bjh.16549.
Fan YH, Cheng J, Vasudevan SA, Dou J, Zhang H, Patel RH, et al. USP7 inhibitor P22077 inhibits neuroblastoma growth via inducing p53-mediated apoptosis. Cell Death Dis. 2013;4:e867. https://doi.org/10.1038/cddis.2013.400.
Colland F, Formstecher E, Jacq X, Reverdy C, Planquette C, Conrath S, et al. Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol Cancer Ther. 2009;8(8):2286–95. https://doi.org/10.1158/1535-7163.MCT-09-0097.
Lamberto I, Liu X, Seo HS, Schauer NJ, Iacob RE, Hu W, et al. Structure-guided development of a potent and selective non-covalent active-site inhibitor of USP7. Cell Chem Biol. 2017;24(12):1490–500 e11. https://doi.org/10.1016/j.chembiol.2017.09.003.
Turnbull AP, Ioannidis S, Krajewski WW, Pinto-Fernandez A, Heride C, Martin ACL, et al. Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature. 2017;550(7677):481–6. https://doi.org/10.1038/nature24451.
Wang F, Wang L, Wu J, Sokirniy I, Nguyen P, Bregnard T, et al. Active site-targeted covalent irreversible inhibitors of USP7 impair the functions of Foxp3+ T-regulatory cells by promoting ubiquitination of Tip60. PLoS One. 2017;12(12):e0189744. https://doi.org/10.1371/journal.pone.0189744.
Schauer NJ, Liu X, Magin RS, Doherty LM, Chan WC, Ficarro SB, et al. Selective USP7 inhibition elicits cancer cell killing through a p53-dependent mechanism. Sci Rep. 2020;10(1):5324. https://doi.org/10.1038/s41598-020-62076-x.
Colombo M, Vallese S, Peretto I, Jacq X, Rain JC, Colland F, et al. Synthesis and biological evaluation of 9-oxo-9H-indeno [1,2-b]pyrazine-2,3-dicarbonitrile analogues as potential inhibitors of deubiquitinating enzymes. ChemMedChem. 2010;5(4):552–8. https://doi.org/10.1002/cmdc.200900409.
Reverdy C, Conrath S, Lopez R, Planquette C, Atmanene C, Collura V, et al. Discovery of specific inhibitors of human USP7/HAUSP deubiquitinating enzyme. Chem Biol. 2012;19(4):467–77. https://doi.org/10.1016/j.chembiol.2012.02.007.
Bartholomeusz G, Talpaz M, Bornmann W, Kong LY, Donato NJ. Degrasyn activates proteasomal-dependent degradation of c-Myc. Cancer Res. 2007;67(8):3912–8. https://doi.org/10.1158/0008-5472.CAN-06-4464.
Pham LV, Tamayo AT, Li C, Bornmann W, Priebe W, Ford RJ. Degrasyn potentiates the antitumor effects of bortezomib in mantle cell lymphoma cells in vitro and in vivo: therapeutic implications. Mol Cancer Ther. 2010;9(7):2026–36. https://doi.org/10.1158/1535-7163.MCT-10-0238.
Zhou B, Jin X, Jin W, Huang X, Wu Y, Li H, et al. WT1 facilitates the self-renewal of leukemia-initiating cells through the upregulation of BCL2L2: WT1-BCL2L2 axis as a new acute myeloid leukemia therapy target. J Transl Med. 2020;18(1):254. https://doi.org/10.1186/s12967-020-02384-y.
Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011;147(1):223–34. https://doi.org/10.1016/j.cell.2011.08.037.
Shao S, Li S, Qin Y, Wang X, Yang Y, Bai H, et al. Spautin-1, a novel autophagy inhibitor, enhances imatinib-induced apoptosis in chronic myeloid leukemia. Int J Oncol. 2014;44(5):1661–8. https://doi.org/10.3892/ijo.2014.2313.
Xu W, Huang Z, Gan Y, Chen R, Huang Y, Xue B, et al. Casein kinase 1alpha inhibits p53 downstream of MDM2mediated autophagy and apoptosis in acute myeloid leukemia. Oncol Rep. 2020;44(5):1895–904. https://doi.org/10.3892/or.2020.7760.
Burkhart RA, Peng Y, Norris ZA, Tholey RM, Talbott VA, Liang Q, et al. Mitoxantrone targets human ubiquitin-specific peptidase 11 (USP11) and is a potent inhibitor of pancreatic cancer cell survival. Mol Cancer Res. 2013;11(8):901–11. https://doi.org/10.1158/1541-7786.MCR-12-0699.
Cahill KE, Karimi YH, Karrison TG, Jain N, Green M, Weiner H, et al. A phase 1 study of azacitidine with high-dose cytarabine and mitoxantrone in high-risk acute myeloid leukemia. Blood Adv. 2020;4(4):599–606. https://doi.org/10.1182/bloodadvances.2019000795.
Wang WS, Tzeng CH, Chiou TJ, Liu JH, Hsieh RK, Yen CC, et al. High-dose cytarabine and mitoxantrone as salvage therapy for refractory non-Hodgkin's lymphoma. Jpn J Clin Oncol. 1997;27(3):154–7. https://doi.org/10.1093/jjco/27.3.154.
Feng X, Holmlund T, Zheng C, Fadeel B. Proapoptotic effects of the novel proteasome inhibitor b-AP15 on multiple myeloma cells and natural killer cells. Exp Hematol. 2014;42(3):172–82. https://doi.org/10.1016/j.exphem.2013.11.010.
Kropp KN, Maurer S, Rothfelder K, Schmied BJ, Clar KL, Schmidt M, et al. The novel deubiquitinase inhibitor b-AP15 induces direct and NK cell-mediated antitumor effects in human mantle cell lymphoma. Cancer Immunol Immunother. 2018;67(6):935–47. https://doi.org/10.1007/s00262-018-2151-y.
Wang X, Mazurkiewicz M, Hillert EK, Olofsson MH, Pierrou S, Hillertz P, et al. The proteasome deubiquitinase inhibitor VLX1570 shows selectivity for ubiquitin-specific protease-14 and induces apoptosis of multiple myeloma cells. Sci Rep. 2016;6:26979. https://doi.org/10.1038/srep26979.
Rowinsky EK, Paner A, Berdeja JG, Paba-Prada C, Venugopal P, Porkka K, et al. Phase 1 study of the protein deubiquitinase inhibitor VLX1570 in patients with relapsed and/or refractory multiple myeloma. Investig New Drugs. 2020;38(5):1448–53. https://doi.org/10.1007/s10637-020-00915-4.
Pellegrini P, Selvaraju K, Faustini E, Mofers A, Zhang X, Ternerot J, et al. Induction of ER Stress in Acute Lymphoblastic Leukemia Cells by the Deubiquitinase Inhibitor VLX1570. Int J Mol Sci. 2020;21(13). https://doi.org/10.3390/ijms21134757.
Liu N, Li X, Huang H, Zhao C, Liao S, Yang C, et al. Clinically used antirheumatic agent auranofin is a proteasomal deubiquitinase inhibitor and inhibits tumor growth. Oncotarget. 2014;5(14):5453–71. https://doi.org/10.18632/oncotarget.2113.
Tessoulin B, Descamps G, Dousset C, Amiot M, Pellat-Deceunynck C. Targeting oxidative stress with Auranofin or Prima-1(met) to circumvent p53 or Bax/Bak deficiency in myeloma cells. Front Oncol. 2019;9:128. https://doi.org/10.3389/fonc.2019.00128.
Nakaya A, Sagawa M, Muto A, Uchida H, Ikeda Y, Kizaki M. The gold compound auranofin induces apoptosis of human multiple myeloma cells through both down-regulation of STAT3 and inhibition of NF-kappaB activity. Leuk Res. 2011;35(2):243–9. https://doi.org/10.1016/j.leukres.2010.05.011.
