
Abstract
N6-methyladenosine (m6A) modification is the most prevalent internal mRNA modification and is involved in many biological processes in eukaryotes. Accumulating evidence has demonstrated that m6A may play either a promoting or suppressing role in breast cancer, including in tumorigenesis, metastasis and angiogenesis. In this review, we summarize the latest research progress on the biological function and prognostic value of m6A modification in breast cancer, as well as potential related therapeutic strategies.
Similar content being viewed by others
Highlights
-
1.
N6-methyladenosine (m6A) modification can promote or suppress cell growth in breast cancer tissues and cells.
-
2.
m6A modification is associated with the clinicopathological features and prognosis of breast cancer.
-
3.
m6A modification is involved in drug efficacy and may be a potential selective therapeutic target in breast cancer.
Background
According to GLOBOCAN 2018, breast cancer is the most commonly diagnosed cancer and the leading cause of cancer death in females [1]. Recently, novel types of drugs have provided effective treatment strategies in the prevention and control of breast cancer, including immune checkpoint inhibitors for PD-1/PD-L1/TMB/CTLA4, PARP inhibitors for BRCA mutation and HRD, CDK4/6 inhibitors against CDK4/6, PI3Kα inhibitors and AKT inhibitors targeting the PI3K/AKT/PTEN pathway, and antibody-drug conjugates targeting HER2 [2,3,4,5,6]. In addition, great progress has been made in whole-exome sequencing, miRNA sequencing, and single nucleotide polymorphism, DNA methylation, and reverse-phase protein array analyses in recent years, which provide some new methods for the diagnosis and treatment of breast cancer [7,8,9,10]. However, global cancer statistics still demonstrate that there were 2,088,849 new cases and 626,679 cancer deaths worldwide in 2018 [1]. In addition, WHO CANCER TOMORROW predicted that approximately 817,361 females could die from breast cancer by 2030. That is, the prognosis of breast cancer remains concerning. Therefore, there is still an urgent need for effective therapies or regimens.
To enhance the efficacy of breast cancer treatment, it is necessary to understand the occurrence, development and molecular biological characteristics of breast cancer. In recent years, many studies have been conducted, but additional research is still required to determine the potential mechanism of oncogenesis and progression of breast cancer. RNA modification, especially N6-methyladenosine (m6A) modification, has provided a more effective method and new prospects in the treatment of breast cancer [11].
Emerging evidence suggests that m6A modification is associated with tumour proliferation, differentiation, tumorigenesis, invasion and metastasis and functions as either an oncogene or anti-oncogene in breast cancer [12,13,14,15]. Despite its functional significance in tumour generation processes, the critical roles of m6A modifications in breast cancer remains uncertain. This article briefly reviews the mechanism, potential therapeutic strategies and possible prognostic implications of m6A modification in breast cancer.
The molecular mechanisms of m6A modification
According to the MODOMICS database, 163 different RNA posttranscriptional modifications had been identified by the end of 2017 [16]. More importantly, over 60% of all RNA modifications are methylation modifications, and m6A is a highly abundant and conserved mRNA modification in mammals. Previous experiments have shown that m6A modification mainly occurs in the adenine of the RRACH sequence (the m6A consensus motif), and the average mRNA contains approximately 3–5 m6A modifications [17, 18].
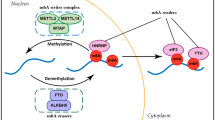
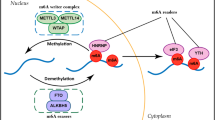
In breast cancer, m6A modification regulates RNA termination codons, the 5′ cap structure, and the 3′ untranslated terminal region (UTR), and in this way affects RNA transcription, RNA processing, RNA splicing, RNA degradation and RNA translation (Fig. 1) [19, 20]. In general, m6A modifiers comprise three vital components: writers, also termed methyltransferases; erasers, or demethylases, which remove m6A modifications; and readers, which recognize m6A-modified sites and further regulate m6A modification [20, 21]. To date, known writers are METTL3, METTL14, METTL16, and WTAP. Known erasers include only demethylases, including FTO, ALKBH5 and ALKBH3. Readers mainly includemembers of the YTH family, HNRNP family, FXR family, IGF2BP family, eIF family, and G3BPs.

The mechanism of action of m6A modification in breast cancer. m6A modification consists of three vital components: writers, also termed methyltransferases; erasers, or demethylases, which remove the m6A modification; and readers, which recognize m6A-modified sites and further regulate m6A modification. m6A modifications affect RNA transcription, RNA processing, RNA splicing, RNA degradation, RNA export and RNA translation
Previous studies have demonstrated that m6A modification indirectly/directly regulates target genes, thereby promoting or inhibiting tumour growth, proliferation, migration, invasion, and metastasis in breast cancers (Fig. 1, Table 1). Moreover, m6A modification patterns have therapeutic implications and correlate with drug resistance. In addition, m6A modification is associated with the clinicopathological features and prognosis of breast cancer (Table 2).
M6A modification modulates biological processes in breast cancer
Methyltransferases/writers in breast cancer
Methyltransferase-like 3 (METTL3) and methyltransferase-like 14 (METTL14) form a stable methyltransferase complex and play a key role in localization recognition [92,93,94]. MELLT3 acts as a catalyst in the cell nucleus and rapidly initiates the formation of the methyltransferase complex. MELLT14 prompts complex binding to relevant RNA sites and recognizes the substrate. In addition, METTL14 induces serine phosphorylation on METTL3.
Previous studies indicated that METTL3 is significantly highly expressed in breast tissues and cells [14, 17, 22, 23, 26, 95]. Hong Wang et al. showed that silencing METTL3 reduces methylation levels, represses proliferation and induces apoptosis by targeting Bcl-2 [23]. Hepatitis B X-interacting protein (HBXIP) upregulates METTL3 expression and then accelerates the proliferation of breast cancer cells by inhibiting the tumour suppressor let-7 g [22]. Similarly, silencing METTL3 inhibits cell growth and proliferation and accelerates cell apoptosis [22]. LINC00942 (LNC942), an oncogene, promotes METTL14 expression and induces cell proliferation and growth by targeting CXCR4 and CYP1B1 [28]. In addition, METTL14 affects hsa-miR-146a-5p expression and promotes migration and invasion but has little effect on cell proliferation [29].
In contrast to previous studies, Wu et al. revealed that METTL3 and METTL14 expressions are lower in breast tissues than in normal tissues [26]. Interestingly, however, the expressions of METTL3 and METTL14 were still higher in normal and luminal types of breast cancer. In addition, overexpression of METTL14 and/or knockdown of ALKBH5 remarkably suppressed cell migratory abilities [26]. A recent study of triple-negative breast cancer (TNBC) also found that METTL3 expression is lower in breast cancer cells. METTL3 inhibits the metastasis of TNBC cells by downregulating the expression of COL3A1 [24]. Nate J. Fry et al. demonstrated that tumour cell proliferation and migration are stimulated by upregulating the expression of METTL3 and METTL14 or downregulating the expression of ALKBH5 [30]. Another study indicated that the expression of METTL14 and METTL16 was not significantly enhanced in MDA-MB-231, MDA-MB-468 and MCF-7 cell lines [31].
Wilms tumour 1-associating protein (WTAP) regulates G2/M cell cycle transition by binding to the 3′ UTR of CCNA2 and promotes cell growth and migration by combining with METTL3/METTL14 [26]. Keratinocyte growth factor enhances the expression of WTAP and thereby increases the proliferation of breast cells [27]. However, another study showed that WTAP expression was not significantly different in 20 breast cancer tissue samples compared with paired normal tissues [26].
KIAA1429, also called VIRMA, is mainly expressed in the cytoplasm in most human breast cancer cell lines and regulates m6A methylation modification as an RNA binding protein. Studies have shown that KIAA1429 is highly expressed in breast cancer tissues and expressed at low levels in pericarcinous tissues [31, 32]. In the MCF-7, MDAMB-231 and SUM1315 cell lines, KIAA1429 facilitates cell proliferation, colony formation, migration, invasion, and metastasis by regulating the expression of CDK1 [32]. KIAA1429 overexpression predicts poor overall survival in breast cancer patients [31]. Another study showed that KIAA1429 is expressed at lower levels in HER2-positive breast cancer [26].
RBM15/RBM15B, RNA binding motif protein 15/15B, promotes the methylation of RNAs by binding to uracil-enriched regions [25]. RBM15, as an oestrogen-responsive gene, regulates cell growth in MCF7 and T47D cell lines [25]. Further research found that BARX2 and oestrogen receptor-a (ESR1) co-ordinately modulate cell growth and cell invasion by affecting the expression of RBM15 [25]. It is well known that RBM15 impacts diseases, and how RBM15 is regulated is an important question; thus far, nothing is known about this issue. RBM15B, located in close proximity to the 3p21 tumour suppressor region, is highly positively correlated with BRCA1-associated protein-1 expression in both black and white female patients with invasive breast carcinoma [96].
Eun-Yeung Gong et al. showed that CBLL1, also named HAIKI or RNF188, is mainly expressed in the cytoplasm in most human breast cancer cell lines [33]. CBLL1, as an E3 ubiquitin protein ligase, competes with ERa coactivators and plays a negative role in the development and progression of breast cancers [34]. A study published in 2009 that enrolled 22 samples found that CBLL1 expression has no correlation with invasive ductal breast carcinomas and normal adjacent tissue [35]. However, CBLL1 mRNA expression was higher (samples 83.3% ER positive) in these breast cancers than in adjacent tissues.
Demethylases/erasers in breast cancer
FTO, as a member of the AlkB family, is the first identified m6A demethylase/eraser, which maps to chromosome 16q12.2 and is highly expressed in most breast cancers [36,37,38, 88, 89]. For MDA-MB-231, MCF-7 and 4 T1 breast cell lines, upregulated FTO promotes tumour proliferation and metastasis or reduces cell apoptosis by targeting BNIP3. Furthermore, the article highlighted that silencing FTO can inhibit the growth of breast tumours in vivo [88]. In HER2-positive breast cancer, FTO also reinforces cell invasion and migration in vitro through the FTO/miR-181b-3p/ARL5B signalling pathway [89]. Recently, one study also illustrated that FTO expression may play an essential role in the development and aggressiveness of breast cancer, especially HER2-overexpressing breast cancer [37]. One researcher showed that FTO expression is low in HER2-positive breast cancer [26].
ALKBH5, another member of the AlkB family, is highly expressed in most breast cancers [26]. ALKBH5 may function as an m6A reader in the cytoplasm and cytosol and promote the translation of its target mRNA [14, 95]. Recently, it was reported that ALKBH5 functions as a tumour promoter in the pathogenesis of breast cancer [39, 95]. ALKBH5 plays a crucial role in tumorigenicity and lung metastasis. Furthermore, under anaerobic conditions, HIF1α and HIF2α significantly increase ALKBH5 mRNA expression [40, 97]. Moreover, knockdown of ALKBH5 expression significantly inhibits tumour formation and decreases breast cancer stem cell number in breast tumours [40]. In addition, ALKBH5, as an oncogene, regulates the self-renewal and proliferation of breast cancer tumour stem cells.
Readers in breast cancer
The YTH family
Previous studies found that members of the YTH family play a crucial role in RNA stability, RNA splicing, RNA translation, and RNA processing in cancers [17, 31, 74, 77, 88, 98, 99]. The YTH family mainly includes the following members: YTHDF1, YTHDF2, YTHDF3, YTHDC1, and YTHDC2. YTHDF2 is the first reader to be associated with m6A modification and mediate mRNA processing [74, 77]. In the cytosol, YTHDF2 regulates mRNA stability and degradation via interaction with CCR4-NOT.
YTHDF1 and YTHDF3 can promote RNA translation in HeLa cells. In the nucleus, YTHDC1 regulates mRNA splicing, mRNA export, RNA degradation and partly RNA transcripts by recruiting certain binding sites [17, 31, 74, 77, 88, 99]. In breast cell lines, YTHDF1 and YTHDF2 are mainly located in the cytoplasm, while YTHDC1 is mainly located in the nucleus [31]. In addition, YTHDF1, YTHDF2 and YTHDF3 are highly expressed in breast cancer and enhance cell proliferation, migration and invasion [31, 75, 98]. Notably, YTHDF3 expression was higher in brain metastases than in parental cells [75]. Furthermore, knockdown of YTHDF3 significantly decreased brain metastasis in mouse models. YTHDF3 copy number gain is higher in breast cancer with brain metastases than in primary breast tumours. Interestingly, in a separate study, miR-106b-5p gene transfection downregulated the expression of YTHDF3, which enhanced cell proliferation, migration and invasion of oestrogen-induced breast cancer [99]. Furthermore, in cells transfected with mimics, researchers detected that miR-106b-5p regulates YTHDF3 expression by suppressing translation or inducing mRNA degradation.
The HNRNP family
HNRNP family members, including HNRNPC, HNRNPG and HNRNPA2B1, also modulate RNA stability, RNA splicing, RNA translation, and RNA processing. HNRNPC, a splicing factor, is highly expressed in breast cancer and promotes tumour cell proliferation and growth [68, 76]. Moreover, HNRNPC is involved in aberrant splicing and implicated in the formation of the tumour transcriptome [69]. In MCF7 and T47D, but not MCF10A, BT549, and MDA-MB-231 cells, even partial suppression of HNRNPC function can result in a type I interferon response, an increase in endogenous dsRNA, and upregulation of ISG expression via activation of the ISGF3 complex. Knockdown of HNRNPC inhibits cell proliferation and tumour growth of these breast cancer cells [68]. Another study found that HNRNPC exhibits considerably higher expression in LvBr2 than MDA-MB-435 cells [68, 70]. In LvBr2 cells, miR-146a inhibits the activation of Akt by downregulating HNRNPC and thereby suppresses tumour migration and invasion.
HNRNPA2B1, another splicing factor of breast cancer, is also significantly upregulated in human breast cancer tissues and cell lines [71]. Emerging evidence has shown that HNRNPC plays a major role in cancer development, progression, gene expression, and signal transduction. Knockdown of HNRNPA2B1 inhibits cell proliferation and cell growth in both the MCF-7 and MDA-MB-231 cell lines. In addition, in MCF-7/H1 and 231/H2 cells with HNRNPA2B1 knockdown, the levels of phospho-STAT3 and phospho-ERK1/2 expression were significantly decreased. That is, HNRNPA2B1 regulates the STAT3 and ERK1/2 signalling pathways in breast cancers. Notably, HNRNPA2B1 expression is higher in LCC9 tamoxifen-resistant cells than in parental tamoxifen-sensitive MCF-7 cells [72]. Conversely, HNRNPA2B1 expression is significantly decreased in invasive breast cancer (GSE59246) and MDAMB-231 and MCF-7 cells [73]. HNRNPA2B1 inhibits the growth of xenograft tumours but promotes spontaneous lung metastasis. HNRNPA2B1 knockdown promotes F-actin cytoskeleton formation, activates the downstream genes of the Wnt pathway, activates the ERK/MAPK/Twist pathway, upregulates PFN2 expression, and inhibits the epithelial-mesenchymal transition and metastasis (EMT)-promoting genes and signalling pathways [73]. HNRNPG, as a specific regulator of ERa exon 7 splicing, regulates oestrogen receptor alpha expression in endometrial cancer [100]. However, little is understood about the molecular characteristics of HNRNPG in breast cancer. Based on these findings, HNRNPG may play a potential role in breast cancer with oestrogen receptor expression.
The FXR family
Three members of the FXR family, FMR1, FXR1 and FXR2, participate in Fragile X Mental Retardation Syndrome by forming multiprotein complexes [41, 90]. Previous research found that all FXR family proteins contain two KH domains and one RCG box and associate with polyribosomes, predominantly with 60S large ribosomal subunits [90].
