
Abstract
Background
Heart failure (HF) is one of the most common complications of cardiovascular diseases (CVDs) and among the leading causes of death in the US. Many other CVDs can lead to increased mortality as well. Investigating the genetic epidemiology and susceptibility to CVDs is a central focus of cardiology and biomedical life sciences. Several studies have explored expression of key CVD genes specially in HF, yet new targets and biomarkers for early diagnosis are still missing to support personalized treatment. Lack of gender-specific cardiac biomarker thresholds in men and women may be the reason for CVD underdiagnosis in women, and potentially increased morbidity and mortality as a result, or conversely, an overdiagnosis in men. In this context, it is important to analyze the expression and enrichment of genes with associated phenotypes and disease-causing variants among high-risk CVD populations.
Methods
We performed RNA sequencing focusing on key CVD genes with a great number of genetic associations to HF. Peripheral blood samples were collected from a broad age range of adult male and female CVD patients. These patients were clinically diagnosed with CVDs and CMS/HCC HF, as well as including cardiomyopathy, hypertension, obesity, diabetes, asthma, high cholesterol, hernia, chronic kidney, joint pain, dizziness and giddiness, osteopenia of multiple sites, chest pain, osteoarthritis, and other diseases.
Results
We report RNA-seq driven case–control study to analyze patterns of expression in genes and differentiating the pathways, which differ between healthy and diseased patients. Our in-depth gene expression and enrichment analysis of RNA-seq data from patients with mostly HF and other CVDs on differentially expressed genes and CVD annotated genes revealed 4,885 differentially expressed genes (DEGs) and regulation of 41 genes known for HF and 23 genes related to other CVDs, with 15 DEGs as significantly expressed including four genes already known (FLNA, CST3, LGALS3, and HBA1) for HF and CVDs with the enrichment of many pathways. Furthermore, gender and ethnic group specific analysis showed shared and unique genes between the genders, and among different races. Broadening the scope of the results in clinical settings, we have linked the CVD genes with ICD codes.
Conclusions
Many pathways were found to be enriched, and gender-specific analysis showed shared and unique genes between the genders. Additional testing of these genes may lead to the development of new clinical tools to improve diagnosis and prognosis of CVD patients.
Similar content being viewed by others


Introduction
Cardiovascular diseases (CVDs) are among the leading causes of morbidity and mortality in the US [1,2,3]. Among all CVDs, ischemic and nonischemic heart failure (HF) and stroke are the most common causes of death [4, 5]. According to the Centers for Disease Control and Prevention (CDC), a person with a CVD dies every 36 s in the US, totaling 655,000 deaths each year [6]. Numerous studies have reported that age and gender are the socio-demographic characteristics most frequently associated with CVDs [7,8,9], yet the molecular underpinnings of these findings are not yet clear.
Establishing a deeper understanding of CVDs by investigating human genetic epidemiology and susceptibility to CVDs is a central focus of cardiology and biomedical life sciences today [10]. Our evolving understanding of CVD has led to the realization that to effectively diagnose and treat CVD patients, a precision medicine approach is essential [11]. To identify patients during the preclinical stages of CVD and provide the most efficacious personalized treatment, it is essential to analyze the expression of human genes with disease-causing variants, along with associated phenotypes among high-risk CVD populations, mainly those with hypertension, obesity, type 2 diabetes mellitus, asthma, high cholesterol, hernia, chronic kidney, joint pain, myalgia, dizziness and giddiness, osteopenia of multiple sites, chest pain, osteoarthritis, and related diseases [12]. The apparent challenge here is to identify and quantify the genes that contribute to major CVD etiologies specifically HF [13].
Heart diseases like HF happens gradually over time when the muscles of the heart become weak and have difficulty pumping enough blood to nourish your body's many cells. HF and most other CVD clinical phenotypes exist due to complicated relations between genetic and ecological factors [14]. Several recently published studies have shown that gene expression analysis is a proven method for understanding and discovering novel and sensitive biomarkers of CVDs [15]. Gene expression and classification analysis have shown strong correlations of age and gender with obstructive coronary arterial disease (CAD) [16], differentiated ischemic and non-ischemic cardiomyopathy conditions [17], identified genes related to HF [18], and discovered differentially regulated genes linked with recurrent cardiovascular outcomes in first-time acute myocardial infarction (AMI) patients [19]. The susceptibility to heart failure depends on complex and heterogeneous genetic predisposition [20]. This genetic and therefore heritable component has been determined in many HF studies [21,22,23,24]. These studies clearly demonstrated the presence of genetic factors as determinants of heart failure. They also showed the relevance of genetic factors as independent risk factors for heart failure.
In this study, we investigated genes responsible for pathophysiological processes in CVDs with a focus on HF. In addition, our expression profiling revealed new gene-disease associations that may lead to the development of new clinical tools to improve diagnosis and prognosis of patients. RNA sequencing (RNA-seq) analyses are used to quantify expressed genes [25]. We performed an RNA-seq analysis from peripheral blood of diverse CVD patients and focusing on HF and other CVD genes. We used gene expression analysis to identify changes in mRNA abundance [26] that correlate with CVDs to precisely stratify, classify, and distinguish gender- and age-based patient populations to CVD risks and subtypes by using genomic phenotypes [27].
Material and methods
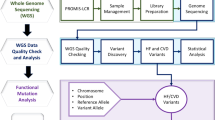
Overall study methodology is divided among four major steps, (1) CVD sample collection, RNA extraction, and high-throughput sequencing, (2) RNA-seq data processing, quality checking, analysis, and visualization, (3) CVD gene-disease annotation and phenotyping, and (4) gene differential expression and pathway enrichment analysis (Fig. 1).

Research methodology divided among four major steps. Steps include, (1) CVD sample collection, RNA extraction, and high-throughput sequencing, (2) RNA-seq data processing, quality checking, analysis, and visualization, (3) CVD gene-disease annotation and phenotyping, and (4) Gene differential expression and pathway enrichment analysis
CVD sample collection, RNA extraction, and high-throughput sequencing
Supporting this study, we have developed an efficient data management system (PROMIS-LCR) for patient recruitment and consent, and for collecting, storing, and tracking of the original and current quantities of biospecimens under standardized conditions for preservation of critical metabolites. This system has been successfully deployed and is operational at the outpatient pavilion (OP) to support establishment of a biobank and a precision medicine initiative (PMI) at UConn Health. Highly heterogeneous and complex clinical terminologies have made electronic health records (EHRs) and diversified public content processing extremely arduous [28]. Addressing this challenge, we have developed an intelligent and dynamic data extract, transform, and loading (ETL) system for efficiently pulling clinical data from different health systems (EPIC and NextGen) and academic data models [29]. We implemented cutting-edge technologies utilizing artificial intelligence (AI) and machine learning (ML) approaches for multimodal data security, aggregation, classification, and examine granularities from population studies to subgroups stratification within the data continuum [28]. We investigated patient’s data centered on medical details, symptoms, age, race, gender, and demographics, and implemented healthcare data analytics process with features to build CVD cohort and from the population data [29]. This system, fully integrated with the PROMIS-LCR system, is tested and operational to efficiently extract and link de-identified medical details of the consented CVDs and even other patients participating in the PMI study with their collected biospecimens at UConn Health.
For high-throughput sequencing, peripheral blood was randomly extracted from 61 CVD patients. Table 1 presents details of all CVD patients (Sample IDs: 1059–1083) and that includes information about their gender (40 male and 21 female), ethnic groups (56 Not Hispanic, 4 Hispanic, and 1 Decline to Answer), and self-described race (42 Whites, 7 Blacks: Blacks or African Americans, 1 Asian, and 1 Decline to Answer, 2 other and 8 NA). These patients were clinically diagnosed with CVDs, and Systolic and Diastolic HF (CMS/HCC), including both heart failure with preserved ejection fraction (HFpEF) and heart failure with reduced ejection fraction (HFrEF). Additional reported diagnoses include cardiomyopathy, hypertension, obesity, type 2 diabetes mellitus, asthma, high cholesterol, hernia, chronic kidney, joint pain, myalgia, dizziness and giddiness, osteopenia of multiple sites, chest pain, and osteoarthritis. Built cohort is based on diverse individuals aged between 45 and 92. All ten healthy (control sample ids 648, 649, 650, 651, 652, 653, 655, 656, 657, 658) individuals (5 male and 5 female patients) had no clinical manifestation of any CVD and were aged between 28 and 78. Among control samples, three patients are self-described Hispanics (651, 656, 653), and the rest of the seven were categorized as non-Hispanic. Nine of them are from White race, and one was unknown (651). Further details are attached in the Additional file 1: Gender and age-based population data classification.
Written informed consent was obtained from all subjects. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. All human samples were used in accordance with relevant guidelines and regulations, and all experimental protocols were approved by Institutional Review Board (IRB), UConn Health. Samples were curated, and all sequencing was done using the Illumina platform. Total RNA was extracted according to the manufacturer’s instructions. RNA quality was assessed for RNA integrity number. For all samples, RNA integrity number was > 7. An Illumina NovaSeq 6000-S4 was used for sequencing. An RNA Sample Preparation kit (Illumina, Inc.) was used for the preparation of cDNA libraries; cDNA libraries that passed size and purity check were retained for the following sequencing. Paired-end 150 bp short sequences (reads, pool across 2 lanes) with 30X coverage were generated for the blood samples, including the Illumina-compatible library (TruSeq Stranded mRNA).
RNA-seq data processing, quality checking, analysis, and visualization
To process and check the quality of RNA-seq data, we developed a pipeline with four operating modules: data pre-processing; data quality checking; data storage and management; and data visualization (Fig. 2). Quality control of raw reads was conducted using FastQC [30], which showed that all raw reads were qualified for downstream analysis. The reads were trimmed using Trimmomatic [31]. We used SAMtools for sorting sequences [32], MarkDuplicates for removing duplicates [33], and CollectInsertSizeMetrics by Picard to compute size distribution and read orientation of paired-end libraries. Afterward, the paired-end raw reads were aligned to the human reference genome (hg38) using HISAT [34] with Bowtie2 [35] software. RNA-seq by expectation maximization (RSEM) [26] was then applied for quantification and identification of identify differentially expressed genes (DEGs) by aligning reads to reference de novo transcriptome assemblies, based on transcript per million mapped reads (TPM). We used TPM as it is the best performing normalization method because it increases the proportion of variation attributable to biology compared to the raw data [36]. The decide-tests were performed to identify DEGs with Benjamini & Hochberg adjustment. Genes with P < 0.05 were selected as the criteria for significant differences (statistical values of all the DEGs are available in the Additional file 3: All DEGs Expression). Hierarchical clustering of DEGs was performed using the “pheatmap” function of the R/Bioconductor package. Expression analysis was also performed to see that the main source of variation is due to biological effects. This analysis was done on genes with an expression level higher than 50 TPM in at least one sample remained as high confidence genes (expression values of all the DEGs are available in the Additional file 5: All DEGs Stats 42 Genes). All computational results were stored in a designated database, using an in-house programmed command line data parser. The expression data were illustrated using the Gene Variant Visualization (GVViZ) environment, another bioinformatics application [37] developed in-house for efficient high-volume sequence data visualization.

RNA-seq data processing pipeline. Used FastQC for quality checking; Trimmomatic to remove adapters and low-quality sequences; SAMtools to sort and index sequences; MarkDuplicates to remove duplicates; CollectInsertSizeMetrics to compute size distribution and read orientation of paired-end libraries; HISAT with Bowtie2 to align sequences to the human reference genome; and RSEM to quantify and identify differentially expressed genes by aligning reads to reference de novo transcriptome assemblies
CVD gene-disease annotation and phenotyping
We have modelled and published a comprehensive knowledgebase of annotated disease-gene-variant data based on multiple clinical and genomics databases, including but not limited to ClinVar, GeneCards, MalaCard, DISEASES, HGMD, Disease Ontology, DiseaseEnhancer, DisGeNET, eDGAR, GTR, OMIM, miR2Disease, DNetDB, The Cancer Genome Atlas, International Cancer Genome Consortium, OMIM, GTR, CNVD, Ensembl, GenCode, Novoseek, Swiss-Prot, LncRNADisease, Orphanet, WHO, FDA, Catalogue Of Somatic Mutations In Cancer (COSMIC), and Genome-wide Association Studies (GWAS) [27, 38, 39]. We used this repository to perform gene-disease annotation for this study and found 43 genes associated with HF. They are TNF, IL6, ACE, MMP2, NOS3, AGT, EDN1, REN, MYH7, AGTR1, AGTR1, NPPA, ADRB2, NR3C2, NR3C2, MME, CRP, MYH6, EPO, CST3, EDNRA, AQP2, MYBPC3, KNG1, VCL, HOTAIR, CDKN2B-AS1, ANKRD1, ADM, AMPD1, PLN, LGALS3, NPPB, ADRB1, UTS2, PIK3C2A, NPPC, CORIN, NPR1, LSINCT5, TUSC7, HSPB7, and RP11-451G4.2 (Table 2). Twenty-three genes associated with other CVDs phenotypes were: SLC2A1, FGF2, FLNA, HBA1, GJB6, ATP2A2, CD40LG, FGF23, TEK, TAC1, DDX41, FADD, ENO2, LEMD3, CD34, TRPV1, GLMN, MB, SMUG1, PDPN, CALD1, KANTR, ZBTB8OS (Table 3). Additional information about these genes is provided in Tables 1 and 2, including names, Ensembl ids, categories, diseases, and chromosomes.
