
Abstract
Background
Long non-coding RNAs (lncRNAs) are a sub-class within non-coding RNA repertoire that have emerged as crucial regulators of the gene expression in various pathophysiological conditions. lncRNAs display remarkable versatility and wield their functions through interactions with RNA, DNA, or proteins. Accumulating body of evidence based on multitude studies has highlighted the role of lncRNAs in many autoimmune and inflammatory diseases, including type 1 diabetes (T1D).
Main body of abstract
This review highlights emerging roles of lncRNAs in immune and islet β cell function as well as some of the challenges and opportunities in understanding the pathogenesis of T1D and its complications.
Conclusion
We accentuate that the lncRNAs within T1D-loci regions in consort with regulatory variants and enhancer clusters orchestrate the chromatin remodeling in β cells and thereby act as cis/trans-regulatory determinants of islet cell transcriptional programs.
Similar content being viewed by others


Background
Type 1 diabetes (T1D) is a chronic immune-mediated disease resulting from selective destruction of insulin-producing pancreatic islet β cells. A complex interplay between several environmental and genetic risk factors contribute to the onset of T1D [1, 2]. Defects in both immune system and β cells play an active role in T1D pathogenesis [3]. In recent years, efforts have been accelerated to gain insights into the molecular mechanisms of pathogenesis of T1D and to determine how genetic loci contribute to the T1D risk [1, 4]. Based on genome-wide association studies (GWAS), currently more than 50 genomic risk loci have been identified for T1D [2, 5–7]. Approximately, 50% of the genetic risk for T1D is known to reside within the human leukocyte antigen (HLA) region; however, other non-HLA disease susceptibility loci have been identified based on their direct influence on the risk. Some of the well-established candidate genes in the non-HLA risk loci including INS (11p15), CTLA4 (2q33), PTPN22 (1p13), PTPN2 (18p11), ERBB3 (12q13), IL2RA (10p15), and IFIH1 (2q24) have been associated with immune response, insulin expression, and β cell function [1, 4, 8]. The risk alleles for several T1D susceptibility genes are not exclusively confined to T1D but have been shown to confer risk in other prevalent autoimmune disorders, including multiple sclerosis (MS), systemic lupus erythematosus (SLE), and rheumatoid arthritis (RA) [7]. Furthermore, most of these risk variants are located in non-coding genomic regions including long non-coding RNAs (lncRNAs) and are enriched in distal regulatory elements such as enhancers and promoters. Non-coding variants affecting regulatory elements have the potential to perturb chromatin folding leading to mis-expression of the target gene. These facts suggest that the regulatory landscape of human genome plays an important role in pathology of a disease and newer approaches are needed to identify putative regulatory risk variants affecting gene regulation and immune function.
Recent advances in our understanding of lncRNA biology has offered new perspectives on gene regulation and has allowed us to unveil the regulatory potential of these versatile molecules in a spectrum of biological processes and pathologies, including autoimmune and inflammatory disorders. High-throughput technologies such as ChIP-seq and chromosome conformation capture techniques have also opened new possibilities to investigate in detail potential regulatory roles of non-coding genome in gene regulation and 3D chromatin folding. In this review, we discuss the recent discoveries in the field of lncRNAs, regulatory elements, 3D genome architecture, and their implications for T1D and β cell function. We further highlight the role of active enhancers associated with T1D–loci lncRNAs and protein-coding genes in regulating β cell gene expression programs through both cis- and trans-regulatory mechanisms involving structural remodeling of chromatin in human islets.