FMR1, containing a CGG trinucleotide repeat element in its 5′ untranslated region, is highly expressed in human breast cancer and distal metastasis and affects cell-cell adhesion, cell shape and invasion of 4 T1 cell lines [41, 42]. FMR1 also mediates E-cadherin and Vimentin expression. Meanwhile, CHIP interacts with FMR1 via a nontetratricopeptide repeat domain and upregulates FMR1 expression [43].
FXR1, as an RNA-binding protein, plays a vital role in breast cancer progression and cell invasion [44, 45]. In TNBC, FXR1, as a novel prognostic biomarker, stimulates lung metastasis FXR1 was associated with better pCR to neoadjuvant chemotherapy in TNBC [44]. FXR1, FXR2 and hnRNPK are required for these treRNA functions, while their expression promotes tumour invasion in vitro and metastasis in vivo [46]. FXR1 or FXR2 did not mediate cell proliferation in MCF7 cells. Moreover, knockdown of hnRNPK or FXR1 but not FXR2 increased the tumour growth of mouse model with MCF7 cells, whereas double knockdown or triple knockdown of the RNA-binding proteins significantly decreased primary tumour growth. That is, FXR1 mediated tumour growth not cell proliferation. Briefly, the combination of hnRNPK and FXR2 affects tumour metastasis in vivo, however, the curative mechanism need to be evaluated.
The IGF2BP family (KH domain proteins)
Insulin-like growth factor 2 mRNA binding proteins (IGF2BPs) recruit target transcripts to cytoplasmic protein-RNA complexes [47, 53, 54, 58,59,60, 101,102,103]. IGF2BP1, IGF2BP2 and IGF2BP3, members of the IGF2BP family, modulate RNA splicing, translation, processing, and stability and thereby affect cell proliferation, differentiation, invasion and metastasis in breast cancer. IGF2BPs, as blinding proteins of FGF13-AS1, prevent targeted Myc mRNA degradation to regulate breast cancer cell proliferation, invasion and metastasis [102].
IGF2BP1, as a binding protein, regulates cell apoptosis by inhibiting the accumulation of eIF5A [104]. Previous studies found that IGF2BP1 is highly expressed in breast cancer [47,48,49,50]. IGF2BP1 promotes the stability of β-catenin mRNA via the β-catenin/TCF4 response element and then accelerates cell adhesion and transcription of breast tumours [47]. Research in mouse xenograft models demonstrated that IGF2BP1 inhibits cell proliferation, tumour growth and tumour metastasis of breast cancer by binding to the 3′UTR of GDF15 mRNA [49, 50], while other research showed that IGF2BP1 does not affect tumour growth or size [51]. In T47D and MDA231 cells, IGF2BP1 plays a crucial role in repressing cell migration and metastasis, partly through the regulation of E-cadherin mRNA expression [50]. In T47D cells, silencing IGF2BP1 resulted in increased levels of UCA1 and thereafter increased the expression of miR-122-5p target mRNAs, eventually prompting breast cancer cell invasion [52].
IGF2BP2, as an oncofoetal protein, is located on chromosome 3q27 and is highly overexpressed in breast cancer cells and tissues [53, 55, 56]. Previous studies demonstrated that knockdown of IGF2BP2 significantly reduced metastatic endothelial density and functional vessel content and thus suppressed lung metastasis of breast tumour cells. IGF2BP2 overexpression increases cell migration and reduces cell adhesion by targeting CTGF mRNA in vitro and stimulates cell proliferation and influences growth and differentiation [56]. Additionally, in MDA-MB-231 and MCF-7 cells, miR-1193 suppresses the expression and activation of the ERK and PI3K/Akt signalling pathways by inhibiting IGF2BP2 translation and reducing the proliferation and invasion of human breast cancer cells [54]. CCN6 directly reduces IGF2BP2 and HMGA2 expressions and further reduces the tumour growth of MDA-MB-231 cells [53].
Emerging evidence has shown that IGF2BP3 is highly expressed in breast cancer [25, 57,58,59,60,61,62,63,64,65,66,67]. Interestingly, IGF2BP3 expression is higher in mesenchymal cells than in epithelial cells [105]. Hye-Youn Kim et al. elucidated that IGF2BP2 and IGF2BP3 cooperate to provoke cell migration, invasion, and invasion by negatively targeting microRNA-200a and partially determining the characteristic phenotype [61]. In breast cancer cells, IGF2BP3 has a direct correlation with E-cadherin, vimentin, and Slug (P < 0.05) and further promotes invasion and migration by prompting the EMT83 and reducing cell apoptosis by activating the Hedgehog signalling pathway CD44/CD44+Fbs/IGF2 [59, 106]. Moreover, IGF2BP3 mediates cell migration, invasion and stem cell properties through EGFR signalling and repression by ERβ, SLUG mRNA, and miRNA-34a [105]. IGF2BP3 increases TRIM25 expression by inhibiting miRNA-3614 maturation, thereby prompting cell growth and proliferation [58]. IGF2BP3, as an RNA-binding protein for CERS6-AS1, prompts CERS6 mRNA stability and then accelerates cell proliferation and suppresses cell apoptosis [60].
The eIF family with C-terminal readers
EIF3A, an oncogene in human breast cancer, was first described as the largest subunit of the eIF family by Bachmann et al. [78]. It has been reported that eIF3A is significantly highly expressed in breast cancer [79,80,81]. Dong et al. found that eIF3A regulates the synthesis of M2 protein, and decreasing eIF3A expression in MCF7 cells significantly reversed their malignant growth phenotype [107]. TG2/TIS21 inhibited translational initiation by depleting eIF availability by inhibiting 4EBP1 phosphorylation [80]. In addition, rs10787899 and rs3824830 SNPs in eIF3A were associated with an increased risk of breast cancer (all P < 0.01) [81]. EIF3A, also described as an oestrogen-responsive gene, can target ER-mediated signals and facilitate the cell proliferation and/or migration of ERα-positive breast cancer cells [79]. More experiments are needed to identify whether downregulation of eIF3A expression can change the malignant phenotype of breast cells.
In MCF7 cells, suppressing eIF4G1 facilitates the expression of eIF4G2, while suppressing eIF4G2 partially increases sensitivity to ionizing radiation-mediated DNA damage [108]. Columba et al. revealed that eIF4G1 has a very weak interaction with DAP5 [109]. Additionally, ERα − breast cancer patients have higher expression of eIF4G2 than ERα + patients. Moreover, eIF4G2 is abundantly expressed in proliferating cells, and the downregulation of eIF4G2 levels decreases the rate of global protein translation and inhibits cell proliferation [110].
Other readers in breast cancer
G3BP1 is dramatically overexpressed in breast cancer [82,83,84,85,86]. G3BP1 promotes cell proliferation by inhibiting PMP22 mRNA expression in breast cells and facilitates tumour metastasis and invasion by inhibiting the expression of E-cadherin and by increasing the expression of TGF-β signalling genes, Smad target genes, vimentin, Snail, Slug, fibronectin and ZEB1 [83, 84, 111]. Moreover, overexpression of G3BP1 induces the EMT in MCF-7 cells, thereby increasing cell migration and invasion [86]. In addition, downregulated G3BP1 restricts the invasion and migration of MDA-MB-231 cells compared to upregulated G3BP1, facilitating the tumour invasion and migration of MCF-7 cells [83, 84]. In addition, G3BP1 inhibits cell apoptosis by restraining nuclear p53 levels [112]. More interestingly, G3BP1 is also involved in vesicle trafficking [113].
The relationship between m6A modification and clinical features
Writers in breast cancer
Recent research by Hong Wang et al. found that high METTL3 expression was associated with worse prognosis in breast cancer (P = 0.002) [23]. Patients with METTL3, METTL14, WTAP and FTO overexpression have better metastasis relapse-free survival [26]. METTL3 overexpression was associated with better distant metastasis-free survival (P = 0.023) and extended OS (P = 0.042) [24]. In addition, METTL14 is associated with T staging, without molecular typing, microvascular invasion, nerve invasion and metastasis [26]. The 5-year overall survival of patients with KIAA1429 and CDK1 overexpression was significantly lower than that of patients with low expression of KIAA1429, CDK1 or both (P = 0.021) [32].
One study also found that surviving patients, regardless of race, have a higher level of RBM15B expression than those who do not survive [96]. That is, RBM15B may suppress tumour growth in these breast patients [96]. ZC3H13 had no association with the OS of TNBC from TCGA downloaded dates (n = 115, P = 0.9623) [87].
Erasers in breast cancer
FTO upregulation is significantly associated with shorter overall survival in patients with advanced-stage breast cancer [88]. Additionally, FTO overexpression correlates with tumour size, nuclear grade, peritumoral lymphovascular invasion, lymph node metastasis, and TNM stage. FTO overexpression implies poor DFS/OS/RFS for HER2-positive breast cancer [89]. In breast cancer, the percentage of FTO-positive expression in the HER2 overexpression subtype (97.1%) was significantly higher than that in the triple-negative (76.2%) and luminal subtypes (52.2%) (P < 0.001) [37]. Moreover, FTO expression has a positive correlation with HER2 status and ER/PR status. However, FTO has no association with age, tumour size, lymph node status, TNM stage, histological grade, Ki67, or BMI in breast cancer [37]. Additionally, ALKBH5 expression has no association with metastasis relapse-free survival [26].
Readers in breast cancer
Both YTHDF1 and YTHDF3 overexpression correlated with poor overall survival in breast cancer patients (all P values < 0.05) [31, 98]. YTHDF3 overexpression is correlated with lower relapse-free survival rates [31]. In the BCIP breast cancer database, patients with high HNRNPA2B1 expression (n = 30) had longer survival times than patients with low HNRNPA2B1 expression (n = 288) [73]. In contrast, breast cancer patients with high HNRNPA2B1 expression have worse survival [72].
FMR1 correlates with HER2 status, ER status, higher tumour grade (G3) and higher Ki67 expression. FMR1 overexpression correlated with an increased probability of breast cancer progression, particularly metastasis to the lungs [41]. FXR1 overexpression is associated with worse disease-specific disease-free survival in the METABRIC breast cancer cohort and worse overall survival in the TCGA cohort [45]. FXR1 overexpression is associated with worse distant metastasis-free survival in TNBC patients (n = 16, P = 0.01, HR = 9.63, 95% CI = 1.7–43.96) but not in TNBC patients [44].
Research in mouse xenograft models demonstrated that IGF2BP1 inhibits pulmonary metastatic tumours of breast cancer [49, 50], while other research showed that IGF2BP1 does not affect tumour growth or size [51]. In patients with early-stage breast cancer, IGF2BP2 overexpression is also correlated with short survival, and therefore IGF2BP2 may be a useful serum biomarker for breast cancer screening and diagnosis [55, 91]. From one cohort of 96 breast cancer patients, IGF2BP2 expression was significantly increased in patients with stages III and IV versus stages I and II. In TNBC, IGF2BP3 is associated with a larger tumour size, higher grade, higher clinical stage, necrosis, and CK5/6 expression [63]. In metaplastic breast carcinoma and TNBC, IGF2BP3 is associated with reduced DFS and OS in patients [62, 63]. Based on previous findings, eIF3A can be a tumour marker of invasive ductal breast carcinomas, but its expression had no association with age, tumour size, differentiation grade, nodal status, or DNA index of breast cancer patients [78].
The role of m6A in lncRNA regulation and cancer stem cells in breast cancer
Emerging evidence demonstrates that long noncoding RNAs (lncRNAs) and cancer stem cells play critical roles in human breast cancer (BC) by interacting with m6A. A previous study indicated that lncRNA KB-1980E6.3 recruited IGF2BP1 to maintain breast cancer stem cell stemness by targeting c-Myc [114]. In breast cancer, METTL3-induced LINC00958 upregulation affects tumorigenesis via the miR-378a-3p/YY1 axis [115]. In addition, ALKBH5 overexpression decreased the percentage of breast cancer stem cells [40]. In MDA-MB-231 cells, the knockdown of ALKBH5 significantly reduced the number of breast cancer stem cells [40].
Impact of m6A modification on therapy resistance
Moreover, 5′-fluorouracil (5-FU), but not doxorubicin, paclitaxel, and cyclophosphamide, reduces the expression of KIAA1429 in breast cancer cells [32]. YTHDF1 also has a positive correlation with molecular subtypes and nodal metastasis [98]. In MCF-7 cells, HNRNPA2B1 overexpression reduces sensitivity to 4-hydroxytamoxifen and fulvestrant [72]. IGF2BP2 could be a pathway and specific activity of MEK1/2 [101]. In TNBC cells, IGF2BP3 promotes chemoresistance to doxorubicin and mitoxantrone by regulating breast cancer resistance protein (ABCG2) [64]. In addition, a previous study identified that G3BP1 is associated with the tumour response to the Akt inhibitor MK-2206 [82].
Discussion and future perspectives
This article describes the role, mechanism and clinical application of m6A in breast cancer. RNA m6A modification plays an important role in promoting or inhibiting tumour growth, proliferation, migration, invasion, specific metastasis, drug resistance and prognosis of breast cancer through three effectors: writers, erasers and readers.
For breast cancer, different researchers have presented inconsistent results on the expression levels of m6A-related proteins, including MELLT3, MELLT14, KIAA1429, and HNRNPA2B1. MELLT3 and MELLT14 were upregulated in breast cancer [23] and downregulated in TNBC [24]. KIAA1429 was upregulated in luminal A and basal breast cancer and downregulated in Her2+ breast cancer. The expression of HNRNPA2B1 was decreased in invasive breast cancers in the GEO database (GSE59246) [73] and was increased in LCC9 breast cancer cells and in Asian patients [71, 72]. These differences have been explored by previous researchers, and there are several reasons that might account for the inconsistent results. First, the enrolled patients had different molecular typing. Second, these studies included patients of different races.
In addition, although m6A modification showed biological effects against breast cancers, there are still some questions that require further investigation and clarification. First, m6A modification has potential as a biomarker in early diagnosis. In patients with early-stage breast cancer, IGF2BP2 may be a useful serum biomarker for breast cancer screening and diagnosis [55, 91]. However, the application of this marker is often restricted by difficulty in obtaining sufficient samples for testing. Thus, large-scale clinical trials on m6A modification are necessary. Second, until now, most studies on m6A modification have tended to focus on its mechanism in breast cancer cell lines. Therefore, more conversion studies are needed to clarify whether m6A alone or in combination with other therapies could be used to treat breast cancers, even other solid tumours. Third, SNPs in eIF3A were associated with an increased risk of breast cancer [81]. It is also necessary to further speculate the impact how the mutations mediate m6A modification. Furthermore, m6A modifications can be targeted by specific drugs to improve drug resistance to breast cancer. Finally, the interactions between m6A and gene mutation frequency, genome copy number variation, and specific molecular subtypes remain unclear and need to be explored.
Conclusion
Recently, with the rapid development of bioinformatics and translational medicine methods and improvements in m6A editing tools and RNA sequencing, it is hopeful that RNA m6A modification can be developed as tumour markers for diagnosis and as drug targets for breast cancer treatment. There are three major types of m6A modification effectors, namely, writers, erasers and readers, in the human body. Thus, m6A modification is also likely to become a new multitargeting inhibitor that is efficacious against breast cancer.
Availability of data and materials
Not applicable.
Abbreviations
- m6A:
-
N6-methyladenosine
- TNBC:
-
Triple-negative breast cancer
- METTL3:
-
Methyltransferase-like 3
- METTL14:
-
Methyltransferase-like 14
- RFS:
-
Relapse-free survival
- DFS:
-
Disease-free survival
- OS:
-
Overall survival
- DMFS:
-
Distant metastasis-free survival
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. https://doi.org/10.3322/caac.21492.
Ochi T, Bianchini G, Ando M, Nozaki F, Kobayashi D, Criscitiello C, et al. Predictive and prognostic value of stromal tumour-infiltrating lymphocytes before and after neoadjuvant therapy in triple negative and HER2-positive breast cancer. Eur J Cancer. 2019;118:41–8. https://doi.org/10.1016/j.ejca.2019.05.014.
Tamura K, Tsurutani J, Takahashi S, Iwata H, Krop IE, Redfern C, et al. Trastuzumab deruxtecan (DS-8201a) in patients with advanced HER2-positive breast cancer previously treated with trastuzumab emtansine: a dose-expansion, phase 1 study. Lancet Oncol. 2019;20(6):816–26. https://doi.org/10.1016/s1470-2045(19)30097-x.
Andre F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N Engl J Med. 2019;380(20):1929–40. https://doi.org/10.1056/NEJMoa1813904.
Bardia A, Mayer IA, Vahdat LT, Tolaney SM, Isakoff SJ, Diamond JR, et al. Sacituzumab govitecan-hziy in refractory metastatic triple-negative breast cancer. N Engl J Med. 2019;380(8):741–51. https://doi.org/10.1056/NEJMoa1814213.
Litton JK, Rugo HS, Ettl J, Hurvitz SA, Goncalves A, Lee KH, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. 2018;379(8):753–63. https://doi.org/10.1056/NEJMoa1802905.
Shahi RB, De Brakeleer S, Caljon B, Pauwels I, Bonduelle M, Joris S, et al. Identification of candidate cancer predisposing variants by performing whole-exome sequencing on index patients from BRCA1 and BRCA2-negative breast cancer families. BMC Cancer. 2019;19(1):313. https://doi.org/10.1186/s12885-019-5494-7.
Chung W, Eum HH, Lee HO, Lee KM, Lee HB, Kim KT, et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun. 2017;8(1):15081. https://doi.org/10.1038/ncomms15081.
Ennour-Idrissi K, Dragic D, Durocher F, Diorio C. Epigenome-wide DNA methylation and risk of breast cancer: a systematic review. BMC Cancer. 2020;20(1):1048. https://doi.org/10.1186/s12885-020-07543-4.
O'Brien NA, McDermott MSJ, Conklin D, Luo T, Ayala R, Salgar S, et al. Targeting activated PI3K/mTOR signaling overcomes acquired resistance to CDK4/6-based therapies in preclinical models of hormone receptor-positive breast cancer. Breast Cancer Res. 2020;22(1):89. https://doi.org/10.1186/s13058-020-01320-8.
Shulman Z, Stern-Ginossar N. The RNA modification N(6)-methyladenosine as a novel regulator of the immune system. Nat Immunol. 2020;21(5):501–12. https://doi.org/10.1038/s41590-020-0650-4.
He L, Li H, Wu A, Peng Y, Shu G, Yin G. Functions of N6-methyladenosine and its role in cancer. Mol Cancer. 2019;18(1):176. https://doi.org/10.1186/s12943-019-1109-9.
Pan Y, Ma P, Liu Y, Li W, Shu Y. Multiple functions of m(6)a RNA methylation in cancer. J Hematol Oncol. 2018;11(1):48. https://doi.org/10.1186/s13045-018-0590-8.
Deng X, Su R, Weng H, Huang H, Li Z, Chen J. RNA N(6)-methyladenosine modification in cancers: current status and perspectives. Cell Res. 2018;28(5):507–17. https://doi.org/10.1038/s41422-018-0034-6.
Huang H, Weng H, Chen J. m(6)A Modification in coding and non-coding RNAs: roles and therapeutic implications in cancer. Cancer Cell. 2020;37(3):270–88. https://doi.org/10.1016/j.ccell.2020.02.004.
Boccaletto P, Machnicka MA, Purta E, Piatkowski P, Baginski B, Wirecki TK, et al. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018;46(D1):D303–D7. https://doi.org/10.1093/nar/gkx1030.
Zhang C, Chen Y, Sun B, Wang L, Yang Y, Ma D, et al. m(6)A modulates haematopoietic stem and progenitor cell specification. Nature. 2017;549(7671):273–6. https://doi.org/10.1038/nature23883.
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–6. https://doi.org/10.1038/nature11112.
Krol A, Branlant C, Lazar E, Gallinaro H, Jacob M. Primary and secondary structures of chicken, rat and man nuclear U4 RNAs. Homologies with U1 and U5 RNAs. Nucleic Acids Res. 1981;25(9):2699–716.
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149(7):1635–46. https://doi.org/10.1016/j.cell.2012.05.003.
Shen H, Lan Y, Zhao Y, Shi Y, Jin J, Xie W. The emerging roles of N6-methyladenosine RNA methylation in human cancers. Biomark Res. 2020;8(1):24. https://doi.org/10.1186/s40364-020-00203-6.
Cai X, Wang X, Cao C, Gao Y, Zhang S, Yang Z, et al. HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7g. Cancer Lett. 2018;415:11–9. https://doi.org/10.1016/j.canlet.2017.11.018.
Wang H, Xu B, Shi J. N6-methyladenosine METTL3 promotes the breast cancer progression via targeting Bcl-2. Gene. 2020;722:144076. https://doi.org/10.1016/j.gene.2019.144076.
Shi Y, Zheng C, Jin Y, Bao B, Wang D, Hou K, et al. Reduced expression of METTL3 promotes metastasis of triple-negative breast cancer by m6A methylation-mediated COL3A1 up-regulation. Front Oncol. 2020;10:1126. https://doi.org/10.3389/fonc.2020.01126.
Stevens TA, Meech R. BARX2 and estrogen receptor-alpha (ESR1) coordinately regulate the production of alternatively spliced ESR1 isoforms and control breast cancer cell growth and invasion. Oncogene. 2006;25(39):5426–35. https://doi.org/10.1038/sj.onc.1209529.
Wu L, Wu D, Ning J, Liu W, Zhang D. Changes of N6-methyladenosine modulators promote breast cancer progression. BMC Cancer. 2019;19(1):326. https://doi.org/10.1186/s12885-019-5538-z.
Zang X-P, Pento JT, Tari AM. Wilms’ tumor 1 protein and focal adhesion kinase mediate keratinocyte growth factor signaling in breast cancer cells. Anticancer Res. 2008;28:133–7.
Sun T, Wu Z, Wang X, Wang Y, Hu X, Qin W, et al. LNC942 promoting METTL14-mediated m(6)A methylation in breast cancer cell proliferation and progression. Oncogene. 2020;39(31):5358–72. https://doi.org/10.1038/s41388-020-1338-9.
Yi D, Wang R, Shi X, Xu L, Yilihamu Y, Sang J. METTL14 promotes the migration and invasion of breast cancer cells by modulating N6methyladenosine and hsamiR146a5p expression. Oncol Rep. 2020;43(5):1375–86. https://doi.org/10.3892/or.2020.7515.
NJF L, Holley BA, Law ORI, Carraway KR, Holley CL, Mansfield KD. N6-methyladenosine contributes to cellular phenotype in a genetically-defined model of breast cancer progression. Oncotarget. 2018;9(58):31231–43.
Liu L, Liu X, Dong Z, Li J, Yu Y, Chen X, et al. N6-methyladenosine-related genomic targets are altered in breast cancer tissue and associated with poor survival. J Cancer. 2019;10(22):5447–59. https://doi.org/10.7150/jca.35053.
Qian J-Y, Gao J, Sun X, Cao M-D, Shi L, Xia T-S, et al. KIAA1429 acts as an oncogenic factor in breast cancer by regulating CDK1 in an N6-methyladenosine-independent manner. Oncogene. 2019;38(33):6123–41. https://doi.org/10.1038/s41388-019-0861-z.
Aparicio LA, Valladares M, Blanco M, Alonso G, Figueroa A. Biological influence of Hakai in cancer: a 10-year review. Cancer Metastasis Rev. 2012;31(1–2):375–86. https://doi.org/10.1007/s10555-012-9348-x.
Gong EY, Park E, Lee K. Hakai acts as a coregulator of estrogen receptor alpha in breast cancer cells. Cancer Sci. 2010;101(9):2019–25. https://doi.org/10.1111/j.1349-7006.2010.01636.x.
Makdissi FBA, Machado LVST, Oliveira AGC, Oliveira TT, Benvenuti MLH, Katayama MM, et al. Expression of E-cadherin, snail and Hakai in epithelial cells isolated from the primary tumor and from peritumoral tissue of invasive ductal breast carcinomas. Braz J Med Biol Res. 2009;42(12):1128–37. https://doi.org/10.1590/S0100-879X2009001200002.
Chen J, Du B. Novel positioning from obesity to cancer: FTO, an m(6)A RNA demethylase, regulates tumour progression. J Cancer Res Clin Oncol. 2019;145(1):19–29. https://doi.org/10.1007/s00432-018-2796-0.
Tan A, Dang Y, Chen G, Mo Z. Overexpression of the fat mass and obesity associated gene (FTO) in breast cancer and its clinical implications. Int J Clin Exp Pathol. 2015;8(10):13405–10.
Melnik BC. Milk: an epigenetic amplifier of FTO-mediated transcription? Implications for Western diseases. J Transl Med. 2015;13(1):385. https://doi.org/10.1186/s12967-015-0746-z.
Shen C, Sheng Y, Zhu AC, Robinson S, Jiang X, Dong L, et al. RNA demethylase ALKBH5 selectively promotes tumorigenesis and cancer stem cell self-renewal in acute myeloid leukemia. Cell Stem Cell. 2020;27(1):64–80 e9. https://doi.org/10.1016/j.stem.2020.04.009.
Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m(6)A-demethylation of NANOG mRNA. Proc Natl Acad Sci U S A. 2016;113(14):E2047–56. https://doi.org/10.1073/pnas.1602883113.
Luca R, Averna M, Zalfa F, Vecchi M, Bianchi F, La Fata G, et al. The fragile X protein binds mRNAs involved in cancer progression and modulates metastasis formation. EMBO Mol Med. 2013;5(10):1523–36. https://doi.org/10.1002/emmm.201302847.
Adamsheck HC, Petty EM, Hong J, Baker MW, Brilliant MH, Mailick MR. Is low FMR1 CGG repeat length in males correlated with family history of BRCA-associated cancers? An exploratory analysis of medical records. J Genet Couns. 2017;26(6):1401–10. https://doi.org/10.1007/s10897-017-0116-5.
Choi YN, Jeong DH, Lee JS, Yoo SJ. Regulation of fragile X mental retardation 1 protein by C-terminus of Hsc70-interacting protein depends on its phosphorylation status. Biochem Biophys Res Commun. 2014;453(1):192–7. https://doi.org/10.1016/j.bbrc.2014.09.099.
Qian J, Chen H, Ji X, Eisenberg R, Chakravarthy AB, Mayer IA, et al. A 3q gene signature associated with triple negative breast cancer organ specific metastasis and response to neoadjuvant chemotherapy. Sci Rep. 2017;7(1):45828. https://doi.org/10.1038/srep45828.
Qian J, Hassanein M, Hoeksema MD, Harris BK, Zou Y, Chen H, et al. The RNA binding protein FXR1 is a new driver in the 3q26-29 amplicon and predicts poor prognosis in human cancers. Proc Natl Acad Sci U S A. 2015;112(11):3469–74. https://doi.org/10.1073/pnas.1421975112.
Gumireddy K, Li A, Yan J, Setoyama T, Johannes GJ, Orom UA, et al. Identification of a long non-coding RNA-associated RNP complex regulating metastasis at the translational step. EMBO J. 2013;32(20):2672–84. https://doi.org/10.1038/emboj.2013.188.
Gu W, Wells AL, Pan F, Singer RH. Feedback regulation between zipcode binding protein 1 and beta-catenin mRNAs in breast cancer cells. Mol Cell Biol. 2008;28(16):4963–74. https://doi.org/10.1128/MCB.00266-08.
Tang L, Chen Y, Tang X, Wei D, Xu X, Yan F. Long noncoding RNA DCST1-AS1 promotes cell proliferation and metastasis in triple-negative breast cancer by forming a positive regulatory loop with miR-873-5p and MYC. J Cancer. 2020;11(2):311–23. https://doi.org/10.7150/jca.33982.
Wang G, Huang Z, Liu X, Huang W, Chen S, Zhou Y, et al. IMP1 suppresses breast tumor growth and metastasis through the regulation of its target mRNAs. Oncotarget. 2016;7(13):15690–702. https://doi.org/10.18632/oncotarget.7464.
Gu W, Katz Z, Wu B, Park HY, Li D, Lin S, et al. Regulation of local expression of cell adhesion and motility-related mRNAs in breast cancer cells by IMP1/ZBP1. J Cell Sci. 2012;125(Pt 1):81–91. https://doi.org/10.1242/jcs.086132.
Nwokafor CU, Sellers RS, Singer RH. IMP1, an mRNA binding protein that reduces the metastatic potential of breast cancer in a mouse model. Oncotarget. 2016;7(45):72662–71.
Zhou Y, Meng X, Chen S, Li W, Li D, Singer R, et al. IMP1 regulates UCA1-mediated cell invasion through facilitating UCA1 decay and decreasing the sponge effect of UCA1 for miR-122-5p. Breast Cancer Res. 2018;20(1):32. https://doi.org/10.1186/s13058-018-0959-1.
McMullen ER, Gonzalez ME, Skala SL, Tran M, Thomas D, Djomehri SI, et al. CCN6 regulates IGF2BP2 and HMGA2 signaling in metaplastic carcinomas of the breast. Breast Cancer Res Treat. 2018;172(3):577–86. https://doi.org/10.1007/s10549-018-4960-2.
Li X, Li Y, Lu H. miR-1193 suppresses proliferation and invasion of human breast cancer cells through directly targeting IGF2BP2. Oncol Res. 2017;25(4):579–85. https://doi.org/10.3727/97818823455816X14760504645779.
Liu W, Li Y, Wang B, Dai L, Qian W, Zhang JY. Autoimmune response to IGF2 mRNA-binding protein 2 (IMP2/p62) in breast cancer. Scand J Immunol. 2015;81(6):502–7. https://doi.org/10.1111/sji.12285.
Li Y, Francia G, Zhang J-Y. p62/IMP2 stimulates cell migration and reduces cell adhesion in breast cancer. Oncotarget. 2015;6(32):32656–68. https://doi.org/10.18632/oncotarget.5328.
Samanta S, Sun H, Goel HL, Pursell B, Chang C, Khan A, et al. IMP3 promotes stem-like properties in triple-negative breast cancer by regulating SLUG. Oncogene. 2016;35(9):1111–21. https://doi.org/10.1038/onc.2015.164.
Wang Z, Tong D, Han C, Zhao Z, Wang X, Jiang T, et al. Blockade of miR-3614 maturation by IGF2BP3 increases TRIM25 expression and promotes breast cancer cell proliferation. EBioMedicine. 2019;41:357–69. https://doi.org/10.1016/j.ebiom.2018.12.061.
Liu Y, Yu C, Wu Y, Sun X, Su Q, You C, et al. CD44(+) fibroblasts increases breast cancer cell survival and drug resistance via IGF2BP3-CD44-IGF2 signalling. J Cell Mol Med. 2017;21(9):1979–88. https://doi.org/10.1111/jcmm.13118.
Bao G, Huang J, Pan W, Li X, Zhou T. Long noncoding RNA CERS6-AS1 functions as a malignancy promoter in breast cancer by binding to IGF2BP3 to enhance the stability of CERS6 mRNA. Cancer Med. 2020;9(1):278–89. https://doi.org/10.1002/cam4.2675.
Kim HY, Ha Thi HT, Hong S. IMP2 and IMP3 cooperate to promote the metastasis of triple-negative breast cancer through destabilization of progesterone receptor. Cancer Lett. 2018;415:30–9. https://doi.org/10.1016/j.canlet.2017.11.039.
Ohashi R, Sangen M, Namimatsu S, Takei H, Naito Z. IMP3 contributes to poor prognosis of patients with metaplastic breast carcinoma: a clinicopathological study. Ann Diagn Pathol. 2017;31:30–5. https://doi.org/10.1016/j.anndiagpath.2017.05.015.
Walter O, Prasad M, Lu S, Quinlan RM, Edmiston KL, Khan A. IMP3 is a novel biomarker for triple negative invasive mammary carcinoma associated with a more aggressive phenotype. Hum Pathol. 2009;40(11):1528–33. https://doi.org/10.1016/j.humpath.2009.05.005.
Samanta S, Pursell B, Mercurio AM. IMP3 protein promotes chemoresistance in breast cancer cells by regulating breast cancer resistance protein (ABCG2) expression. J Biol Chem. 2013;288(18):12569–73. https://doi.org/10.1074/jbc.C112.442319.
Ohashi R, Sangen M, Namimatsu S, Yanagihara K, Yamashita K, Sakatani T, et al. Prognostic value of IMP3 expression as a determinant of chemosensitivity in triple-negative breast cancer. Pathol Res Pract. 2017;213(9):1160–5. https://doi.org/10.1016/j.prp.2017.07.002.
Sjekloca N, Tomic S, Mrklic I, Vukmirovic F, Vuckovic L, Lovasic IB, et al. Prognostic value of IMP3 immunohistochemical expression in triple negative breast cancer. Medicine (Baltimore). 2020;99(7):e19091. https://doi.org/10.1097/MD.0000000000019091.
Samanta S, Sharma VM, Khan A, Mercurio AM. Regulation of IMP3 by EGFR signaling and repression by ERbeta: implications for triple-negative breast cancer. Oncogene. 2012;31(44):4689–97. https://doi.org/10.1038/onc.2011.620.
Zhang J, Lin Y, Sun XJ, Wang BY, Wang ZH, Luo JF, et al. Biomarker assessment of the CBCSG006 trial: a randomized phase III trial of cisplatin plus gemcitabine compared with paclitaxel plus gemcitabine as first-line therapy for patients with metastatic triple-negative breast cancer. Ann Oncol. 2018;29(8):1741–7. https://doi.org/10.1093/annonc/mdy209/5037891.
Wen J, Toomer KH, Chen Z, Cai X. Genome-wide analysis of alternative transcripts in human breast cancer. Breast Cancer Res Treat. 2015;151(2):295–307. https://doi.org/10.1007/s10549-015-3395-2.
Hwang SJ, Seol HJ, Park YM, Kim KH, Gorospe M, Nam DH, et al. MicroRNA-146a suppresses metastatic activity in brain metastasis. Mol Cells. 2012;34(3):329–34. https://doi.org/10.1007/s10059-012-0171-6.
Hu Y, Sun Z, Deng J, Hu B, Yan W, Wei H, et al. Splicing factor hnRNPA2B1 contributes to tumorigenic potential of breast cancer cells through STAT3 and ERK1/2 signaling pathway. Tumour Biol. 2017;39(3):1010428317694318. https://doi.org/10.1177/1010428317694318.
Klinge CM, Piell KM, Tooley CS, Rouchka EC. HNRNPA2/B1 is upregulated in endocrine-resistant LCC9 breast cancer cells and alters the miRNA transcriptome when overexpressed in MCF-7 cells. Sci Rep. 2019;9(1):9430. https://doi.org/10.1038/s41598-019-45636-8.
Liu Y, Li H, Liu F, Gao LB, Han R, Chen C, et al. Heterogeneous nuclear ribonucleoprotein A2/B1 is a negative regulator of human breast cancer metastasis by maintaining the balance of multiple genes and pathways. EBioMedicine. 2020;51:102583. https://doi.org/10.1016/j.ebiom.2019.11.044.
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161(6):1388–99. https://doi.org/10.1016/j.cell.2015.05.014.
Chang G, Shi L, Ye Y, Shi H, Zeng L, Tiwary S, et al. YTHDF3 induces the translation of m(6)A-enriched gene transcripts to promote breast cancer brain metastasis. Cancer Cell. 2020;38(6):857–871.e7. https://doi.org/10.1016/j.ccell.2020.10.004.
Sarbanes SL, Le Pen J, Rice CM. Friend and foe, HNRNPC takes on immunostimulatory RNAs in breast cancer cells. EMBO J. 2018;37(23). https://doi.org/10.15252/embj.2018100923.
Xu C, Liu K, Ahmed H, Loppnau P, Schapira M, Min J. Structural basis for the discriminative recognition of N6-Methyladenosine RNA by the human YT521-B homology domain family of proteins. J Biol Chem. 2015;290(41):24902–13. https://doi.org/10.1074/jbc.M115.680389.
Bachmann F, Bänziger R, Burger MM. Cloning of a novel protein overexpressed in human mammary carcinoma. Cancer Res. 1997;57:988–94.
Yamaga R, Ikeda K, Horie-Inoue K, Ouchi Y, Suzuki Y, Inoue S. RNA sequencing of MCF-7 breast cancer cells identifies novel estrogen-responsive genes with functional estrogen receptor-binding sites in the vicinity of their transcription start sites. Horm Cancer. 2013;4(4):222–32. https://doi.org/10.1007/s12672-013-0140-3.
Devanand P, Sundaramoorthy S, Ryu MS, Jayabalan AK, Ohn T, Lim IK. Translational downregulation of Twist1 expression by antiproliferative gene, B-cell translocation gene 2, in the triple negative breast cancer cells. Cell Death Dis. 2019;10(6):410. https://doi.org/10.1038/s41419-019-1640-z.
Olson JE, Wang X, Goode EL, Pankratz VS, Fredericksen ZS, Vierkant RA, et al. Variation in genes required for normal mitosis and risk of breast cancer. Breast Cancer Res Treat. 2010;119(2):423–30. https://doi.org/10.1007/s10549-009-0386-1.
Chien AJ, Cockerill A, Fancourt C, Schmidt E, Moasser MM, Rugo HS, et al. A phase 1b study of the Akt-inhibitor MK-2206 in combination with weekly paclitaxel and trastuzumab in patients with advanced HER2-amplified solid tumor malignancies. Breast Cancer Res Treat. 2016;155(3):521–30. https://doi.org/10.1007/s10549-016-3701-7.
Zhang CH, Wang JX, Cai ML, Shao R, Liu H, Zhao WL. The roles and mechanisms of G3BP1 in tumour promotion. J Drug Target. 2019;27(3):300–5. https://doi.org/10.1080/1061186X.2018.1523415.
Tong D, Heinze G, Pils D, Wolf A, Singer CF, Concin N, et al. Gene expression of PMP22 is an independent prognostic factor for disease-free and overall survival in breast cancer patients. BMC Cancer. 2010;10(1):682. https://doi.org/10.1186/1471-2407-10-682.
Barnes CJ, Li F, Mandal M, Yang Z, Sahin AA, Kumar R. Heregulin induces expression, ATPase activity, and nuclear localization of G3BP, a ras signaling component, in human breast tumors. Cancer Res. 2002;62:1251–5.
Wu Y, Zhu J, Huang X, Du Z. Crystal structure of a dimerization domain of human Caprin-1: insights into the assembly of an evolutionarily conserved ribonucleoprotein complex consisting of Caprin-1, FMRP and G3BP1. Acta Crystallogr D Struct Biol. 2016;72(Pt 6):718–27. https://doi.org/10.1107/S2059798316004903.
Funakoshi Y, Wang Y, Semba T, Masuda H, Hout D, Ueno NT, et al. Comparison of molecular profile in triple-negative inflammatory and non-inflammatory breast cancer not of mesenchymal stem-like subtype. PLoS One. 2019;14(9):e0222336. https://doi.org/10.1371/journal.pone.0222336.
Niu Y, Lin Z, Wan A, Chen H, Liang H, Sun L, et al. RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol Cancer. 2019;18(1):46. https://doi.org/10.1186/s12943-019-1004-4.
Xu Y, Ye S, Zhang N, Zheng S, Liu H, Zhou K, et al. The FTO/miR-181b-3p/ARL5B signaling pathway regulates cell migration and invasion in breast cancer. Cancer Commun (Lond). 2020;40(10):484–500. https://doi.org/10.1002/cac2.12075.
Zhang Y, O'Connor JP, Siomi MC, Srinivasan S, Dutra A, Nussbaum RL, et al. The fragile X mental retardation syndrome protein interacts with novel homologs FXR1 and FXR2. EMBO J. 1995;14(21):5358–66. https://doi.org/10.1002/j.1460-2075.1995.tb00220.x.
Barghash A, Helms V, Kessler SM. Overexpression of IGF2 mRNA-binding protein 2 (IMP2/p62) as a feature of basal-like breast cancer correlates with short survival. Scand J Immunol. 2015;82(2):142–3. https://doi.org/10.1111/sji.12307.
Xiang Y, Laurent B, Hsu CH, Nachtergaele S, Lu Z, Sheng W, et al. RNA m(6)A methylation regulates the ultraviolet-induced DNA damage response. Nature. 2017;543(7646):573–6. https://doi.org/10.1038/nature21671.
Wang P, Doxtader Katelyn A, Nam Y. Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol Cell. 2016;63(2):306–17. https://doi.org/10.1016/j.molcel.2016.05.041.
Śledź P, Jinek M. Structural insights into the molecular mechanism of the m6A writer complex. eLife. 2016;5. https://doi.org/10.7554/eLife.18434.
Deng X, Su R, Feng X, Wei M, Chen J. Role of N(6)-methyladenosine modification in cancer. Curr Opin Genet Dev. 2018;48:1–7. https://doi.org/10.1016/j.gde.2017.10.005.
Shahriyari L, Abdel-Rahman M, Cebulla C. BAP1 expression is prognostic in breast and uveal melanoma but not colon cancer and is highly positively correlated with RBM15B and USP19. PLoS One. 2019;14(2):e0211507. https://doi.org/10.1371/journal.pone.0211507.
Zhang C, Zhi WI, Lu H, Samanta D, Chen I, Gabrielson E, et al. Hypoxia-inducible factors regulate pluripotency factor expression by ZNF217- and ALKBH5-mediated modulation of RNA methylation in breast cancer cells. Oncotarget. 2016;7(40):64527–42.
Anita R, Paramasivam A, Priyadharsini JV, Chitra S. The m6A readers YTHDF1 and YTHDF3 aberrations associated with metastasis and predict poor prognosis in breast cancer patients. Am J Cancer Res. 2020;10(8):2546–54.
Liu M, Zhou S, Wang J, Zhang Q, Yang S, Feng J, et al. Identification of genes associated with survival of breast cancer patients. Breast Cancer. 2019;26(3):317–25. https://doi.org/10.1007/s12282-018-0926-9.
Hirschfeld M, Ouyang YQ, Jaeger M, Erbes T, Orlowska-Volk M, Zur Hausen A, et al. HNRNP G and HTRA2-BETA1 regulate estrogen receptor alpha expression with potential impact on endometrial cancer. BMC Cancer. 2015;15(1):86. https://doi.org/10.1186/s12885-015-1088-1.
Liu G, Zhu T, Cui Y, Liu J, Liu J, Zhao Q, et al. Correlation between IGF2BP2 gene polymorphism and the risk of breast cancer in Chinese Han women. Biomed Pharmacother. 2015;69:297–300. https://doi.org/10.1016/j.biopha.2014.12.017.
Ma F, Liu X, Zhou S, Li W, Liu C, Chadwick M, et al. Long non-coding RNA FGF13-AS1 inhibits glycolysis and stemness properties of breast cancer cells through FGF13-AS1/IGF2BPs/Myc feedback loop. Cancer Lett. 2019;450:63–75. https://doi.org/10.1016/j.canlet.2019.02.008.
Fakhraldeen SA, Clark RJ, Roopra A, Chin EN, Huang W, Castorino J, et al. Two isoforms of the RNA binding protein, coding region determinant-binding protein (CRD-BP/IGF2BP1), are expressed in breast epithelium and support clonogenic growth of breast tumor cells. J Biol Chem. 2015;290(21):13386–400. https://doi.org/10.1074/jbc.M115.655175.
Miyake T, Pradeep S, Wu SY, Rupaimoole R, Zand B, Wen Y, et al. XPO1/CRM1 inhibition causes antitumor effects by mitochondrial accumulation of eIF5A. Clin Cancer Res. 2015;21(14):3286–97. https://doi.org/10.1158/1078-0432.CCR-14-1953.
Huang Q-D, Zheng S-R, Cai Y-J, Chen DL, Shen Y-Y, Lin C-Q, et al. IMP3 promotes TNBC stem cell property through miRNA-34a regulation. Eur Rev Med Pharmacol Sci. 2019;22:2688–96.
Su P, Hu J, Zhang H, Li W, Jia M, Zhang X, et al. IMP3 expression is associated with epithelial-mesenchymal transition in breast cancer. Int J Clin Exp Pathol. 2014;7(6):3008–17.
Dong Z, Liu LH, Han B, Pincheira R, Zhang JT. Role of eIF3 p170 in controlling synthesis of ribonucleotide reductase M2 and cell growth. Oncogene. 2004;23(21):3790–801. https://doi.org/10.1038/sj.onc.1207465.
Badura M, Braunstein S, Zavadil J, Schneider RJ. DNA damage and eIF4G1 in breast cancer cells reprogram translation for survival and DNA repair mRNAs. Proc Natl Acad Sci U S A. 2012;109(46):18767–72. https://doi.org/10.1073/pnas.1203853109.
de la Parra C, Ernlund A, Alard A, Ruggles K, Ueberheide B, Schneider RJ. A widespread alternate form of cap-dependent mRNA translation initiation. Nat Commun. 2018;9(1):3068. https://doi.org/10.1038/s41467-018-05539-0.
Rezaul K, Thumar JK, Lundgren DH, Eng JK, Claffey KP, Wilson L, et al. Differential protein expression profiles in estrogen receptor-positive and -negative breast cancer tissues using label-free quantitative proteomics. Genes Cancer. 2010;1(3):251–71. https://doi.org/10.1177/1947601910365896.
Winslow S, Leandersson K, Larsson C. Regulation of PMP22 mRNA by G3BP1 affects cell proliferation in breast cancer cells. Mol Cancer. 2013;12:1–10.
Mao C, Wang X, Liu Y, Wang M, Yan B, Jiang Y, et al. A G3BP1-interacting lncRNA promotes ferroptosis and apoptosis in cancer via nuclear sequestration of p53. Cancer Res. 2018:canres.3454.2017. https://doi.org/10.1158/0008-5472.Can-17-3454.
Alshaker H, Wang Q, Brewer D, Pchejetski D. Transcriptome-wide effects of sphingosine kinases knockdown in metastatic prostate and breast cancer cells: implications for therapeutic targeting. Front Pharmacol. 2019;10:303. https://doi.org/10.3389/fphar.2019.00303.
Zhu P, He F, Hou Y, Tu G, Li Q, Jin T, et al. A novel hypoxic long noncoding RNA KB-1980E6.3 maintains breast cancer stem cell stemness via interacting with IGF2BP1 to facilitate c-Myc mRNA stability. Oncogene. 2021;40(9):1609–27. https://doi.org/10.1038/s41388-020-01638-9.
Rong D, Dong Q, Qu H, Deng X, Gao F, Li Q, et al. m(6)A-induced LINC00958 promotes breast cancer tumorigenesis via the miR-378a-3p/YY1 axis. Cell Death Discov. 2021;7(1):27. https://doi.org/10.1038/s41420-020-00382-z.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Key R&D Program of China (2018YFC0115204), Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences Clinical and Translational Medicine Research Fund (12019XK320071), CSCO Pilot Oncology Research Fund (Y-2019AZMS-0377) and Capital Health Development Research Project (2018-2-4023).
Author information
Authors and Affiliations
Contributions
Fangchao Zheng, Feng Du, Jiuda Zhao and Peng Yuan conceived of the study and drafted the manuscript. Xue Wang, Yiran Si, Peng Jin, Haili Qian and Binghe Xu participated in its modification. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors agree to publish this article.
Competing interests
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zheng, F., Du, F., Zhao, J. et al. The emerging role of RNA N6-methyladenosine methylation in breast cancer. Biomark Res 9, 39 (2021). https://doi.org/10.1186/s40364-021-00295-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-021-00295-8