Gene differential expression and pathway enrichment analysis
To associate cellular functions with the DSGs, Gene Set Enrichment Analysis (GSEA) [40] was performed to verify the differences between comparisons. GSEA was carried out by using the curated gene sets of the Molecular Signature Database v7.0. The gene lists of hallmark gene sets (H), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database (C2), and REACTOME pathway database (C2) were used to run GSEA, following the standard procedure described by GSEA user guide. Significantly enriched terms with similar descriptions and functions were further grouped into distinct biological categories (to better reflect the biology in context) and top categories were schematically projected on the network of enriched terms.
Results
Cardiovascular disease is the most important cause of morbidity and mortality in developed countries, causing twice as many deaths as cancer in the USA. The underlying molecular pathogenic mechanisms for these disorders are still largely unknown, but gene expression may play a central role in the development and progression of cardiovascular disease. In this context, we have performed a comprehensive expression study comprising of two types of expression analysis between healthy controls and CVD patients diagnosed with HF and other cardiovascular phenotypes. We started with a global differential gene expression analysis based on TPM count for protein genes to identify significantly differentiated genes (Fig. 3A). We generated a multidimensional scaling (MDS) [41] plot of biological coefficient of variation (BCV) [42] to identify biological variation between case and control groups (Fig. 3B). There were no outliers seen in the MDS plot. We identified 4,712 DEGs between the controls and the CVD group (Fig. 3A) which can be grouped into two clusters (kmeans row clustering) (Fig. 3A). Statistical significance of P value < 0.05 and |log2FC| ≥ 2 showed 42 genes with greater than twofold change. Some of these highly significant genes have already been reported in multiple CVDs (APOD, PIGR, CELSR1, COBLL1, FCRL5, TEAD2, ABCA6, COL4A3, CYP4F2, FMOD, GNG8, IGF2R, PEG10, RAPGEF3, RASGRF1, SCARNA17, TCF4), while some genes (ADAM29, ARHGAP44, CD200, CLEC17A, CLNK, CNTNAP1, CNTNAP2, CTC-454I21.3, DMD, FAM129C, FAM3C, FCRL1, FCRL2, FCRLA, GPM6A, KLHL14, MTRNR2L3, NPIPB5, OSBPL10, PAX5, PCDH9, PHYHD1, POU2AF1, RALGPS2, ZNF888) have shown a novel expression in CVD. Statistical difference in expression for these genes can be seen in the Additional file 4: All DEGs Stats. Gene enrichment of all the DEGs revealed 190 pathways upregulated in the CVD patients and 408 pathways were found to be down-regulated (Fig. 3E). Figure 3C shows top 20 up-regulated and down-regulated pathways in CVD patients. Major up-regulated pathways were protein translation and localization, cardiac muscle contraction, oxidative phosphorylation, mitochondrial translation and protein import, electron transport and citric acid cycle. The pathways involved in down-regulation included FGFR1, FGFR2, FGFR3, EGFR, TGF beta, MET mediated signaling, estrogen-dependent gene expression, NR1H2, NR1H3 mediated cholesterol transport and efflux, and regulation of white adipocytes differentiation. By default, gene sets are ordered by normalized enrichment score (NES). More details on all the enriched pathways are available in the Additional file 6: CVD Enrichments. From the list of annotated CVD genes, 15 genes showed a differentiated expression (Fig. 3D). Among them, 7 are HF genes (CST3, LGALS3, MME, NR3C2, PIK3C2A, TNF, VCL), and 8 are other CVD genes (ATP2A2, FADD, FLNA, HBA1, LEMD3, SLC2A1, SMUG1, ZBTB8OS). Enrichment of these genes showed down-regulation was seen in NR3C2, LEMD3, PIK3C2A, FLNA, MME, ATP2A2, and VCL, while a pattern of upregulation was observed in FADD, SLC2A1, TNF, ZBTB8OS, HBA1, LGALS3, CST3, and SMUG1, suggesting that intrinsic biological differences account for, at least, part of CVD.

Differentially regulated gene expression and enrichment. A Differential gene expression of protein coding genes with two major clusters. B MDS plot showing biological distance between case–control samples based on BCV. C Top 20 enriched pathways showing up-regulation and down-regulation in CVD based on their normalized enrichment scores (NES). D Differential gene expression of annotated CVD genes. E Gene enrichment heatmap of differentially expressed genes
The second type of analysis was based on expression analysis to compare expression of all 48 CVD genes between CVD patients and healthy controls. We used our in-house developed GVViZ platform to perform expression analysis using TPM counts of the protein coding genes computed from RNA-seq data. Furthermore, the expression data were linked to gene-disease annotation databases [27, 38, 39] to classify and differentiate between CVD and other disease-based functional and non-functional genes. A heatmap of all the CVD genes was constructed (Fig. 4) and annotated with their associated clinical CVD phenotype. In GVViZ-generated Fig. 4, the X-axis signifies samples (healthy ids: 648, 649, 650, 651, 652, 653, 655, 656, 657, 658, and CVD ids: 1058–1118), the right Y-axis shows genes, and the left Y-axis presents genes associated with the CVDs. There were apparent differences in the filtered expression counts for healthy controls and CVD patients mapped to visualize the variations across the cohort. The analysis showed clear separation of a subset of CVD patients with significantly variable expression for a cluster of genes (details attached in the Additional file 7: Original Raw Data).

Gene expression analysis of all CVD genes. Genes-disease heatmap for the expression analysis of CVDs among all diseased and healthy control patients. The X-axis signifies samples (healthy ids: 648, 650, 651, 652, 653, 655, 656, and CVD ids: 1058–1118), the right Y-axis shows genes, and the left Y-axis presents genes associated with the CVDs
To systematically inspect gene expression in this dataset, CVD patients were mainly stratified into condition, control, and gender for further analysis (Figs. 5 and 6). With a focus on HF and all other CVDs grouped together, we analyzed the expression of all protein coding genes (Fig. 5A), and only highly expressed protein-coding genes (Fig. 5B) related to HF disease, as well as expression analysis of protein coding genes (Fig. 5C), and only highly expressed protein coding genes (Fig. 5D) related to other CVDs. In GVViZ-generated Fig. 5, the X-axis signifies samples (healthy patient ids 648, 649, 650, 651, 652, 653, 655, 656, 657, 658; CVD patient ids 1058–1118), and the Y-axis shows genes associated with HF (Fig. 5A, B) and CVDs (Fig. 5C, D).

Gene expression analysis of HF and other CVD genes. A Expression analysis of protein-coding genes in HF. B Highly expressed protein-coding genes related to HF disease. C Expression analysis of protein-coding genes in other CVD genes. D Highly expressed protein-coding genes related to other CVDs

Gender-based gene expression analysis of HF and other CVD genes. A Protein-coding genes related to HF in males, B Highly expressed protein-coding genes related to HF in males, C Protein-coding genes related to CVD in males, D Highly expressed protein-coding genes related to CVD in males. E Protein-coding genes related to HF in females, F Highly expressed protein-coding genes related to HF in females, G Protein-coding genes related to other CVD sin females, and H highly expressed protein-coding genes related to other CVDs in females
During this disease stratification (Fig. 5), we found patterns that significantly differentiate the HF and CVD groups from the healthy control group. Three clusters were identified in the HF expression analysis, which showed altered expression between the condition and the control groups (Fig. 5A). The first cluster consisted of five genes (ADRB2, TNF, ADM, MME, and CST3), the second cluster included three genes (IL6, MYBPC3, NPPA), and the third cluster contained seven genes (PIK3C2A, EDN1, NR3C2, NMP2, ACE, NOS3, and CORIN). Among these three clusters, all HF genes showed low expression compared to the healthy control group, indicating their down regulation. However, four HF protein-coding genes (LGALS3, CST3, MME, and ADM) showed high expression in one or more patients (Fig. 5B).
Expression analysis of genes accounting for other CVDs showed four clusters between healthy and disease groups (Fig. 5C). The first cluster included nine genes (TEK, GJB6, CD34, ENO2, CALD1, LEMD3, GLMN, ATP2A2, and TRPV1), the second cluster showed four genes (KANTR, CD40LG, ZBTB8OS, and DDX41), the third cluster consisted of three genes (SLC2A1, FADD, and FLNA), and the fourth cluster had only one gene (HBA1). Genes in the first cluster had over 80% of patients showing low expression in comparison with the healthy control group, indicating their down regulation. However, genes in the second and third clusters had over 50% patients with low expression compared to the control group. On the contrary, HBA1 showed high expression during analysis. Other CVD protein-coding genes that had the highest expressed were HBA1, FLNA, and DDX41 (Fig. 5D).
To further classify the groups, we performed gender-based gene expression analysis of HF and other CVD genes (Fig. 6). We compared gender-matched case and control groups (male CVD vs male controls, and female CVD vs female controls). The results illustrated for HF protein-coding genes in the male group (Fig. 6A, B) with genes showing a relatively low expression in comparison with the control group (ADM, MME, VCL, MYBPC3, IL6, MMP2, ACE, NR3C2, EDN1, and PIK3C2A). Some genes showed a rise in expression in comparison with the control group (NPR1, ANKRD1, NPPC, and UTS2). Looking at the HF protein-coding genes in the female group (Fig. 6E, F), gene LGALS3 was found to be highly regulated among diseased samples in comparison with healthy controls, whereas some genes showed a down regulated expression (ADM, MME, ADRB2, TNF, VCL, MYBPC3, MYH7, HDPB7, MMP2, NPR1, and EDN1). Interestingly similar protein-coding genes related to HF were found to be highly expressed in both males and females (CST3, LGALS3, MME). However, ADM was only found in males.
Likewise, gender-based gene expression analysis of other CVD genes revealed altered expression in the male group (Fig. 6C, D). We identified several CVD genes with low expression in the male cohort (ELNA, FADD, DDX41, CD34, SMUG1, GJB6, TEK, TRPV1, ATP2A2, GLMN, LEMD3, CALD1, ENO2, and FGF2). In the female group, we also observed low expression in CVD genes (FLNA, FADD, SLC2A1, CD40LG, LEMD3, DDX41, ENO2, ATP2A2, KANTR, MB, GLMN, TRPV1, CALD1, CD34, GJB6, TEK, and FGF2) (Fig. 6G, H). HBA1, FLNA, and DDX41 were found as the highly expressed protein-coding CVD genes in both gender groups, and ENO2 was the only highly expressed gene in the female group.
We investigated HF and other CVD associated protein coding genes and their expression levels among difference races (Fig. 7). We observed MME, CST3 and LGALS3 HF genes with high expression among White Americans (Fig. 7A), Blacks/African Blacks (Fig. 7B), and all other races (Fig. 7C). When ADM was only located within White Americans. We commonly found DDX41, FLNA and HB1 CVD genes with high expression among white Americans (Fig. 7D), Blacks/African Blacks (Fig. 7E), and all other races (Fig. 7F). However, we have also presented all differentially expressed HF and other CVD genes among these all races in Fig. 7. High resolution figures are attached in Additional file 2. To incorporate produced results in clinical settings, and to get given recommendations back into EHRs, we have linked HF and other CVD genes (Ensembl) with the International Classification of Disease (ICD) codes (Table 4).

Race-based gene expression analysis of HF and other CVD genes. All and highly expressed protein-coding genes related to HF in self-described Whites (A), Blacks/African Americans (B), and all other races (C). All and highly expressed protein-coding genes related to other CVDs in Whites (D), Blacks/African Americans (E), and all other races (F)
Discussion
Over the past few years, genomic-sequencing technologies have emerged to improve the clinical diagnosis of genetic disorders and continuing to expand the potential of basic sciences in developing biological insights of human genetic variations and their biologic consequences [43]. Several clinically established cardiovascular circulating biomarkers are measured to help diagnose, stratify risk, and monitor people with suspected CVDs. Use of one or more of these biomarkers can help physicians identify a heart condition and initiate appropriate therapy, as well as follow the course of disease. CVD presents differently in women and men both symptomatically and biochemically [44]. However, some studies have failed to detect a heart condition in women with elevated death rates [45]. Lack of gender-specific cardiac biomarker thresholds in men and women may be the reason for CVD underdiagnosis in women, and potentially increased morbidity and mortality as a result, or conversely, an overdiagnosis in men.
Here, we report a peripheral blood gene expression analysis focused on HF- and CVD genes to identify gender-specific differences in patients aged between 45 and 95 years old. Our major findings include disease specific up- and down-regulated differentially expressed protein-coding genes in HF and CVDs and categorized their major signaling pathways involved in disease physiology. This analysis also revealed 25 novel gene expression in CVD patients. Our results on gender-specific differences in expression of protein-coding genes related to HF and other CVDs show that it is important to systematically investigate gender-differences in high-impact genes in HF and CVDs [46, 47]. We found differentially altered expression of FLNA, CST3, LGALS3, and HBA1, potentially responsible for HF and other CVDs in both male and female populations. FLNA is a gene known for CVDs, as mutations in FLNA can lead to cardiological phenotypes with aortic or mitral regurgitation [48]. High expression and mutations in the CST3 (Cystatin C) gene have been reported in systolic HF, ischemic stroke, and CAD [49, 50]. The LGALS3 gene encodes the galectin-3 (35-kDa) protein, and single nucleotide polymorphisms (SNPs) and promoter-regulated expression of LGALS3 are considered potential candidates that cause CVDs, especially CAD, dilated cardiomyopathy, and HF [51,52,53,54]. The HBA1 (glycated hemoglobin A1c) gene (chromosome 16) is considered a prognostic marker responsible for the increased cardiovascular mortality risk in age- and gender-classified populations [55, 56]. Mutations in HBA1 can cause myocardial infarction, stroke, coronary heart disease, and HF [56]. The differential expression of ENO2 (Enolase 2) gene in CVDs also highlighted gender-specific (female) alterations, which has been reported in other conditions [57].
RNA-seq driven gene expression analysis is an advancement in the field clinical genomics to analyze chromatin and patterns of expression in genes and differentiating the pathways, which differ between healthy and diseased people [43]. Our study aimed to investigate the clinical significance of gene expression in HF and CVDs using RNA-seq data. We analyzed the differences between healthy and diseased states to understand the pathology of disease [58]. The risk for and the course of heart failure also depends on genomic variants and mutations underlying the so‐called genetic predisposition. Several studies have demonstrated that only about half of all DNA genetic variants are detectable by RNA sequencing of human tissue and cell lines [59,60,61]. However, this approach has some potential limitations. Accurate capture of DNA variants using the RNA-seq data requires high coverage and sufficient samples per population as it has already been tested in cancer [62, 63], which we expect will be mitigated by generating whole genome sequencing (WGS) data to perform variant analysis of the genes responsible for HF (Table 2) and CVDs (Table 3). Nonetheless, with a need to expand the cohort of healthy controls to investigate DEGs with significantly regulated expression and increase the power to substantiate association with related variables in the CVD populations will help to scale down to clinically important genetic variations. Also, PCR validation of the differentially regulated genes will add prognostic value to the study and consolidate the role of specific genes as important biomarkers in HF. Our future plans involve application of AI and ML techniques [28] to advance investigating correlation and overlapping of reported diagnoses of HF and CVD patients in clinical data. Finally, assessment of genotype and phenotype associations to find potentially high-risk indistinct results for patient care from highly regulated genes and disease-causing variants [11].
Conclusion
Our analysis identified four altered expression of HF- and other CVD genes (FLNA, CST3, LGALS3, and HBA1) with gender differences in middle-aged to frail patients and revealed differential regulation of 41 genes related to HF and 23 genes related to other CVDs. Furthermore, many pathways were found to be enriched, and gender-specific analysis showed shared and unique genes between the genders. Additional testing of these genes may lead to the development of new clinical tools to improve diagnosis and prognosis of CVD patients.
Availability of data and materials
The data analyzed in the current study are available from the corresponding author on reasonable request.
Abbreviations
- AMI:
-
Acute myocardial infarction
- AI:
-
Artificial intelligence
- BCV:
-
Biological coefficient of variation
- CVDs:
-
Cardiovascular diseases
- COSMIC:
-
Catalogue of somatic mutations in cancer
- CDC:
-
Center for disease control and prevention
- DEGs:
-
Differentially expressed genes
- HER:
-
Electronic health record
- EMR:
-
Electronic medical record
- EM:
-
Expectation–maximization
- ETL:
-
Extract, transform, and loading
- GWAS:
-
Genome-wide Association Studies
- GVViZ:
-
Gene variant visualization
- GSEA:
-
Gene set enrichment analysis
- HF:
-
Heart failure
- HFpEF:
-
Heart failure with preserved ejection fraction
- HFrEF:
-
Heart failure with reduced ejection fraction
- hg38:
-
Human reference genome
- ICD:
-
International Classification of Disease codes
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- ML:
-
Machine learning
- mRNAs:
-
Messenger ribonucleic acids
- MDS:
-
Multidimensional scaling
- OP:
-
Outpatient pavilion
- RNA-seq:
-
RNA sequencing
- RSEM:
-
RNA-seq by expectation maximization
- CMS/HCC:
-
Systolic and diastolic HF
- TPM:
-
Transcript per million mapped reads
- WGS:
-
Whole genome sequencing
References
Mc Namara K, Alzubaidi H, Jackson JK. Cardiovascular disease as a leading cause of death: how are pharmacists getting involved? Integr Pharm Res Pract. 2019;8:1–11. https://doi.org/10.2147/IPRP.S133088.
Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, Alvarado M, Anderson HR, Anderson LM, Andrews KG, Atkinson C, Baddour LM, Barker-Collo S, Bartels DH, Bell ML, Benjamin EJ, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet (London, England). 2012;380(9859):2095–128. https://doi.org/10.1016/S0140-6736(12)61728-0.
Stewart J, Manmathan G, Wilkinson P. Primary prevention of cardiovascular disease: a review of contemporary guidance and literature. JRSM Cardiovasc Dis. 2017;6:2048004016687211. https://doi.org/10.1177/2048004016687211.
Roth GA, Johnson C, Abajobir A, Abd-Allah F, Abera SF, Abyu G, Ahmed M, Aksut B, Alam T, Alam K, Alla F, Alvis-Guzman N, Amrock S, Ansari H, Ärnlöv J, Asayesh H, Atey TM, Avila-Burgos L, Awasthi A, Banerjee A, et al. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J Am Coll Cardiol. 2017;70(1):1–25. https://doi.org/10.1016/j.jacc.2017.04.052.
Walden R, Tomlinson B, et al. Cardiovascular disease. In: Benzie I, et al., editors. Herbal medicine: biomolecular and clinical aspects, vol. 2. Boca Raton: CRC Press; 2011.
Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Delling FN, Djousse L, Elkind M, Ferguson JF, Fornage M, Khan SS, Kissela BM, Knutson KL, Kwan TW, Lackland DT, Lewis TT, American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee, et al. Heart disease and stroke statistics-2020 update: a report from the American Heart Association. Circulation. 2020;141(9):e139–596. https://doi.org/10.1161/CIR.0000000000000757.
Ruan Y, Guo Y, Zheng Y, Huang Z, Sun S, Kowal P, Shi Y, Wu F. Cardiovascular disease (CVD) and associated risk factors among older adults in six low-and middle-income countries: results from SAGE Wave 1. BMC Public Health. 2018;18(1):778. https://doi.org/10.1186/s12889-018-5653-9.
Ingles J, Semsarian C. Sudden cardiac death in the young: a clinical genetic approach. Intern Med J. 2007;37(1):32–7. https://doi.org/10.1111/j.1445-5994.2006.01241.x.
Novelli G, Predazzi IM, Mango R, Romeo F, Mehta JL. Role of genomics in cardiovascular medicine. World J Cardiol. 2010;2(12):428–36. https://doi.org/10.4330/wjc.v2.i12.428.
Kathiresan S, Srivastava D. Genetics of human cardiovascular disease. Cell. 2012;148(6):1242–57. https://doi.org/10.1016/j.cell.2012.03.001.
Ahmed Z. Practicing precision medicine with intelligently integrative clinical and multi-omics data analysis. Hum Genomics. 2020;14(1):35. https://doi.org/10.1186/s40246-020-00287-z.
Wung SF, Hickey KT, Taylor JY, Gallek MJ. Cardiovascular genomics. J Nursing Scholarsh. 2013;45(1):60–8. https://doi.org/10.1111/jnu.12002.
Seo D, Ginsburg GS, Goldschmidt-Clermont PJ. Gene expression analysis of cardiovascular diseases: novel insights into biology and clinical applications. J Am Coll Cardiol. 2006;48(2):227–35. https://doi.org/10.1016/j.jacc.2006.02.070.
Skrzynia C, Berg JS, Willis MS, Jensen BC. Genetics and heart failure: a concise guide for the clinician. Curr Cardiol Rev. 2015;11(1):10–7. https://doi.org/10.2174/1573403x09666131117170446.
Kazmi N, Gaunt TR. Diagnosis of coronary heart diseases using gene expression profiling; stable coronary artery disease, cardiac ischemia with and without myocardial necrosis. PLoS ONE. 2016;11(3):e0149475. https://doi.org/10.1371/journal.pone.0149475.
Elashoff MR, Wingrove JA, Beineke P, Daniels SE, Tingley WG, Rosenberg S, Voros S, Kraus WE, Ginsburg GS, Schwartz RS, Ellis SG, Tahirkheli N, Waksman R, McPherson J, Lansky AJ, Topol EJ. Development of a blood-based gene expression algorithm for assessment of obstructive coronary artery disease in non-diabetic patients. BMC Med Genomics. 2011;4:26. https://doi.org/10.1186/1755-8794-4-26.
Kittleson MM, Ye SQ, Irizarry RA, Minhas KM, Edness G, Conte JV, Parmigiani G, Miller LW, Chen Y, Hall JL, Garcia JG, Hare JM. Identification of a gene expression profile that differentiates between ischemic and nonischemic cardiomyopathy. Circulation. 2004;110(22):3444–51. https://doi.org/10.1161/01.CIR.0000148178.19465.11.
Min KD, Asakura M, Liao Y, Nakamaru K, Okazaki H, Takahashi T, Fujimoto K, Ito S, Takahashi A, Asanuma H, Yamazaki S, Minamino T, Sanada S, Seguchi O, Nakano A, Ando Y, Otsuka T, Furukawa H, Isomura T, Takashima S, et al. Identification of genes related to heart failure using global gene expression profiling of human failing myocardium. Biochem Biophys Res Commun. 2010;393(1):55–60. https://doi.org/10.1016/j.bbrc.2010.01.076.
Suresh R, Li X, Chiriac A, Goel K, Terzic A, Perez-Terzic C, Nelson TJ. Transcriptome from circulating cells suggests dysregulated pathways associated with long-term recurrent events following first-time myocardial infarction. J Mol Cell Cardiol. 2014;74:13–21. https://doi.org/10.1016/j.yjmcc.2014.04.017.
Czepluch FS, Wollnik B, Hasenfuß G. Genetic determinants of heart failure: facts and numbers. ESC heart failure. 2018;5(3):211–7. https://doi.org/10.1002/ehf2.12267.
Lee DS, Pencina MJ, Benjamin EJ, Wang TJ, Levy D, O’Donnell CJ, Nam BH, Larson MG, D’Agostino RB, Vasan RS. Association of parental heart failure with risk of heart failure in offspring. N Engl J Med. 2006;355(2):138–47. https://doi.org/10.1056/NEJMoa052948.
Lindgren MP, Smith JG, Li X, Sundquist J, Sundquist K, Zöller B. Sibling risk of hospitalization for heart failure—a nationwide study. Int J Cardiol. 2016;223:379–84. https://doi.org/10.1016/j.ijcard.2016.08.067.
Towbin JA, Lowe AM, Colan SD, Sleeper LA, Orav EJ, Clunie S, Messere J, Cox GF, Lurie PR, Hsu D, Canter C, Wilkinson JD, Lipshultz SE. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA. 2006;296(15):1867–76. https://doi.org/10.1001/jama.296.15.1867.
Monserrat L, Hermida M, Bouzas B, Mosquera I, Mahon N, Peteiro J, Alvarez N, Penas-Lado M, Crespo M, Castro-Beiras A. Miocardiopatía dilatada familiar en pacientes trasplantados por miocardiopatía dilatada idiopática [Familial dilated cardiomyopathy in patients transplanted for idiopathic dilated cardiomyopathy]. Rev Esp Cardiol. 2002;55(7):725–32. https://doi.org/10.1016/s0300-8932(02)76691-8.
Lovén J, Orlando DA, Sigova AA, Lin CY, Rahl PB, Burge CB, Levens DL, Lee TI, Young RA. Revisiting global gene expression analysis. Cell. 2012;151(3):476–82. https://doi.org/10.1016/j.cell.2012.10.012.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. https://doi.org/10.1186/1471-2105-12-323.
Zeeshan S, Xiong R, Liang BT, Ahmed Z. 100 Years of evolving gene-disease complexities and scientific debutants. Brief Bioinform. 2020;21(3):885–905. https://doi.org/10.1093/bib/bbz038.
Ahmed Z, Mohamed K, Zeeshan S, Dong X. Artificial intelligence with multi-functional machine learning platform development for better healthcare and precision medicine. Database. 2020;2020:baa010. https://doi.org/10.1093/database/baaa010.
Ahmed Z, Kim M, Liang BT. MAV-clic: management, analysis, and visualization of clinical data. JAMIA open. 2018;2(1):23–8. https://doi.org/10.1093/jamiaopen/ooy052.
Trivedi UH, Cézard T, Bridgett S, Montazam A, Nichols J, Blaxter M, Gharbi K. Quality control of next-generation sequencing data without a reference. Front Genet. 2014;5:111. https://doi.org/10.3389/fgene.2014.00111.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics (Oxford, England). 2014;30(15):2114–20. https://doi.org/10.1093/bioinformatics/btu170.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics (Oxford, England). 2009;25(16):2078–9. https://doi.org/10.1093/bioinformatics/btp352.
Ebbert MT, Wadsworth ME, Staley LA, Hoyt KL, Pickett B, Miller J, Duce J, Alzheimer’s Disease Neuroimaging Initiative, Kauwe JS, Ridge PG. Evaluating the necessity of PCR duplicate removal from next-generation sequencing data and a comparison of approaches. BMC Bioinformatics 2016; 17 Suppl 7(Suppl 7): 239. https://doi.org/10.1186/s12859-016-1097-3
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–60. https://doi.org/10.1038/nmeth.3317.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9. https://doi.org/10.1038/nmeth.1923.
Abrams ZB, Johnson TS, Huang K, Payne P, Coombes K. A protocol to evaluate RNA sequencing normalization methods. BMC Bioinformatics. 2019;20(Suppl 24):679. https://doi.org/10.1186/s12859-019-3247-x.
Ahmed Z, Renart EG, Zeeshan S, Dong X. Advancing clinical genomics and precision medicine with GVViZ: FAIR bioinformatics platform for variable gene-disease annotation, visualization, and expression analysis. Hum Genomics. 2021;15(1):37. https://doi.org/10.1186/s40246-021-00336-1.
Ahmed Z, Zeeshan S, Mendhe D, Dong X. Human gene and disease associations for clinical-genomics and precision medicine research. Clin Transl Med. 2020;10(1):297–318. https://doi.org/10.1002/ctm2.28.
Ahmed Z, Zeeshan S, Xiong R, Liang BT. Debutant iOS app and gene-disease complexities in clinical genomics and precision medicine. Clin Transl Med. 2019;8(1):26. https://doi.org/10.1186/s40169-019-0243-8.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–50. https://doi.org/10.1073/pnas.0506580102.
Hout MC, Papesh MH, Goldinger SD. Multidimensional scaling. Wiley Interdiscip Rev Cogn Sci. 2013;4(1):93–103. https://doi.org/10.1002/wcs.1203.
McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40(10):4288–97. https://doi.org/10.1093/nar/gks042.
Collins FS, Doudna JA, Lander ES, Rotimi CN. Human molecular genetics and genomics—important advances and exciting possibilities. N Engl J Med. 2021;384(1):1–4. https://doi.org/10.1056/NEJMp2030694.
Sobhani K, Nieves Castro DK, Fu Q, Gottlieb RA, Van Eyk JE, Merz CNB. Sex differences in ischemic heart disease and heart failure biomarkers. Biol Sex Differ. 2018;9(1):43. https://doi.org/10.1186/s13293-018-0201-y.
Shah AS, Griffiths M, Lee KK, McAllister DA, Hunter AL, Ferry AV, Cruikshank A, Reid A, Stoddart M, Strachan F, Walker S, Collinson PO, Apple FS, Gray AJ, Fox KA, Newby DE, Mills NL. High sensitivity cardiac troponin and the under-diagnosis of myocardial infarction in women: prospective cohort study. BMJ. 2015;350: g7873. https://doi.org/10.1136/bmj.g7873.
McNally EM, O’Donnell CJ. Genetic contribution to common heart failure—not so rare? JAMA Cardiol. 2020. https://doi.org/10.1001/jamacardio.2020.6508.
Povysil G, Chazara O, Carss KJ, Deevi S, Wang Q, Armisen J, Paul DS, Granger CB, Kjekshus J, Aggarwal V, Haefliger C, Goldstein DB. Assessing the role of rare genetic variation in patients with heart failure. JAMA cardiology. 2021;6(4):379–86. https://doi.org/10.1001/jamacardio.2020.6500.
de Wit MC, de Coo IF, Lequin MH, Halley DJ, Roos-Hesselink JW, Mancini GM. Combined cardiological and neurological abnormalities due to filamin A gene mutation. Clin Res Cardiol. 2011;100(1):45–50. https://doi.org/10.1007/s00392-010-0206-y.
van der Laan SW, Fall T, Soumaré A, Teumer A, Sedaghat S, Baumert J, Zabaneh D, van Setten J, Isgum I, Galesloot TE, Arpegård J, Amouyel P, Trompet S, Waldenberger M, Dörr M, Magnusson PK, Giedraitis V, Larsson A, Morris AP, Felix JF, et al. Cystatin C and cardiovascular disease: a Mendelian randomization study. J Am Coll Cardiol. 2016;68(9):934–45. https://doi.org/10.1016/j.jacc.2016.05.092.
Dupont M, Wu Y, Hazen SL, Tang WH. Cystatin C identifies patients with stable chronic heart failure at increased risk for adverse cardiovascular events. Circ Heart Fail. 2012;5(5):602–9. https://doi.org/10.1161/CIRCHEARTFAILURE.112.966960.
Dong R, Zhang M, Hu Q, Zheng S, Soh A, Zheng Y, Yuan H. Galectin-3 as a novel biomarker for disease diagnosis and a target for therapy (Review). Int J Mol Med. 2018;41(2):599–614. https://doi.org/10.3892/ijmm.2017.3311.
Gleissner CA, Erbel C, Linden F, Domschke G, Akhavanpoor M, Helmes CM, Doesch AO, Kleber ME, Katus HA, Maerz W. Galectin-3 binding protein, coronary artery disease and cardiovascular mortality: Insights from the LURIC study. Atherosclerosis. 2017;260:121–9. https://doi.org/10.1016/j.atherosclerosis.2017.03.031.
Zhang Y, Wang Y, Zhai M, Gan T, Zhao X, Zhang R, An T, Huang Y, Zhou Q, Zhang J. Influence of LGALS3 gene polymorphisms on susceptibility and prognosis of dilated cardiomyopathy in a Northern Han Chinese population. Gene. 2018;642:293–8. https://doi.org/10.1016/j.gene.2017.11.026.
Suthahar N, Meijers WC, Silljé H, Ho JE, Liu FT, de Boer RA. Galectin-3 activation and inhibition in heart failure and cardiovascular disease: an update. Theranostics. 2018;8(3):593–609. https://doi.org/10.7150/thno.22196.
Cavero-Redondo I, Peleteiro B, Álvarez-Bueno C, Rodriguez-Artalejo F, Martínez-Vizcaíno V. Glycated haemoglobin A1c as a risk factor of cardiovascular outcomes and all-cause mortality in diabetic and non-diabetic populations: a systematic review and meta-analysis. BMJ Open. 2017;7(7): e015949. https://doi.org/10.1136/bmjopen-2017-015949.
Cavero-Redondo I, Peleteiro B, Álvarez-Bueno C, Rodríguez-Artalejo F, Martínez-Vizcaíno V. Glycosylated haemoglobin as a predictor of cardiovascular events and mortality: a protocol for a systematic review and meta-analysis. BMJ Open. 2016;6(7): e012229. https://doi.org/10.1136/bmjopen-2016-012229.
Liu D, Mao Y, Chen C, Zhu F, Lu W, Ma H. Expression patterns and clinical significances of ENO2 in lung cancer: an analysis based on Oncomine database. Annals Transl Med. 2020;8(10):639. https://doi.org/10.21037/atm-20-3354.
Rodriguez-Esteban R, Jiang X. Differential gene expression in disease: a comparison between high-throughput studies and the literature. BMC Med Genomics. 2017;10:59. https://doi.org/10.1186/s12920-017-0293-y.
Ku CS, Wu M, Cooper DN, Naidoo N, Pawitan Y, Pang B, Iacopetta B, Soong R. Exome versus transcriptome sequencing in identifying coding region variants. Expert Rev Mol Diagn. 2012;12(3):241–51. https://doi.org/10.1586/erm.12.10.
Cirulli ET, Singh A, Shianna KV, Ge D, Smith JP, Maia JM, Heinzen EL, Goedert JJ, Goldstein DB, Center for HIV/AIDS Vaccine Immunology (CHAVI). Screening the human exome: a comparison of whole genome and whole transcriptome sequencing. Genome Biol. 2010;11(5):R57. https://doi.org/10.1186/gb-2010-11-5-r57.
Piskol R, Ramaswami G, Li JB. Reliable identification of genomic variants from RNA-seq data. Am J Hum Genet. 2013;93(4):641–51. https://doi.org/10.1016/j.ajhg.2013.08.008.
O’Brien TD, Jia P, Xia J, Saxena U, Jin H, Vuong H, Kim P, Wang Q, Aryee MJ, Mino-Kenudson M, Engelman JA, Le LP, Iafrate AJ, Heist RS, Pao W, Zhao Z. Inconsistency and features of single nucleotide variants detected in whole exome sequencing versus transcriptome sequencing: a case study in lung cancer. Methods. 2015;83:118–27. https://doi.org/10.1016/j.ymeth.2015.04.016.
Angell TE, Wirth LJ, Cabanillas ME, Shindo ML, Cibas ES, Babiarz JE, Hao Y, Kim SY, Walsh PS, Huang J, Kloos RT, Kennedy GC, Waguespack SG. Analytical and clinical validation of expressed variants and fusions from the whole transcriptome of thyroid FNA samples. Front Endocrinol. 2019;10:612. https://doi.org/10.3389/fendo.2019.00612.
Acknowledgements
We appreciate great support by the Pat and Jim Calhoun Cardiology Center, and Department of Genetics and Genome Sciences, at the UConn School of Medicine, UConn Health; Rutgers Institute for Health, Health Care Policy and Aging Research (IFH), and Rutgers Robert Wood Johnson Medical School (RWJMS), Rutgers Biomedical and Health Sciences (RBHS) at the Rutgers, The State University of New Jersey. We appreciate all colleagues and institutions who provided direct and indirect insight and expertise that greatly assisted the research and development of this project. We would like to give special thanks to Dr. Christopher Bonin and Dr. Geneva Hargis for stylistic and native speaker corrections. We thank members and collaborators of Ahmed Lab at the Rutgers (IFH, RWJMS, RBHS) for their support, participation, and contribution to this study.
Funding
This work was supported by the School of Medicine, UConn Health, CT.
Author information
Authors and Affiliations
Contributions
ZA lead this study. ZA did RNA-seq data processing, quality checking, and downstream analysis. ZA developed PROMIS-LCR, supervised GVViZ implementation, and performed cohort building and integrative clinical data analysis of consented patients. SZ supported computational analysis and data visualization. ZA drafted the paper, and all authors have participated in writing, review, and revisions. BL proposed, supervised, and supported the study. All authors read and approved the final manuscript.
Authors' information
ZA is the Assistant Professor of Medicine—Tenure Track and Core Member at the Rutgers Institute for Health, Health Care Policy and Aging Research; and Department of Medicine—Division of General Internal Medicine, Rutgers Robert Wood Johnson Medical School, Rutgers Biomedical and Health Sciences, Rutgers University-New Brunswick, NJ. ZA is the Adjunct Assistant Professor at the Department of Genetics and Genome Sciences, and Pat and Jim Calhoun Cardiology Center, UConn School of Medicine, UConn Health, CT; and Full Academic Member of the Rutgers Microbiology and Molecular Genetics; Center for Cancer Health Equity, Rutgers Cancer Institute of New Jersey; Rutgers Human Genetics Institute of New Jersey, NJ.
SZ is the Senior Postdoctoral Research Associate at the Rutgers Cancer Institute of New Jersey.
BL is the Professor, Dean UConn School of Medicine, Director Pat and Jim Calhoun Cardiology Center, and Ray Neag Distinguished Professor of Cardiovascular Biology and Medicine, UConn Health, CT, USA.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
Informed consent was obtained from all subjects. All human samples were used in accordance with relevant guidelines and regulations, and all experimental protocols were approved by Institutional Review Board, UConn Health.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing financial or non-financial interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Gender and age-based population data classification.
Additional file 2.
High resolution figures.
Additional file 3.
All DEGs Expression
Additional file 4.
All DEGs Stats
Additional file 5.
All DEGs Stats 42 Genes
Additional file 6.
CVD Enrichments
Additional file 7.
Original Raw Data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ahmed, Z., Zeeshan, S. & Liang, B.T. RNA-seq driven expression and enrichment analysis to investigate CVD genes with associated phenotypes among high-risk heart failure patients. Hum Genomics 15, 67 (2021). https://doi.org/10.1186/s40246-021-00367-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-021-00367-8