Main text
lncRNAs in T1D and other immune-mediated diseases
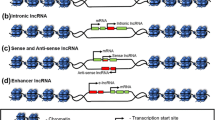
lncRNAs are non-coding RNAs that are more than 200 nucleotides in length, and are capped, polyadenylated, and spliced like their well-characterized “cousins,” protein-coding transcripts, with one exception; lncRNAs do not code for proteins [9]. Most of the lncRNAs are expressed in a cell-specific manner and are usually expressed in lower abundance than the protein-coding transcripts. In terms of genomic location, lncRNAs have been often categorized as long intergenic non-coding RNAs (lincRNAs), intronic lncRNAs, antisense lncRNAs, divergent lncRNAs and enhancer-derived lncRNAs (lncRNAs arising from enhancer-like regions) [10–12] (Fig. 1). lncRNAs have emerged as important players of gene regulation and have been implicated in various human pathologies [13]. lncRNAs regulate various cellular and biological processes including heterochromatin formation, histone modifications, DNA methylation targeting, and gene silencing [14, 15]. The lncRNA-recruited regulatory complexes orchestrate development and differentiation of various immune cell lineages and actively regulate expression programs within these cells.

Bio-types of lncRNAs and enhancer-derived lncRNA function. a Different bio-types of lncRNAs based on their genomic location include antisense, intergenic, intronic, divergent and enhancer-derived lncRNAs. lncRNAs are depicted in blue, while protein-coding genes are shown in green. b The postulated role of enhancer-derived lncRNAs for both cis- and trans-mediated regulation of target genes is shown via chromatin loop formation. Figure modified from Ref. [10, 11]
Multiple studies have highlighted the potential roles of lncRNAs in pancreatic islets and T1D pathogenesis [10, 16, 17]. Based on transcriptome profiling studies of islets and β cells, more than 1000 islet-specific lncRNAs have been identified in both human and mouse islets [18, 19]. The ability of lncRNAs to regulate gene expression and cell-specific identity provides an exciting opportunity to advance our understanding of T1D pathogenesis. Table 1 lists examples of lncRNAs that have been implicated in β cell function and T1D. lncRNA MALAT1 has been associated with diabetes-induced microvascular dysfunction in STZ-induced diabetic rats and db/db mice [20]. Knockdown of MALAT1 prevents the hyper-proliferation of retinal endothelial cells through p38 MAPK signaling and might serve as a potential target for anti-angiogenic therapy for diabetic retinopathy. lncRNA MEG3 has been associated with paternally inherited risk of T1D [21] and its downregulation affects insulin synthesis and secretion in mouse β cells [22]. The knockdown of trans-acting islet-specific lncRNA HI-LNC25 (LINC01370) in mature β cells resulted in downregulation of GLIS3 gene [18]. GLIS3 encodes an islet transcription factor (TF) and is a candidate gene for both type 1 and type 2 diabetes. Another islet-specific lncRNA βlinc1 (previously known as HI-LNC15) has been shown to be essential for proper specification and function of β cells [23]. Knockdown of βlinc1 in mouse MIN6 cells and human insulin-producing EndoC-βH1 cells resulted in downregulation of several islet-specific TFs, including Nkx2.2, Pax6, and Mafb [23]. Also, deletion of βlinc1 resulted in defective islet development and disrupted glucose homeostasis in the adult mice [23]. It is particularly intriguing that βlinc1 specifically regulates three essential islet TFs (Nkx2.2, Pax6, and MafB) and additional β cell genes on chromosome 2, all of which are associated with endocrine development and maintaining islet morphology [23]. Nuclear-enriched β cell lncRNA PLUTO (PLUT) (previously known as HI-LNC71) regulates the transcription of PDX1 gene which is a key pancreatic β cell transcriptional regulator [24]. PLUT encompasses a cluster of enhancers that make 3D contacts with the PDX1 promoter in human islets and in human β cell line EndoC-βH1. Loss of PLUT was associated with downregulation of PDX1 at both mRNA and protein levels in EndoC-βH1 cells, primary islet cells, and a similar effect was observed for the mouse lncRNA ortholog in mouse β cell line MIN6 [24]. Knockdown of lncRNA TUNAR (HI-LNC78) resulted in reduced insulin content and impaired glucose-stimulated insulin secretion in T antigen-excised EndoC-βH3 cells [24]. lncRNA TUG1 is a highly conserved lncRNA in mammals and is highly expressed in mouse pancreatic tissues [25]. In mouse β cells, downregulation of TUG1 as a consequence of hyperglycemia resulted in increased apoptosis and reduced insulin synthesis and secretion [25]. These findings suggest that TUG1 may partially contribute to the impairment of β cell function and could therefore be implicated in diabetes pathogenesis.
In recent years, growing body of evidence has linked dysregulation of lncRNA expression to a spectrum of autoimmune disease [26, 27]. Hrdlickova and colleagues found enrichment of lincRNAs in autoimmune disease-associated loci in a subset of immune cells [28]. A number of studies describe the emerging role of lncRNAs in transcriptional regulation of inflammatory gene expression [29, 30]. For example, in human monocytes, lncRNA THRIL has been shown to interact with hnRNP-L and regulate the expression of TNFα [31]. Correspondingly, additional examples of lncRNAs involvement in inflammatory signaling cascades and regulation of innate immune responses includes (1) lncRNA PACER (PACERR) which has been shown to bind p50 subunit of NFκB and control the basal expression levels of Cox2 (PTGS2) [32]; (2) in primary human monocytes, knockdown of NFκB regulated, enhancer-RNA (eRNA) IL1β-eRNA and region of bidirectional transcription (RBT) IL1β-RBT46, mitigated bacterial lipopolysaccharide (LPS) pro-inflammatory cytokine IL1β induction and release [33]; (3) overexpression of natural antisense transcript anti-IL1β alters the chromatin structure around the IL1β promoter and consequently inhibits the IL1β expression [34, 35]. In a murine model, lncRNA NeST (Nettoie Salmonella pas Theiler’s; cleanup Salmonella not Theiler’s) was shown to epigenetically regulate the interferon-γ (IFN-γ) locus and control the susceptibility to Theiler’s virus and Salmonella infection [36, 37]. Together, these findings indicate that lncRNAs play etiological role in autoimmune diseases. lncRNAs have also been shown to play pivotal roles in the Toll-like receptor (TLR) signaling pathway. For example, when macrophages and dendritic cells (DCs) were stimulated with TLR ligands, the lincRNA-Cox2A was found to be highly inducible and also controlled the basal expression levels of interferon-stimulated genes (ISGs) and pro-inflammatory cytokines [29]. Intriguingly, pseudogenes lncRNAs have been identified to act as functional regulators of inflammatory signaling with their expression being actively regulated. Stimulation of mouse embryonic fibroblast cells by TNFα has been found to induce expression of lncRNAs. Lethe, a pseudogene lncRNA, has been shown to function as a novel negative regulator of NF-κB. Lethe, wields its regulatory function by binding directly RelA, a subunit of NF-κB heterodimeric complex, preclude NF-κB binding to the promoter regions of target genes [38]. Another pseudogene lncRNA Lnc-dendritic cell (DC) (WFDC21P) has been shown to be involved in monocyte to DC differentiation [39].
Systemic cell-mediated immunity is known to play a central role in the apoptotic β cell destruction that culminates in T1D. The T helper 17 (Th17) cells are known to protect mucosal barriers from opportunistic infections and are also associated with number of autoimmune inflammatory diseases. Like many other autoimmune diseases, T1D is also a T cell-mediated malady, and imbalance between the Th17 cells and T regulatory (Treg) has been implicated in development of the T1D [40, 41]. Recently, Huang et al. [42] demonstrated role of DEAD-box protein 5 (DDX5) as a binding partner of RORγt, a well-known ligand-regulated nuclear receptor that controls the differentiation of Th17 cells. Interestingly, DDX5 coordinates the transcription of selective Th17 genes through its interaction with RORγt, and it is also required for Th17 cell-mediated inflammatory diseases. The interaction between DDX5 and RORγt is dependent on the inherent RNA helicase activity of DDX5 and the binding of Rmrp, an evolutionarily conserved nuclear lncRNA. Furthermore, Rmrp was found mutated in patients with cartilage-hair hypoplasia, and corresponding mutation in Rmrp in mice resulted in altered chromatin interaction, and diminished interaction between the DDX5 and RORγt, and also downregulated expression of selective Th-17 genes [42].
These examples highlight the importance of lncRNAs in regulating gene expression in immune cells and underscore yet another layer of complexity in gene regulation. Future studies should be focused towards elucidating their molecular functions which in turn could provide crucial insights into novel mechanisms of gene regulation, autoimmune and inflammation-mediated disorders, including T1D.
Genome-wide interactions between T1D SNPs, lncRNAs, enhancers, and other distal regulatory elements
More than 90% of disease-associated single-nucleotide polymorphisms (SNPs) are located within the non-coding regions of the genome such as promoters, enhancers, intergenic regions, and ncRNA genes [43]. The disease-associated SNPs have the potential to be regulatory in nature, particularly if they are significantly enriched in functional regulatory elements such as transcription factor binding sites (TFBSs), histone modification marks, DNase-I hypersensitive sites, and expression quantitative trait loci (eQTLs) [44, 45]. These disease-associated regulatory SNPs are also referred to as “functional SNPs” [44]. Approximately, 10% of the autoimmune disease-associated SNPs are present within lncRNAs and some of these SNPs are also known to act as cis-eQTLs [46]. It has been shown that 75% of the lincRNA cis-eQTLs specifically alter the expression of lincRNAs in a tissue-dependent fashion but does not affect the nearby protein-coding genes, and many of these cis-eQTLs SNPs are known to be associated with complex genetic diseases [47]. Since the expression of protein-coding genes can be regulated by lincRNAs either in cis [48] or trans [49] manner, this suggests a link between disease-associated SNPs within the non-coding regions with the regulation of protein-coding gene expression.
Distal regulatory elements such as enhancers, locus control regions (LCRs), and insulators are highly abundant in the human genome and play an important role in transcriptional control. These elements represent the primary mechanism by which cell and developmental specific gene expression is accomplished. Enhancers are regulatory sequences that can activate gene expression independent of their proximity to their target genes in a tissue-specific manner [50]. Multiple enhancer elements arrayed over large regions can synergistically regulate the expression of individual genes or gene clusters by altering the TF binding and chromatin states [51, 52]. Additionally, multiple polymorphisms in linkage disequilibrium (LD) impact clusters of enhancer elements active in the same cell type and cooperatively contribute to altered expression of their gene targets [53]. The multiple enhancer variants within a given locus typically target the same gene which results in either gain- or loss-of-function [53]. Additionally, the genes associated with multiple enhancer variants encode proteins that are often functionally related and enriched in common pathways. Recently, several methods have been developed for genome-wide prediction of enhancers primarily based on chromatin marks such as H3K4 monomethyl (H3K4me1) and H3K27 acetyl (H3K27ac) modifications, bi-directional transcription, and binding of p300 [54–57]. The underlying mechanism for enhancer function has been suggested to involve formation of long-range chromatin loops, bringing enhancers and promoters into proximity and allowing interaction of the necessary co-transcriptional factors [58, 59]. Formation of chromatin loops occurs between two distant genomic sequences that are brought in close vicinity by protein complexes and are assumed to be chemically cross-linked. Various chromosomal conformation capture techniques such as 3C (chromosome conformation capture) [60], 4C (circular chromosome conformation capture) [61], 5C (chromosome conformation capture carbon copy) [62], ChIA-PET (Chromatin Interaction Analysis with Paired-End-Tag sequencing) [63], and Hi-C [64] have been used to detect genome-wide chromosome interactions. Examples of long-range interactions within mammalian gene loci include the locus control region (LCR) and β-globin promoter [65, 66]; the α-globin gene cluster in erythroid cells [67]; the TH2 and MHC loci in T cells [68, 69]; and the imprinted gene clusters Dlx5, Dlx6 [70], and H19-Igf2 [71–73]. Additional example of long-range chromatin loop mediated interaction among regulatory elements on different chromosomes has been observed at the IFNγ and TH2 cytokine loci [74]. The transcriptional regulation of IL-21 gene at the chromatin level was recently uncovered to be mediated through long-range chromatin interaction in CD4+ T cells. IL-21, a pro-inflammatory cytokine with pleiotropic effects, is strongly associated with autoimmunity and inflammation and regulates various immune responses. A study by Park et al. showed that a distal enhancer element within an evolutionary conserved non-coding sequence 49 kb upstream of the IL-21 can upregulate IL-21 gene expression in a STAT3- and NFAT-dependent manner [75]. Stimulation of CD4+ T cells with IL-6 leads to the recruitment of STAT3 to the IL-21 promoter and the distal enhancer region, bringing them in close spatial proximity. As a consequence, this long-range interaction between the promoter and distal enhancer region dependent on IL-6/STAT3 signaling pathway alters the chromatin configuration dynamically, and controls the expression of IL-21 in CD4+ T cells [75].
Based on published genome-wide chromosome conformational capture datasets from various cell-lines and T1D associated SNPs we identified physical interactions between distant regulatory regions in T1D loci. Figure 2 shows an interactive map of T1D loci highlighting the physical interactions between distal regulatory elements and potential functional T1D SNP at each locus. T1D risk SNPs and SNPs in LD (r2 > 0.8, CEU HapMap3 population) were selected and scored based on the original GWAS signal, long range chromosome interactions, overlap with chromatin marks, epigenetic modifications and sequence motifs from various ENCODE cell lines [2, 76–78]. The top most scoring SNP for each region was inferred as the most significant functional SNP and selected for plotting along with the most significant distal chromosomal interaction signal. As an example, in ERBB3 locus, SNP rs4759229 qualified as the most significant variant. rs4759229 is in perfect LD with T1D risk SNP rs2292239, overlaps a known enhancer region and has a long range interaction signal with an antisense lncRNA AC008079.1 (Ensembl ID: ENSG00000187979) located at USP18 locus on chromosome 22. In a recent study, we proposed that the ERBB3 SNP rs2292239 and its proxy SNPs in perfect LD rs3741499 and rs4759229 are putatively functional based on the overlapping open chromatin marks, TFBs and DNase I hypersensitivity peaks [79]. We further showed that SNP rs4759229 overlaps a known enhancer element and is in the vicinity of several lncRNA transcripts overlapping ERBB3 locus. The potential functional T1D SNPs within known T1D candidate genes such as GLIS3, ERBB3, CTRB1, CTSH, FUT2, IL27, SKAP2, TNFAIP3 and PTPN2 had highly significant distal chromosomal interactions including enhancers and lncRNAs that are worthy of specific laboratory investigations (Fig. 2). Intriguingly, the impact of interactions between T1D SNPs and enhancer associated lncRNAs on transcription and ultimately on T1D risk remains to be seen. Based on the above evidence we postulate that the T1D SNPs mapping to enhancer associated lncRNAs could potentially alter the expression of their gene targets through enhancer mediated interactions and thereby significantly impact β cell gene expression.

Distal regulatory elements and potential functional T1D SNPs in T1D susceptibility loci. The inner circle represents T1D susceptibility genes and genomic loci and the outer circle lists the regulatory variants and distal interaction regions. The distal interactive elements are denoted by an asterisk which includes enhancer elements and lncRNAs. The red lines indicate long-range chromosome interaction signals to another locus and the intensity of interaction is represented by the width of the red lines
Many lncRNAs have been reported to be expressed from the enhancer regions that are produced by activity-dependent RNA polymerase II binding of specific enhancers [80]. The expression levels of these enhancer associated lncRNAs positively correlate with the expression of neighboring protein-coding genes, i.e., depletion of enhancer associated lncRNAs led to decreased expression of their neighboring protein-coding genes [48, 80]. In addition, enhancers overlapping lncRNAs have higher H3K4me3/H3K4me1 ratios as compared to enhancers that do not overlap lncRNAs [81]. The enrichment of H3K4me3 marks (which are also associated with active promoters) points towards strong transcriptional capabilities of overlapping lncRNAs. lncRNAs transcribed from active enhancers have been identified as important players in mediating enhancer function [82]. Enhancers associated lncRNA play roles in important physiological processes and influence the activation of protein coding and non-coding genes in both cis and trans mediated mechanism. For example, lncRNA NEST is involved in cis-activation of the neighboring interferon γ locus, whereas lncRNA Jpx regulates trans-activation of another lncRNA, XIST, (which is critical for X inactivation) [36, 83]. While acting in trans, enhancer associated lncRNAs act over long distances by long range chromatin loop mediated interactions and activate transcription at distal promotors. It has also been suggested that bridging factors such as Mediator/Cohesin complex and enhancer associated lncRNAs are involved in establishment of chromatin looping between the lncRNAs and their regulated distal promotors [84]. Knockdown of either lncRNA or mediator subunits has been shown to abolish the chromosomal interactions [84].
It has been shown that islet-specific lncRNAs and TFs co-regulate genes associated with enhancer clusters [24, 85]. lncRNAs regulating enhancer cluster-associated genes bound by multiple islet-specific TFs include HI-LNC12, HI-LNC15, HI-LNC30, HI-LNC78, HI-LNC80, HI-LNC85, and PLUT (HI-LNC71) [24]. However, further studies are warranted that employ chromosomal conformation techniques to identify potential targets of enhancer associated lncRNAs in human islets.
Future challenges and opportunities
Although lncRNAs have achieved formable recognition as key players in gene regulation and disease, there is still a huge gap in our overall understanding of lncRNA regulatory functions and underlying molecular mechanisms, particularly in context of β cell function and development of T1D. The rise of lncRNAs as key regulators of gene expression during normal development and diseases has positioned these pervasive transcripts in the crosshairs of novel disease-specific biomarker discovery. The dysregulation of lncRNA expression not only represents a newfangled layer of intricacy in the molecular architecture of human malady, but it also unveils the potential to use lncRNAs as disease biomarkers. In contrast to their “cousins” mRNA transcripts, the lncRNAs themselves are functional molecules and their expression levels might serve as a better disease indicator. Moreover, expression of lncRNAs is highly tissue-specific and disease-specific which indicates that lncRNA-based expression signatures could effectively be used to accurately diagnose and classify disease. Although, the field of lncRNA-based diagnostics is still in its infancy, the use of distinct lncRNAs in a clinical diagnostics setting has already taken off. Indeed, lncRNAs have already been suggested as potential biomarkers for a number of diseases, including cancer. For example, PCA3, a prostate-specific lncRNA notably overexpressed in prostate cancer, has been developed into diagnostic assay to detect prostate cancer [86]. Nevertheless, there are still many challenges ahead of us that need to be addressed in order to fully appreciate the function of lncRNAs in islet biology and T1D contexts.
Most of the functionally characterized lncRNAs exhibit modular-domain architecture that arises from their well-conserved secondary and tertiary structures and is crucial for their biological functions. This assumes importance as it illustrates the importance of the conservation at secondary and tertiary structure level rather than at primary sequence level [87–89]. Therefore, future studies are needed to identify homologous lncRNAs taking a structure-based evolutionary conservation criterion into consideration rather than relying only on the primary sequence-based conservation in cellular and subclinical models of T1D.
Although, a handful of lncRNAs have been functionally characterized, they have certainly emerged as bona fide players in regulating the gene expression at various levels during all the stages of development and disease. With the advent of next-generation sequencing technologies, thousands of lncRNA genes have already been identified and annotated in human and mouse. According to the recent GENCODE v26 (www.gencodegenes.org), more than 15,000 and 11,000 lncRNA genes have already identified in the human and mouse, respectively. In addition, FANTOM5 cap analysis of gene expression (CAGE) in primary cell types and tissues identified 27,919 human lncRNA genes with accurate 5′ termini [90]. Based on multiple lines of evidence, including genomic, epigenomic features and evolutionary conservation of the lncRNAs, 19,175 were reported as potentially functional in the human genome [90]. Surprisingly, most of the intergenic lncRNAs identified in the FANTOM5 project were transcribed from the enhancers and not from the promoters.
One of the major bottlenecks in studying the lncRNA functions has been their low steady-state levels in the cells [91]. Majority of lncRNAs are expressed at low-levels which makes it more challenging to accurately annotate their gene boundaries. The problem of incomplete annotation of lncRNAs is further compounded by the lack of typical genomic hallmarks of transcription initiation and termination that are often used as primary flag posts for defining the gene boundaries [91]. But, recently many methods have been developed and implemented to improve the annotation of lncRNAs, including RNA Capture Sequencing (CaptureSeq) [92], coupling of rapid amplification of cDNA ends (RACE) technique to long-read sequencing (RACE-Seq) [93], RNA Capture Long Seq (CLS) [94], and genome-scale CRISPR-mediated interference (CRISPRi) [95]. So far, these methods have been used for exploring the transcriptomic structure of lncRNA loci using cell or tissue types that are not relevant to diabetes. Nevertheless, these methods provide an excellent experimental framework for future studies to address the lingering questions regarding the roles and molecular mechanisms of lncRNAs in β cell function and T1D pathogenesis. Therefore, employing methods like CLS, RACE-Seq, CAGE and CRISPRi under the proinflammatory cytokines stimulation and control settings will enable to disentangle the transcriptomic landscape of β cells.
It is known that active β cells exhibit extensive allelic imbalance in gene expression [96]. However, the impact of allelic imbalance on the lncRNA expression has been not studied. A robust reassessment of the regulation of allele-specific gene expression in lncRNA loci overlapping or lying in close proximity of T1D GWAS SNPs in islets and β cells derived from cadaveric pancreas with known genotypes would be highly desirable. This could provide important clues linking disease-associated SNPs with the lncRNA expression. Furthermore, these studies have the potential to unravel the diversity of lncRNA repertoire, including the enhancer RNAs (eRNAs) [97] and novel rare transcripts and many of which might have important functional roles in modulating the β cell function.
Conclusions
In immune-mediated and inflammatory diseases such as T1D, lncRNA-based transcriptional signatures might open new avenues for lncRNA-based diagnostics, classification or personalized therapeutic regimens in near future. Furthermore, assessment of functional implications of T1D SNPs overlapping lncRNAs and enhancers regions is highly warranted from perspective of β cell function and development of T1D. The precise biochemical characteristics and molecular basis of β- and immune cells expressed lncRNA functions are necessary to elucidate how deregulations of immune cell-specific T1D loci-associated lncRNAs, as well islet-specific lncRNAs, potentially contribute to the development of islet autoimmunity to progression of T1D.
Abbreviations
- eQTL:
-
Expression quantitative trait loci
- GWAS:
-
Genome-wide association studies
- LD:
-
Linkage disequilibrium
- lncRNA:
-
Long non-coding RNA
- SNP:
-
Single-nucleotide polymorphism
- TF:
-
Transcription factor
- TFBS:
-
Transcription factor binding site
- T1D:
-
Type 1 diabetes
References
Pociot F, Akolkar B, Concannon P, Erlich HA, Julier C, Morahan G, et al. Genetics of type 1 diabetes: what’s next? Diabetes. 2010;59:1561–71.
Groop L, Pociot F. Genetics of diabetes—are we missing the genes or the disease? Mol Cell Endocrinol. 2014;382:726–39.
Soleimanpour SA, Stoffers DA. The pancreatic β cell and type 1 diabetes: innocent bystander or active participant? Trends Endocrinol Metab. 2013;24:324–31.
Noble JA, Erlich HA. Genetics of type 1 diabetes. Cold Spring Harb Perspect Med. 2012;2:a007732.
Rich SS, Concannon P, Erlich H, Julier C, Morahan G, Nerup J, et al. The type 1 diabetes genetics consortium. Ann N Y Acad Sci. 2006;1079:1–8.
Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41:703–7.
Onengut-Gumuscu S, Chen W-M, Burren O, Cooper NJ, Quinlan AR, Mychaleckyj JC, et al. Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat Genet. 2015;47:381–6.
Santin I, Eizirik DL. Candidate genes for type 1 diabetes modulate pancreatic islet inflammation and β-cell apoptosis. Diabetes Obes Metab. 2013;15(Suppl 3):71–81.
Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–66.
Knoll M, Lodish HF, Sun L. Long non-coding RNAs as regulators of the endocrine system. Nat Rev Endocrinol. 2015;11:151–60.
Lam MTY, Li W, Rosenfeld MG, Glass CK. Enhancer RNAs and regulated transcriptional programs. Trends Biochem Sci. 2014;39:170–82.
Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154:26–46.
Fitzgerald KA, Caffrey DR. Long noncoding RNAs in innate and adaptive immunity. Curr Opin Immunol. 2014;26:140–6.
Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A. 2009;106:11667–72.
Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–7.
Eliasson L, Esguerra JLS. Role of non-coding RNAs in pancreatic beta-cell development and physiology. Acta Physiol (Oxf). 2014;211:273–84.
Motterle A, Gattesco S, Caille D, Meda P, Regazzi R. Involvement of long non-coding RNAs in beta cell failure at the onset of type 1 diabetes in NOD mice. Diabetologia. 2015;58:1827–35.
Morán I, Akerman I, van de Bunt M, Xie R, Benazra M, Nammo T, et al. Human β cell transcriptome analysis uncovers lncRNAs that are tissue-specific, dynamically regulated, and abnormally expressed in type 2 diabetes. Cell Metab. 2012;16:435–48.
Ku GM, Kim H, Vaughn IW, Hangauer MJ, Myung Oh C, German MS, et al. Research resource: RNA-Seq reveals unique features of the pancreatic β-cell transcriptome. Mol Endocrinol. 2012;26:1783–92.
Liu J-Y, Yao J, Li X-M, Song Y-C, Wang X-Q, Li Y-J, et al. Pathogenic role of lncRNA-MALAT1 in endothelial cell dysfunction in diabetes mellitus. Cell Death Dis. 2014;5:e1506.
Wallace C, Smyth DJ, Maisuria-Armer M, Walker NM, Todd JA, Clayton DG. The imprinted DLK1-MEG3 gene region on chromosome 14q32.2 alters susceptibility to type 1 diabetes. Nat Genet. 2010;42:68–71.
You L, Wang N, Yin D, Wang L, Jin F, Zhu Y, et al. Downregulation of long noncoding RNA Meg3 affects insulin synthesis and secretion in mouse pancreatic beta cells. J Cell Physiol. 2016;231:852–62.
Arnes L, Akerman I, Balderes DA, Ferrer J, Sussel L. βlinc1 encodes a long noncoding RNA that regulates islet β-cell formation and function. Genes Dev. 2016;30:502–7.
Akerman I, Tu Z, Beucher A, Rolando DMY, Sauty-Colace C, Benazra M, et al. Human pancreatic β cell lncRNAs control cell-specific regulatory networks. Cell Metab. 2017;25:400–11.
Yin D, Zhang E, You L, Wang N, Wang L, Jin F, et al. Downregulation of lncRNA TUG1 affects apoptosis and insulin secretion in mouse pancreatic β cells. Cell Physiol Biochem. 2015;35:1892–904.
Li J, Xuan Z, Liu C. Long non-coding RNAs and complex human diseases. Int J Mol Sci. 2013;14:18790–808.
Hrdlickova B, de Almeida RC, Borek Z, Withoff S. Genetic variation in the non-coding genome: involvement of micro-RNAs and long non-coding RNAs in disease. Biochim Biophys Acta. 1842;2014:1910–22.
Hrdlickova B, Kumar V, Kanduri K, Zhernakova DV, Tripathi S, Karjalainen J, et al. