
Abstract
Polyadenylation of pre-messenger RNA (pre-mRNA) specific sites and termination of their downstream transcriptions are signaled by unique sequence motif structures such as AAUAAA and its auxiliary elements. Alternative polyadenylation (APA) is an important post-transcriptional regulatory mechanism that processes RNA products depending on its 3′-untranslated region (3′-UTR) specific sequence signal. APA processing can generate several mRNA isoforms from a single gene, which may have different biological functions on their target gene. As a result, cellular genomic stability, proliferation capability, and transformation feasibility could all be affected. Furthermore, APA modulation regulates disease initiation and progression. APA status could potentially act as a biomarker for disease diagnosis, severity stratification, and prognosis forecast. While the advance of modern throughout technologies, such as next generation-sequencing (NGS) and single-cell sequencing techniques, have enriched our knowledge about APA, much of APA biological process is unknown and pending for further investigation. Herein, we review the current knowledge on APA and how its regulatory complex factors (CFI/IIm, CPSF, CSTF, and RBPs) work together to determine RNA splicing location, cell cycle velocity, microRNA processing, and oncogenesis regulation. We also discuss various APA experiment strategies and the future direction of APA research.
Similar content being viewed by others
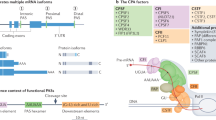

Introduction
Splicing, capping, and polyadenylation are three major steps in processing pre-messenger RNA (pre-mRNA) to mRNA [1, 2]. Polyadenylation (poly(A)) involves in endonucleolytic cleavage of pre-mRNA and addition of the poly(A) tail at the cleavage site [1]. Individual pre-mRNA usually harbors a few cleavage/polyadenylation (C/P) sites (polyA sites or pA) [2]. Alternative polyadenylation (APA) can eventually produce several mRNA polyadenylation isoforms [3].
According to current understanding, APA is a comprehensive process accomplished via coordinative actions of several small molecules. The 3′-processing factors are the major targets of APA regulation [4]. Typical APA processing includes the following steps: (1) CFIm (cleavage factor I) binds to the UGUA field of pre-mRNA upstream of the pA site and attracts CPSF (cleavage and polyadenylation specificity factor) and CSTF (cleavage stimulation factor) to assemble at the end of RNA polymerase II; (2) as RNA polymerase II advances, CPSF binds to the pA signal sequence (e.g. AAUAAA) and CSTF is transferred to the new mRNA precursor, binding to the GU or U-rich sequence; (3) CPSF and CSTF initiate the cleavage of ~ 35 nucleosides after the pA signal sequence, and polyadenylation binding protein (PABPN1) in the nucleus will bind to the polyadenylation tail sequence to begin the PAP process; (4) while PAP-mediated polyadenylation continues, adenosine tails of~ 50–250 nucleotides (nt) are prepared (depending on the species of the organism) and CPSF dissociates from its binding sequence; (5) PABPN1 works as a molecular ruler during this APA progression, defining when the polyadenylation process should stop; (6) PAP begins to dissociate, although PABPN1 continues to maintain its binding status. The combination of above 6 steps in conjunction with the 5′-capping process promotes mRNA maturation and eventual exportation from nucleus to cytoplasm.
Approximately 50 ~ 80% of mammalian pre-mRNA transcripts have more than one pA sites [5, 6]. The 3′-UTRof mRNA harbors key RNA regulatory elements that determine when, where, and how much mRNA transcript will be translated [1]. APA is a crucial 3′-UTR post-transcriptional regulation mechanism. The 3′-UTR APA isoforms play various roles in determining mRNA stability, localization, half-life, and functions. Furthermore, previous studies demonstrated that APA is involved in disease progression and drug sensitivity, especially for drugs targeting chromatin modifiers [2, 7,8,9]. Though APA research is still in its early stage, its unique post-transcriptional regulatory effect makes it potentially both a biomarker for cancer prognosis and diagnosis, and a target for novel target therapy development [10, 11].
How APA modulates pre-mRNA
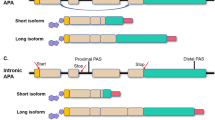
Based on the locations of pAs, APA can be classified into two major categories: UTR-APA (Fig. 1a) and coding region-APA (CR-APA) (Fig. 1b-d). For CR-APA, alternative pAs are located in exons or introns. Therefore, CR-APA affects coding regions via alternative splicing (AS), leading to generation of protein isoforms with distinct C-termini [12, 13]. For UTR-APA, alternative pAs are located in the 3′-UTR, leading to the transcription products containing the same coding frame but variable 3′-UTRs. Previous studies suggested that global UTR-APA events are tissue-specific, with3′-UTR shortening positively correlates to cell proliferation and negatively to cellular differentiation [14,15,16].

Comparison of APA and AS. a-c The patterns of APA can be classified into two main types: UTR-APA and CR-APA. For UTR-APA, alternative PAS resides in the 3′-UTR. Therefore, UTR-APA can generate transcripts with varying UTR lengths without changing the coding sequences. There are major types of CR-APA that may yield transcripts with truncated coding sequence. d Yellow is used to label the extended exon. For AS, e-constitutive splicing; f exon skipping/ inclusion;g alternative 5′-splice sites;h alternative 3′-splice sites; i intron retention; j mutually exclusive exons
The pre-mRNA 3′-processing complex is formed by several elements, including the canonical poly(A) signal sequence AAUAAA or its close variants (e.g. AAAUAA, AUAAAA, AUUAAA, AUAAAU, AUAAAG, CAAUAA, UAAUAA, AUAAAC, AAAAUA, AAAAAA, AAAAAG), which are utilized with varying frequencies throughout the genome, usually within 15–50 nts from the pA site [6, 8, 17,18,19,20]. UGUA elements are often located upstream of the pA site, U-rich elements are located near the pA site, and U/GU-rich elements are located within ~ 100 nts downstream of the pA site [7, 21, 22]. However, ~ 20% of human poly(A) signals are not surrounded by U−/GU-rich regions [23].
Out of 80 core factors in mammalian cells, about 20 of them are involved in the C/P machinery [24,25,26]. Generally, these core factors can be divided into four elements as followings (Fig. 2) [27, 28]:

The APA complex and its machinery. CFIm complex binds to the conserved upstream UGUA region to mediate the cleavage reaction and recruit other proteins, including CPSF and CSTF. After combining with PAP, this complex translocates through the pre-mRNA in a 5′ to 3′ fashion. Upon arrival at the AAUAAA region, the adenosine acidification signal CPSF recognizes the polyadenylation signal AAUAAA and CPSF73 cleaves the mRNA. CSTF then binds to the GU- or U-rich sequence. The U-rich region bound to the FIP1L1 subunit of the CPSF is located between the polyadenylation signal AAUAAA and the cleavage site. Symplekin functions as a scaffold protein and PAPs catalyze the addition of untemplated adenosines. Generally, the usage of the proximal pAs generates short isoforms and the translation can be suppressed, often resulting in less protein
CPSF (cleavage and polyadenylation specificity factor) is composed of CPSF1–CPSF4 (also known as CPSF160, CPSF100, CPSF73 and CPSF30), WDR33 and FIP1L1 (also known as Fip1) [22, 29]. The current understanding is that WDR33 and CPSF4 directly interact with pAs, and CPSF3 carries out the endonucleolytic cleavage [30, 31]. Working as a complex, CPSF recognizes the polyadenylation signal sequence AAUAAA and cleaves the pre-mRNA. This provides sequence specificity that may play an important role in regulating pA site selection, gene expression, cancer cell migration, metastasis, and eventually disease outcome [32]. As a part of CPSF complex, CPSF73 is an endonuclease that cleaves the pre-mRNA at the pA site [33]. However, under oxidative stress, CPSF73 translocates from the nucleus to the cytosol and causes significant inhibition of polyadenylation activity in prostate cancers [34]. Furthermore, Fip1, a member of the CPSF complex, potentially serves as a regulator of cellular self-renewal. Indeed, Fip1 depletion in mouse embryonic stem cells (ESCs) results in loss of cellular undifferentiated states and self-renewal capabilities due to the usage of preferred distal poly(A) site (dpA), ultimately leading to 3′-UTR lengthening of selected genes that determine the cell fate [35].
CSTF (cleavage stimulation factor) is composed of CSTF1, CSTF2, and CSTF3 (50 kDa, 64 kDa, and 77 kDa, respectively), and plays a key role in the cleavage reaction [36, 37]. CSTF complex can bind to the U- or GU-rich field downstream of the cleavage site to boost cleavage. For example, CSTF2, also known as CSTF64, directly interacts with the U/GU-rich region to modulate the 3′-terminal processing efficiency [38, 39]. Some studies reported that CSTF not only promote the usage of pAs, but also affect cell proliferation and potentially act as a biomarker of cancer invasion and prognosis [40, 41]. CSTF64 acts as an essential polyadenylation factor and a master regulator of 3′-UTR shortening across multiple tumor types. The expression of CSTF64 was found to be associated with poor lung cancer prognosis and overexpression of CSTF64 promoted lung cancer cell proliferation and invasion [25].
CFI and CFII (cleavage factors I and II) are consisted of CFIm25 (also known as NUDT21/nudix hydrolase 21/CPSF5), CFIm59 and CFIm68, all of which bind upstream of the conserved UGUA motif to mediate the cleavage reaction [28, 42]. CFIm binding can function as a primary determinant of pA sites by looping out an entire pA region and thereby inducing the selection of an APA site [43]. Other proteins, including symplekin, poly(A) polymerase (PAP), and poly(A) binding protein (PAB), can regulate APA site selection as well. PABs (PABII, RBBP6, PABPN1) bind to the growing poly(A) tail, preventing the interaction between CPSF and the poly(A) polymerase. Those activities primarily occur when the tail is ~ 250 nts and the purpose of which is to control poly(A) tail length while APA in progression [44, 45].
The factors involved in the C/P machinery usually participate in APA regulation. Among them, CFIm25 has been identified as the major global regulator of APA, whose knockdown not only induce a global switch to the use of proximal poly(A) signal, but also enhance target gene stability and expression [41, 46]. Huang et al. reported that CFIm25 depletion significantly increases the transcript levels of CCND1 and GSK3β, in addition to decrease the utilization of dPAS by several oncogenes (IGF1R, CCND1, and GSK3β) [46]. Furthermore, gene ontology analyses (GO) demonstrated that CFIm25 not only modulate APA via MAPK signaling pathways, but is also linked with cancer-associated signaling and protein ubiquitination signaling pathways [47]. Moreover, depletion of CFIm25 and CFIm68, but not CFIm59, leads to proximal polyadenylation site selection in HEK293 cells [48, 49]. However, Xia et al. reported that there are no CFIm25 expression differences between tumor tissue and healthy tissue [3]. Kubo et al. also reported that CFIm may not have a role for poly(A) site selection [27]. Additionally, Takagaki et al. demonstrated that CSTF64 is the first factor in APA 3′-end processing and that IgM can employ APA to activate mouse B-cells [50]. While it appears that CFIm plays a key role in the regulation of APA, its exact role still remains unclear [51].
RNA-binding proteins (RBPs) can also affect APA’s capability to target mRNAs by competing with or enhancing the binding of polyadenylation machinery proteins to their target sites [8]. Xiang et al. analyzed the global APA profiles from a large database across different cancer types and suggested that PABPN1 is the master regulator of APA profiling across different cancer types. A CTRP dataset demonstrated that PABPN1 expression is statistically correlated with the sensitivity towards 31 drugs [52]. RBPs can work alone to prevent the binding of other APA factors to the proximal poly(A) sites or affect APA selection through its role in maintaining RNA stability [53,54,55]. Furthermore, RBPs can regulate the dynamic APA profile and promote mitosis-to-meiosis transition [4].
How APA is regulated
APA is a very comprehensive molecular biological process, involving numerous cellular elements. Currently, we still don’t know much about this unique biologic process. However, the situation has been rapidly improved in a very short period of time after the scientific community sensed the importance of APA in cellular biology and its potential role as a novel cancer therapy target. APA is a dynamically and spatiotemporally coordinated process of numerous core factors. For example, CFIm can bind to the specific RNA sequence in a pre-mRNA and then recruits the core factor CPSF through its interaction with a CPSF subunit, hFip15 [56]. CSTF-64 may interact with CPSF73, but not CFIm25. It was observed that both CSTF64 and CPSF73 levels are elevated in the cells that migrate into the healthy tissue, but not for CFIm25 level [17]. CFIm is involved in the early step of pre-mRNA 3’-processing complexe assembling via alternatively stimulating or suppressing cleavage and poly(A) addition depending on the levels of its own or other core factors, and the RNA sequence surrounding the potential cleavage sites [57].
Besides the core factors, a variety of physiological conditions also participate in APA regulation, such as the local chromatin structure, nucleosome positioning, DNA methylation, and histone modifications [58]. Interestingly, some factors participating in the 5′-terminal capping can also influence the efficiencies of both cleavage and polyadenylation [59].
Additionally, APA can be regulated at the transcription level. The transcription machinery, such as transcription initiation, progression, and splicing, is likely to affect the efficiency and specificity of polyadenylation [60]. Therefore, investigating the association between the specific sequence elements at the promoter region and the poly(A) site selection will greatly aid us in uncovering the mechanism behind this interesting phenomenon, which may potentially help in developing a novel cancer therapy strategy [61].
How APA is methodologically analyzed
Since the effects of pAs in IgM and dihydrofolate reductase (DHFR) gene encoding were observed in 1980, a series of stringent research methods and strategies have been developed to identify and study APA, such as the Poly(A)-ClickSeq next-generation sequencing (NGS) technology [62,63,64,65]. With the support of these novel methodologies, especially with the advancement of NGS technology and the rapid accumulation of sequencing data from those gene expression variants, the experimentally determined genetic pA databases are continuously expanding [66, 67].
Based on 3′-enriched RNA-seq protocols, APA analysis methods can be classified mainly into two categories: oligo (dT) priming-based methods and RNA manipulation-based methods [5, 62, 68, 69]. Because the only reads mapped to the 3′ -termini of the mRNA are useful for APA discovery, the number of reads limited these methods. If the read coverage to 5′- and 3′-termini are low, RNA-seq will not be suitable for identifying pAs precisely and extensively. Moreover, another challenge is to resolve the read mapping ambiguity due to isoform transcripts overlap [18, 70]. Though it has reading length limitation, a range of RNA-seq algorithms have been developed to quantify relative changes in 3′-UTR length, therefore to predict APA events. Several pA detection and APA analytical methods and algorithms also have been developed in the last several years, such as Dynamic Analyses of Alternative PolyA Adenylation (DaPars), 3USS, MISO, Roar, QAPA, and Change Points [3, 71, 72] . A 2019 review by Gruber and Zavolaneloquently compared these methods [73] .
DaPars is the most popular data analysis method among them, although QAPA is more efficient and sensitive [74]. DaPars identifies distal pAs based on RNA-seq data, and then uses a regression model to perform de novo identification and quantification of dynamic APA events between two conditions, regardless of any prior APA annotation [3]. The probability of yielding sequenced reads is unified among individual isoforms. The pAs present at positions along gene locations that exhibit a distinct drop in RNA-seq read coverage [75]. After correcting the potential RNA-seq non-uniformity bias along the gene body, the exact location of proximal APA site can be identified, and the statistically significant dynamic APAs and their activities then will be detected. The key methodological innovation of DaPars is the direct inference of de novo APA events from existing RNA-seq data without relying on any additional experiments. Another advantage of DaPars is that it can resolve the overlapping of neighboring genes that may give false-positive results by increasing the cutoffs. However, due to non-uniform read coverage along loci, this method limits the accuracy of de novo poly(A) site detection by increasing the false positive rate.
QAPA quantitatively infers APA from conventional RNA-seq data by directly estimating the absolute alternative 3′-UTR isoform expression. It then computes the relative expression of each isoform among all isoforms to assess APA [74]. The limitation of QAPA is that it requires pre-defined pAs. However, this problem can be mitigated by the generation of an expanded resource of annotated pAs that incorporate data from 3′-UTR RNA-seq and other resources [74]. Because of reading coverage biases at the 3′-terminus of transcripts, poor yields of non-templated poly(A) tail-containing reads, and ambiguity of read mapping in overlapping transcript isoforms, the methods based on canonical RNA-seq data are limited while attempting to precisely map the pAs [18, 76]. However, with the advance of molecular technology, the methods to study APA have been continuously growing. Wang et al. used CRISPR/Cas9 methodology to study the biological function of APA via editing the weak poly(A) signal to a canonical poly (A) signal and directing the signals to target specific poly(A) sites [77].
In brief, each of current available APA analytic methods has its advantages and limitations. The analytical strategies based on canonical RNA-seq data are utilized most within the APA research community.
Single-cell level study The advantage of single-cell approach is that it can significantly reduce the background noise from bulk cells that contain a mixture of RNA material extracted from cells originating from various tissues or differentiations.
With the development of single-cell analysis technology, APA variations among the cells has been recently investigated [78]. Though single-cell APA research has rarely been conducted on a large scale, this technique works on high-depth and full-length of single-cell RNA-seq (scRNA-seq), which makes it a possible tool to accurately analyze APA. Jingle Bells and scRNA-SeqDB (https://bioinfo.uth.edu/scrnaseqdb/) utilized scRNA-seq datasets to investigate a variety of cancer types [79]. Ye et al. reported the use of scRNA-seq data to investigate dynamic APA usage variations in different bone marrow mononuclear cell types from a large sample collections containing both healthy controls and AML patients. They found that, in comparing to healthy individuals, AML patients appear to have lower APA diversity among eight different cell types. They further revealed extensive involvement of APA regulation in erythropoiesis during leukemia progression at the single-cell level [50]. By analyzing 515 scRNA-seq datasets extracted from 11 breast cancer patients, Kim et al. reported that cell-type-specific APA can be identified in single cell level based on 3′-UTR length variation in combination with gene expression level and APA patterns. Moreover, they demonstrated that immune-specific APA signatures in breast cancer can potentially be utilized as a prognostic marker for early stage breast cancers [31].
APA and alternative splicing: Though there are significant differences between APA and alternative splicing (AS), both APA and AS can generate various isoforms, even interacting with each other during pre-mRNA process. Additionally, while APA has four typical isoforms, AS has six (Fig. 2). Several in-depth analyses of transcriptomic data from various human tissues and cell lines revealed a strong correlation between APA and AS [6, 55, 80]. If the pA is within the terminal exon, the APA can act like a special type of AS, named CR-APA, which cannot possess an in-frame stop codon or 3′-UTR and is likely to be degraded rapidly through the non-stop code mediated mRNA decay process (Fig. 1b) [11, 12, 81]. Shen et al. reported that APA and splicing factor SRSF3 worked together to modulate the cell-aging process [82]. While APA may play a role in some splicing factor-mediated AS, splicing factors may also work with APA elements to assist in this process. For example, U2AF2 and RBPs are capable of interacting and recruiting CFI to facilitate 3′-terminus formation near the polypyrimidine tracts [79, 83]. Furthermore, CPSF complex can interact with splicing factor TFIID (transcription factor II D) in regulating RNA polymerase II [84, 85]. It is also observed that U1 snRNP (small nuclear ribonucleoprotein) can work within introns by suppressing premature cleavage and polyadenylation. U1 depletion also leads to the activation of intron poly(A) signals and causes genome-wide APA [86, 87].
AS and APA also compete each other while in CR-APA. For example, the ablation of the splicing factor 3B subunit1 (a component of U2 snRNP, also named SF3b1) can activate the intron PAS. U1 snRNP can also independently influence APA splicing activities [88]. Since U1 snRNP can bind to the 5′-terminal region of the transcript and block potential cleavage factor recognition, U1 snRNP knockdown increases the utilization of the pA sites within introns close to that transcript area [89, 90]. However, Movassat et al. demonstrated that the association between APA and AS is limited to terminal introns [91]. They also demonstrated that CstF64 knockdown can indirectly influence the AS of hnRNP A2/B1, but not APA, in HeLa cells [92].
How APA regulate cell cycle
There are many genes, including TP53, CDC6 (cell division cycle 6), CyclinD1 (CCND1), and CDK (cyclin-dependent kinase), are associated with cell cycle checkpoints and regulate cell cycle progression. As pre-mRNA usually has more than one pA sites, the cell cycle relevant gene products are modulated by the APA mechanism and generate various isomers. 3′-UTR shortening of CDC6, a major regulator of DNA replication, is linked to higher CDC6 protein levels and increased S-phase entry in breast cancer cells [93]. Cyclin D1, which plays a critical role in promoting G1–S phase transition in many cell types, is subject to APA regulation via both UTR-APA and CR-APA mechanisms [77, 94]. In addition, Xiang et al. examined the top 10% of all 20,532 genes associated with APA events and observed that most of these genes participate in chromatin structure-related activities, suggesting a relationship between APA processing and chromatin structure modification [53]. Mitra et al. found that APA acts as a linkage between cell cycle and tissue migration through analyzing mice dermal excisional wounds [17]. They demonstrated that proliferating cells adjacent to wounds express higher levels of APA factors than quiescent fibroblasts in unwounded skin. PIGN, which regulates cell cycle through interacting with the spindle assembly checkpoint proteins, is found to harbrt 6 pA sites in its 3′-UTR (Fig. 3) [95].

Track of PIGN by Grch37/hg19. PIGN location in chromosome18(q21.33) has three transcripts. There are 6 pAs in the polyA database
How APA interacts with miRNA in post-transcriptional modulation
More than 50% of conserved microRNAs (miRNAs) target sites residing downstream of proximal pAs in mammalian genes. As a result, the UTR-APA plays a key role in regulating the interaction between transcripts and miRNAs [96]. APA is recently identified as a widespread mechanism controlling gene stability and expression. The miRNA targeting sites are mostly located in 3′-UTR [26, 52, 97]. The transcripts with shorter 3′-UTR lengths are usually more stable due to the loss of targeting sites for miRNAs. It was previously demonstrated that APA is a crucial regulatory mechanism in several cancer types, such as glioblastoma tumor, hepatocellular carcinoma, prostate cancer, and breast cancer [1]. However, Gruber et al. reported that 3′-UTR shortening has only a limited impact on murine and human T-lymphocyte proliferation. It also showed that not every APA event relates to higher protein levels [98]. Several studies have reported that the effects of APA on mRNA stability and ribosome loading are marginal, depending on the cell-type-specific miRNA expression and availability of RNA-binding proteins [37, 96]. A typical example is PAX3 gene expression regulation. PAX3 is a major regulator of myogenic differentiation, whose transcript has a miR-206 target site in the 3′-UTR. However, PAX3 isoforms show variant differentiation patterns in different muscle types [98, 99].
APA can also modulate miRNA targets that are located in introns. The ZFR gene is targeted by its intronic miRNA (miR-579) in U87 cell line. Hinske et al. also reported that the APA signal plays a role in delivering miRNA negative feedback to ZFP gene [100].
APA affects gene expression not only by shortening the 3′-UTR to remove the miRNA targeting sites, but also via other molecular mechanisms. Masamha et al. reported that CFIm25 and miR-23 were independent in suppressing the expression of one of the glutaminase isoforms’ 3′-UTRs [22]. Therefore, although mRNA escapes miRNA suppression via shortening 3′-UTR to remove miRNA target site (a canonical APA mechanism), other APA and miRNA interaction mechanisms also coexist.
Prospects
APA is relatively a new biomedical research field. Though we achieved some milestone accomplishments on APA research in the last several years, much remains to be elucidated (Fig. 4). The APA studies have been focusing on the direct actions of various trans-acting factors in the last several years. Future investigations hopefully will concentrate on the signal regulation of those trans-acting factors at molecular and cellular levels. It is known that APA plays crucial roles in editing pre-mRNAs and determining the specificity and stability of the subsequent mRNA isoforms. APA participates in modulating innate antiviral immune response, regulating cancer initiation and prognosis, and developing drug resistance. Meanwhile, APA behaves differently on individual gene, cell type, tissue type, and even disease. Understanding APA and its comprehensive regulatory mechanisms in human diseases will open a new venue for pursuing precision medicine and personalized medicine.

APA Impacts at molecular, cellular, and clinical levels. a APA can affect cell functions through various molecular mechanisms;b&c APA has relationship with many type diseases and the diagnosis, prognosis and treatment of diseases
Conclusion
APA is a crucial post-transcriptional regulatory mechanism that could generate various mRNA isoforms from a single gene. Each mRNA isoform eventually translates into a protein product with unique biological functions. APA could regulate almost every major step of molecular cell biological process, such as cellular genomic stability, proliferation capability, and transformation feasibility. Our journal of understanding APA is just beginning. However, the emerging evidences indicate that APA, at least, could be potentially a biomarker for disease diagnosis, severity stratification, and prognostic forecast; and possibly a novel therapy target.
Availability of data and materials
Yes.
Abbreviations
- APA:
-
Alternative polyadenylation
- NGS:
-
Next generation-sequencing
- CFI/IIm:
-
Cleavage factors I /II
- CFIm25:
-
Homolog of CFIM-25
- CPSF:
-
Cleavage and polyadenylation specificity factor
- CSTF:
-
Cleavage stimulation factor
- RBPs:
-
RNA-binding proteins
- pAs:
-
Cleavage/polyadenylation(C/P) sites
- PABPN1:
-
Polyadenylation binding protein 1
- CR-APA:
-
Coding region-APA
- AS:
-
Alternative splicing
- ESCs:
-
Embryonic stem cells
- PAP:
-
Poly(A) polymerase
- PAB:
-
Poly(A) binding protein
- GO:
-
Gene ontology analyses
- DHFR:
-
Dihydrofolate reductase
- DaPars:
-
Dynamic Analyses of Alternative PolyAAdenylation
- 3USS:
-
A web server for detecting alternative 3′UTRs from RNA-seq experiments
- MISO:
-
mixture-of-isoforms model
- Roar:
-
detecting alternative polyadenylation with standard mRNA sequencing libraries
- QAPA:
-
Quantification of APA
- scRNA-seq:
-
Single-cell RNA-seq
- BMMCs:
-
Bone marrow mononuclear cells
- TFIID:
-
Transcription factor II D
- SF3b1:
-
Splicing factor 3B subunit1
- CDC6 :
-
Division cycle 6
- CDK :
-
Cyclin-depentkinase
- PIGN :
-
Phosphatidylinositol glycan anchor biosynthesis class N
- PAX3 :
-
Paired box 3 gene
- Pre-mRNA:
-
Pre-messenger RNA
References
Yeh HS, Yong J. Alternative Polyadenylation of mRNAs: 3′-Untranslated Region Matters in Gene Expression. Mol Cells. 2016;39(4):281–5.
Proudfoot NJ. Ending the message: poly(A) signals then and now. Genes Dev. 2011;25(17):1770–82.
Xia Z, Donehower LA, Cooper TA, Neilson JR, Wheeler DA, Wagner EJ, et al. Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3′-UTR landscape across seven tumour types. Nat Commun. 2014;5:5274.
Shan L, Wu C, Chen D, Hou L, Li X, Wang L, et al. Regulators of alternative polyadenylation operate at the transition from mitosis to meiosis. J Genet Genome. 2017;44(2):95–106.
Hoque M, Ji Z, Zheng D, Luo W, Li W, You B, et al. Analysis of alternative cleavage and polyadenylation by 3′ region extraction and deep sequencing. Nat Methods. 2013;10(2):133–9.
Derti A, Garrett-Engele P, Macisaac KD, Stevens RC, Sriram S, Chen R, et al. A quantitative atlas of polyadenylation in five mammals. Genome Res. 2012;22(6):1173–83.
Millevoi S, Vagner S. Molecular mechanisms of eukaryotic pre-mRNA 3′ end processing regulation. Nucleic Acids Res. 2010;38(9):2757–74.
Erson-Bensan AE, Can T. Alternative Polyadenylation: Another Foe in Cancer. Mol Cancer Res. 2016;14(6):507–17.
Mitra M, Johnson EL, Swamy VS, Nersesian LE, Corney DC, Robinson DG, et al. Alternative polyadenylation factors link cell cycle to migration. Genome Biol. 2018;19(1):176.
Li W, You B, Hoque M, Zheng D, Luo W, Ji Z, et al. Systematic profiling of poly(A)+ transcripts modulated by core 3′ end processing and splicing factors reveals regulatory rules of alternative cleavage and polyadenylation. PLoS Genet. 2015;11(4):e1005166.
Mayr C. Evolution and Biological Roles of Alternative 3'UTRs. Trends Cell Biol. 2016;26(3):227–37.
Di Giammartino DC, Nishida K, Manley JL. Mechanisms and consequences of alternative polyadenylation. Mol Cell. 2011;43(6):853–66.
Tian B, Manley JL. Alternative cleavage and polyadenylation: the long and short of it. Trends Biochem Sci. 2013;38(6):312–20.
Zhang H, Lee JY, Tian B. Biased alternative polyadenylation in human tissues. Genome Biol. 2005;6(12):R100.
Ji Z, Tian B. Reprogramming of 3′ untranslated regions of mRNAs by alternative polyadenylation in generation of pluripotent stem cells from different cell types. PLoS One. 2009;4(12):e8419.
Ulitsky I, Shkumatava A, Jan CH, Subtelny AO, Koppstein D, Bell GW, et al. Extensive alternative polyadenylation during zebrafish development. Genome Res. 2012;22(10):2054–66.
Beaudoing E, Freier S, JR JRW, Claverie JM, Gautheret D. Patterns of variant polyadenylation signal usage in human genes. Genome Res. 2000;10:1001-10.
Elkon R, Ugalde AP, Agami R. Alternative cleavage and polyadenylation: extent, regulation and function. Nat Rev Genet. 2013;14(7):496–506.
Wang R, Zheng D, Yehia G, Tian BA. Compendium of Conserved Cleavage and Polyadenylation Events in Mammalian Genes. Genome Res. 2018;28(10):1427–41.
Gruber AJ, Schmidt R, Gruber AR, Martin G, Ghosh S, Belmadani M, et al. A comprehensive analysis of 3′ end sequencing data sets reveals novel polyadenylation signals and the repressive role of heterogeneous ribonucleoprotein C on cleavage and polyadenylation. Genome Res. 2016;26(8):1145–59.
Legendre M, Gautheret D. Sequence determinants in human polyadenylation site selection. BMC Genomics. 2003;4(1):7.
Shi Y, Di Giammartino DC, Taylor D, Sarkeshik A, Rice WJ, Yates JR 3rd, et al. Molecular architecture of the human pre-mRNA 3′ processing complex. Mol Cell. 2009;33(3):365–76.
Mandel CR, Kaneko S, Zhang H, Gebauer D, Vethantham V, Manley JL, et al. Polyadenylation factor CPSF-73 is the pre-mRNA 3′-end-processing endonuclease. Nature. 2006;444(7121):953–6.
Mandel CR, Bai Y, Tong L. Protein factors in pre-mRNA 3′-end processing. Cell Mol Life Sci. 2008;65(7–8):1099–122.
Shi Y, Manley JL. The end of the message: multiple protein-RNA interactions define the mRNA polyadenylation site. Genes Dev. 2015;29(9):9.
Masamha CP, Xia Z, Yang J, Albrecht TR, Li M, Shyu AB, et al. CFIm25 links alternative polyadenylation to glioblastoma tumour suppression. Nature. 2014;510(7505):412–6.
Kubo T, Wada T, Yamaguchi Y, Shimizu A, Handa H. Knock-down of 25 kDa subunit of cleavage factor Im in Hela cells alters alternative polyadenylation within 3′-UTRs. Nucleic Acids Res. 2006;34(21):6264–71.
Venkataraman K, Brown KM, Gilmartin GM. Analysis of a noncanonical poly(A) site reveals a tripartite mechanism for vertebrate poly(A) site recognition. Genes Dev. 2005;19(11):1315–27.
Kaufmann I, Martin G, Friedlein A, Langen H, Keller W. Human Fip1 is a subunit of CPSF that binds to U-rich RNA elements and stimulates poly(A) polymerase. EMBO J. 2004;23(3):616–26.
Dominski Z, Yang XC, Marzluff WF. The polyadenylation factor CPSF-73 is involved in histone-pre-mRNA processing. Cell. 2005;123(1):37–48.
Kim N, Chung W, Eum HH, Lee HO, Park WY. Alternative polyadenylation of single cells delineates cell types and serves as a prognostic marker in early stage breast cancer. PLoS One. 2019;14(5):e0217196.
Brown KMGG. A mechanism for the regulation of pre-mRNA 3′processing by human cleavage factor Im. Mol Cell. 2003;12(6):1467–76.
Ye C, Zhou Q, Hong Y, Li QQ. Role of alternative polyadenylation dynamics in acute myeloid leukaemia at single-cell resolution. RNA Biol. 2019;16(6):785–97.
Zhu ZH, Yu YP, Shi YK, Nelson JB, Luo JH. CSR1 induces cell death through inactivation of CPSF3. Oncogene. 2009;28(1):41–51.
Lackford B, Yao C, Charles GM, Weng L, Zheng X, Choi EA, et al. Fip1 regulates mRNA alternative polyadenylation to promote stem cell self-renewal. EMBO J. 2014;33(8):878–89.
Wilusz J, Shenk T. A uridylate tract mediates efficient heterogeneous nuclear ribonucleoprotein C protein-RNA cross-linking and functionally substitutes for the downstream element of the polyadenylation signal. Mol Cell Biol. 1990;10:6397-06.
Spies N, Burge CB, Bartel DP. 3′ UTR-isoform choice has limited influence on the stability and translational efficiency of most mRNAs in mouse fibroblasts. Genome Res. 2013;23(12):2078–90.
Nunes NM, Li W, Tian B, Furger A. A functional human Poly(A) site requires only a potent DSE and an A-rich upstream sequence. EMBO J. 2010;29(9):1523–36.
McCracken S, Fong N, Rosonina E, Yankulov K, Brothers G, Siderovski D, Hessel A, Foster S, Shuman S, Bentley DL. 5′-capping enzymes are targeted to pre-mRNA by binding to the phosphorylated carboxy-terminal domain of RNA polymerase II. Genes Dev. 1997;11:3306-18.
Shell SA, Hesse C, Morris SM Jr, Milcarek C. Elevated levels of the 64-kDa cleavage stimulatory factor (CstF-64) in lipopolysaccharide-stimulated macrophages influence gene expression and induce alternative poly(A) site selection. J Biol Chem. 2005;280(48):39950–61.
Hwang HW, Park CY, Goodarzi H, Fak JJ, Mele A, Moore MJ, et al. PAPERCLIP Identifies MicroRNA Targets and a Role of CstF64/64tau in Promoting Non-canonical poly(A) Site Usage. Cell Rep. 2016;15(2):423–35.
Yang Q, Gilmartin GM, Doublie S. Structural basis of UGUA recognition by the Nudix protein CFI(m)25 and implications for a regulatory role in mRNA 3′ processing. Proc Natl Acad Sci U S A. 2010;107(22):10062–7.
Yang Q, Doublie S. Structural biology of poly(A) site definition. Wiley Interdiscip Rev RNA. 2011;2(5):732–47.
Kuhn U, Gundel M, Knoth A, Kerwitz Y, Rudel S, Wahle E. Poly(A) tail length is controlled by the nuclear poly(A)-binding protein regulating the interaction between poly(A) polymerase and the cleavage and polyadenylation specificity factor. J Biol Chem. 2009;284(34):22803–14.
Lin Y, Li Z, Ozsolak F, Kim SW, Arango-Argoty G, Liu TT, et al. An in-depth map of polyadenylation sites in cancer. Nucleic Acids Res. 2012;40(17):8460–71.
Huang J, Weng T, Ko J, Chen NY, Xiang Y, Volcik K, et al. Suppression of cleavage factor Im 25 promotes the proliferation of lung cancer cells through alternative polyadenylation. Biochem Biophys Res Commun. 2018;503(2):856–62.
Brumbaugh J, BDi S, Wang X, Borkent M, Forouzmand E, Clowers KJ, et al. Nudt21 Controls Cell Fate by Connecting Alternative Polyadenylation to Chromatin Signaling. Cell. 2018;172(1–2):106–20 e21.
Gruber AR, Martin G, Keller W, Zavolan M. Cleavage factor Im is a key regulator of 3′ UTR length. RNA Biol. 2012;9(12):1405–12.
Martin G, Gruber AR, Keller W, Zavolan M. Genome-wide analysis of pre-mRNA 3′ end processing reveals a decisive role of human cleavage factor I in the regulation of 3′ UTR length. Cell Rep. 2012;1(6):753–63.
Takagaki Y SR, Peterson ML, Manley JL. The polyadenylation factor CstF-64 regulates alternative processing of IgM heavy chain pre-mRNA during B cell differentiation. Cell. 1996:87:941-52.
Jafari Najaf Abadi MH, Shafabakhsh R, Asemi Z, Mirzaei HR, Sahebnasagh R, Mirzaei H, et al. CFIm25 and alternative polyadenylation: Conflicting roles in cancer. Cancer Lett. 2019;459:112-21.
Xiang Y, Ye Y, Lou Y, Yang Y, Cai C, Zhang Z, et al. Comprehensive Characterization of Alternative Polyadenylation in Human Cancer. J Natl Cancer Inst. 2018;110(4):379–89.
de Klerk E, Venema A, Anvar SY, Goeman JJ, Hu O, Trollet C, et al. Poly(A) binding protein nuclear 1 levels affect alternative polyadenylation. Nucleic Acids Res. 2012;40(18):9089–101.
Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40(12):1413–5.
Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456(7221):470–6.
Rappsilber J, Ryder U, Lamond AI, Mann M. Large-scale proteomic analysis of the human spliceosome. Genome Res. 2002;12(8):1231–45.
Chan S, Choi EA, Shi Y. Pre-mRNA 3′-end processing complex assembly and function. Wiley Interdiscip Rev RNA. 2011;2(3):321–35.
Huang H, Liu H, Sun X. Nucleosome distribution near the 3′ ends of genes in the human genome. Biosci Biotechnol Biochem. 2013;77(10):2051–5.
Jiao X, Chang JH, Kilic T, Tong L, Kiledjian M. A mammalian pre-mRNA 5′ end capping quality control mechanism and an unexpected link of capping to pre-mRNA processing. Mol Cell. 2013;50(1):104–15.
Proudfoot NJFA, Dye MJ. Integrating mRNA processing with transcription. Cell. 2002;108:501-12.
Mansfield KD, Keene JD, Neuron-specific ELAV. Hu proteins suppress HuR mRNA during neuronal differentiation by alternative polyadenylation. Nucleic Acids Res. 2012;40(6):2734–46.
Jan CH, Friedman RC, Ruby JG, Bartel DP. Formation, regulation and evolution of Caenorhabditis elegans 3'UTRs. Nature. 2011;469(7328):97–101.
Routh A, Ji P, Jaworski E, Xia Z, Li W, Wagner EJ. Poly(A)-ClickSeq: click-chemistry for next-generation 3-end sequencing without RNA enrichment or fragmentation. Nucleic Acids Res. 2017;45(12):e112.
Fullwood MJ, Wei CL, Liu ET, Ruan Y. Next-generation DNA sequencing of paired-end tags (PET) for transcriptome and genome analyses. Genome Res. 2009;19(4):521–32.
Wang L, Dowell RD, Yi R. Genome-wide maps of polyadenylation reveal dynamic mRNA 3′-end formation in mammalian cell lineages. RNA. 2013;19(3):413–25.
Wang R, Nambiar R, Zheng D, Tian B. PolyA_DB 3 catalogs cleavage and polyadenylation sites identified by deep sequencing in multiple genomes. Nucleic Acids Res. 2018;46(D1):D315–D9.
You L, Wu J, Feng Y, Fu Y, Guo Y, Long L, et al. APASdb: a database describing alternative poly(A) sites and selection of heterogeneous cleavage sites downstream of poly(A) signals. Nucleic Acids Res. 2015;43(Database issue):D59–67.
Berkovits BD, Mayr C, et al. Nature. 2015;522(7556):363–7.
Yoon OK, Hsu TY, Im JH, Brem RB, et al. PLoS Genet. 2012;8(8):e1002882.
Ji Z, Luo W, Li W, Hoque M, Pan Z, Zhao Y, et al. Transcriptional activity regulates alternative cleavage and polyadenylation. Mol Syst Biol. 2011;7:534.
Le Pera L, Mazzapioda M, Tramontano A, et al. Bioinformatics. 2015;31(11):1845–7.
Katz Y, Wang ET, Airoldi EM, Burge CB. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods. 2010;7(12):1009–15.
Gruber AJ, Zavolan M. Alternative cleavage and polyadenylation in health and disease. Nat Rev Genet. 2019;20(10):599–614.
KCH H, Blencowe BJ, Morris Q. A new method for the systematic analysis of alternative polyadenylation from RNA-seq data. Genome Biol. 2018;19(1):45.
Wu X, Liu M, Downie B, Liang C, Ji G, Li QQ, et al. Genome-wide landscape of polyadenylation in Arabidopsis provides evidence for extensive alternative polyadenylation. Proc Natl Acad Sci U S A. 2011;108(30):12533–8.
Pickrell JK, Marioni JC, Pai AA, Degner JF, Engelhardt BE, Nkadori E, et al. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature. 2010;464(7289):768–72.
Wang Q, He G, Hou M, Chen L, Chen S, Xu A, et al. Cell Cycle Regulation by Alternative Polyadenylation of CCND1. Sci Rep. 2018;8(1):6824.
Velten L, Anders S, Pekowska A, Jarvelin AI, Huber W, Pelechano V, et al. Single-cell polyadenylation site mapping reveals 3′ isoform choice variability. Mol Syst Biol. 2015;11(6):812.
Millevoi S, Loulergue C, Dettwiler S, Karaa SZ, Keller W, Antoniou M, et al. An interaction between U2AF 65 and CF I(m) links the splicing and 3′ end processing machineries. EMBO J. 2006;25(20):4854–64.
Shepard PJ, Choi EA, Lu J, Flanagan LA, Hertel KJ, Shi Y. Complex and dynamic landscape of RNA polyadenylation revealed by PAS-Seq. RNA. 2011;17(4):761–72.
Masamha CP, Xia Z, Peart N, Collum S, Li W, Wagner EJ, et al. CFIm25 regulates glutaminase alternative terminal exon definition to modulate miR-23 function. RNA. 2016;22(6):830–8.
Shen TLH, Song Y, et al. Alternative polyadenylation dependent function of splicing factor SRSF3 contributes to cellular senescence. Aging. 2019;11(5):1356–88.
Chang JW, Zhang W, Yeh HS, de Jong EP, Jun S, Kim KH, et al. mRNA 3′-UTR shortening is a molecular signature of mTORC1 activation. Nat Commun. 2015;6:7218.
Dantonel JCMK, Manley JL, Tora L. Transcription factor TFIID recruits factor CPSF for formation of 30 end of mRNA. Nature. 1997;389:399-402.
Glover-Cutter K, Kim S, Espinosa J, Bentley DL. RNA polymerase II pauses and associates with pre-mRNA processing factors at both ends of genes. Nat Struct Mol Biol. 2008;15(1):71–8.
Kwon C, Tak H, Rho M, Chang HR, Kim YH, Kim KT, et al. Detection of PIWI and piRNAs in the mitochondria of mammalian cancer cells. Biochem Biophys Res Commun. 2014;446(1):218–23.
Spraggon L, Cartegni L. U1 snRNP-Dependent Suppression of Polyadenylation: Physiological Role and Therapeutic Opportunities in Cancer. Int J Cell Biol. 2013;2013:846510.
Kaida DB, Berg MG, Younis I, Kasim M, Singh LN, Wan L, Dreyfuss G. U1 snrnp protects pre-mRNAs from premature cleavage and poly(A)denylation. Nature. 2010;468:664-8.
Kaida D, Berg MG, Younis I, Kasim M, Singh LN, Wan L, et al. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature. 2010;468(7324):664–8.
Berg MG, Singh LN, Younis I, Liu Q, Pinto AM, Kaida D, et al. U1 snRNP determines mRNA length and regulates isoform expression. Cell. 2012;150(1):53–64.
Movassat M, Crabb TL, Busch A, Yao C, Reynolds DJ, Shi Y, et al. Coupling between alternative polyadenylation and alternative splicing is limited to terminal introns. RNA Biol. 2016;13(7):646–55.
Jia X, Yuan S, Wang Y, Fu Y, Ge Y, Ge Y, et al. The role of alternative polyadenylation in the antiviral innate immune response. Nat Commun. 2017;8:14605.
Akman BH, Can T, Erson-Bensan AE. Estrogen-induced upregulation and 3′-UTR shortening of CDC6. Nucleic Acids Res. 2012;40(21):10679–88.
Wiestner A, Tehrani M, Chiorazzi M, Wright G, Gibellini F, Nakayama K, et al. Point mutations and genomic deletions in CCND1 create stable truncated cyclin D1 mRNAs that are associated with increased proliferation rate and shorter survival. Blood. 2007;109(11):4599–606.
Teye EK, Sido A, Finnberg NK, Xin P, Kawasawa YI, Salzberg AC, et al. PIGN gene expression aberration is associated with genomic instability and leukemic progression in AML with myelodysplastic features. Oncotarget. 2017;8(18):29887–905. https://doi.org/10.18632/oncotarget.15136.
Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science. 2008;320(5883):1643–7.
Morris AR, Bos A, Diosdado B, Rooijers K, Elkon R, Bolijn AS, et al. Alternative cleavage and polyadenylation during colorectal cancer development. Clin Cancer Res. 2012;18(19):5256–66.
Gruber AR, Martin G, Muller P, Schmidt A, Gruber AJ, Gumienny R, et al. Global 3′ UTR shortening has a limited effect on protein abundance in proliferating T cells. Nat Commun. 2014;5:5465.
Boutet SC, Cheung TH, Quach NL, Liu L, Prescott SL, Edalati A, et al. Alternative polyadenylation mediates microRNA regulation of muscle stem cell function. Cell Stem Cell. 2012;10(3):327–36.
Hinske LC, Galante PA, Limbeck E, Mohnle P, Parmigiani RB, Ohno-Machado L, et al. Alternative polyadenylation allows differential negative feedback of human miRNA miR-579 on its host gene ZFR. PLoS One. 2015;10(3):e0121507.
Acknowledgments
None.
Ethics approval and consent for participate
All researches conducted in this study were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Funding
This study was supported by: AA&MDSIF research grant to JJP (146818), American Cancer Society grant to JJP (124171-IRG-13-043-02), NIDA/FDA research grant to JJP (P50 DA036107), JTTai&Co Foundation Cancer Research Grant to JJP, Paige’s Butterfly cancer research grant, and A SUNY Upstate Medical University research grant to JJP.
Author information
Authors and Affiliations
Contributions
JJP initiated the research idea; JJP and FR designed this study, JJP and FR wrote this manuscript; NZ, LZ, and EM participated in manuscript formation by providing comments and suggestions. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflict of interest to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ren, F., Zhang, N., Zhang, L. et al. Alternative Polyadenylation: a new frontier in post transcriptional regulation. Biomark Res 8, 67 (2020). https://doi.org/10.1186/s40364-020-00249-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-020-00249-6