Fidyt K, Pastorczak A, Goral A, Szczygiel K, Fendler W, Muchowicz A, et al. Targeting the thioredoxin system as a novel strategy against B-cell acute lymphoblastic leukemia. Mol Oncol. 2019;13(5):1180–95. https://doi.org/10.1002/1878-0261.12476.
Fiskus W, Saba N, Shen M, Ghias M, Liu J, Gupta SD, et al. Auranofin induces lethal oxidative and endoplasmic reticulum stress and exerts potent preclinical activity against chronic lymphocytic leukemia. Cancer Res. 2014;74(9):2520–32. https://doi.org/10.1158/0008-5472.CAN-13-2033.
Chen X, Shi X, Zhao C, Li X, Lan X, Liu S, et al. Anti-rheumatic agent auranofin induced apoptosis in chronic myeloid leukemia cells resistant to imatinib through both Bcr/Abl-dependent and -independent mechanisms. Oncotarget. 2014;5(19):9118–32. https://doi.org/10.18632/oncotarget.2361.
Kim IS, Jin JY, Lee IH, Park SJ. Auranofin induces apoptosis and when combined with retinoic acid enhances differentiation of acute promyelocytic leukaemia cells in vitro. Br J Pharmacol. 2004;142(4):749–55. https://doi.org/10.1038/sj.bjp.0705708.
Wang J, Wang J, Lopez E, Guo H, Zhang H, Liu Y, et al. Repurposing auranofin to treat TP53-mutated or PTEN-deleted refractory B-cell lymphoma. Blood Cancer J. 2019;9(12):95. https://doi.org/10.1038/s41408-019-0259-8.
Zhang H, Rose BJ, Pyuen AA, Thamm DH. In vitro antineoplastic effects of auranofin in canine lymphoma cells. BMC Cancer. 2018;18(1):522. https://doi.org/10.1186/s12885-018-4450-2.
Lee BH, Lee MJ, Park S, Oh DC, Elsasser S, Chen PC, et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature. 2010;467(7312):179–84. https://doi.org/10.1038/nature09299.
Wang Y, Jiang Y, Ding S, Li J, Song N, Ren Y, et al. Small molecule inhibitors reveal allosteric regulation of USP14 via steric blockade. Cell Res. 2018;28(12):1186–94. https://doi.org/10.1038/s41422-018-0091-x.
Coughlin K, Anchoori R, Iizuka Y, Meints J, MacNeill L, Vogel RI, et al. Small-molecule RA-9 inhibits proteasome-associated DUBs and ovarian cancer in vitro and in vivo via exacerbating unfolded protein responses. Clin Cancer Res. 2014;20(12):3174–86. https://doi.org/10.1158/1078-0432.CCR-13-2658.
Kemp M. Recent advances in the discovery of Deubiquitinating enzyme inhibitors. Prog Med Chem. 2016;55:149–92. https://doi.org/10.1016/bs.pmch.2015.10.002.
Wang SA, Young MJ, Wang YC, Chen SH, Liu CY, Lo YA, et al. USP24 promotes drug resistance during cancer therapy. Cell Death Differ. 2021. https://doi.org/10.1038/s41418-021-00778-z.
Wrigley JD, Gavory G, Simpson I, Preston M, Plant H, Bradley J, et al. Identification and characterization of dual inhibitors of the USP25/28 Deubiquitinating enzyme subfamily. ACS Chem Biol. 2017;12(12):3113–25. https://doi.org/10.1021/acschembio.7b00334.
Yue W, Chen Z, Liu H, Yan C, Chen M, Feng D, et al. A small natural molecule promotes mitochondrial fusion through inhibition of the deubiquitinase USP30. Cell Res. 2014;24(4):482–96. https://doi.org/10.1038/cr.2014.20.
Kluge AF, Lagu BR, Maiti P, Jaleel M, Webb M, Malhotra J, et al. Novel highly selective inhibitors of ubiquitin specific protease 30 (USP30) accelerate mitophagy. Bioorg Med Chem Lett. 2018;28(15):2655–9. https://doi.org/10.1016/j.bmcl.2018.05.013.
Liu Y, Lashuel HA, Choi S, Xing X, Case A, Ni J, et al. Discovery of inhibitors that elucidate the role of UCH-L1 activity in the H1299 lung cancer cell line. Chem Biol. 2003;10(9):837–46. https://doi.org/10.1016/j.chembiol.2003.08.010.
Mermerian AH, Case A, Stein RL, Cuny GD. Structure-activity relationship, kinetic mechanism, and selectivity for a new class of ubiquitin C-terminal hydrolase-L1 (UCH-L1) inhibitors. Bioorg Med Chem Lett. 2007;17(13):3729–32. https://doi.org/10.1016/j.bmcl.2007.04.027.
Davies CW, Chaney J, Korbel G, Ringe D, Petsko GA, Ploegh H, et al. The co-crystal structure of ubiquitin carboxy-terminal hydrolase L1 (UCHL1) with a tripeptide fluoromethyl ketone (Z-VAE (OMe)-FMK). Bioorg Med Chem Lett. 2012;22(12):3900–4. https://doi.org/10.1016/j.bmcl.2012.04.124.
Mtango NR, Sutovsky M, Vandevoort CA, Latham KE, Sutovsky P. Essential role of ubiquitin C-terminal hydrolases UCHL1 and UCHL3 in mammalian oocyte maturation. J Cell Physiol. 2012;227(5):2022–9. https://doi.org/10.1002/jcp.22931.
Shi T, Bao J, Wang NX, Zheng J, Wu D. Identification of small molecule TRABID Deubiquitinase inhibitors by computation-based virtual screen. BMC Chem Biol. 2012;12:4. https://doi.org/10.1186/1472-6769-12-4.
Bednash JS, Weathington N, Londino J, Rojas M, Gulick DL, Fort R, et al. Targeting the deubiquitinase STAMBP inhibits NALP7 inflammasome activity. Nat Commun. 2017;8:15203. https://doi.org/10.1038/ncomms15203.
Li J, Yakushi T, Parlati F, Mackinnon AL, Perez C, Ma Y, et al. Capzimin is a potent and specific inhibitor of proteasome isopeptidase Rpn11. Nat Chem Biol. 2017;13(5):486–93. https://doi.org/10.1038/nchembio.2326.
Lauinger L, Li J, Shostak A, Cemel IA, Ha N, Zhang Y, et al. Thiolutin is a zinc chelator that inhibits the Rpn11 and other JAMM metalloproteases. Nat Chem Biol. 2017;13(7):709–14. https://doi.org/10.1038/nchembio.2370.
Li J, Zhang Y, Da Silva Sil Dos Santos B, Wang F, Ma Y, Perez C, et al. Epidithiodiketopiperazines inhibit protein degradation by targeting proteasome Deubiquitinase Rpn11. Cell Chem Biol. 2018;25(11):1350–8 e9. https://doi.org/10.1016/j.chembiol.2018.07.012.
Acknowledgements
Not applicable.
Funding
This work was supported in part by grants from the National Key Research and Development Program of China (No. 2017YFA0505202), National Natural Science Foundation of China (81700475, 81570118).
Author information
Authors and Affiliations
Contributions
H. Lei conceived, written, illustrated, revised and submitted the review. J. Wang, J. Hu, and Q. Zhu collected some literatures and participating in part of the writing and revision. Y. Wu participated in the conception and revision of the review. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lei, H., Wang, J., Hu, J. et al. Deubiquitinases in hematological malignancies. Biomark Res 9, 66 (2021). https://doi.org/10.1186/s40364-021-00320-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-021-00320-w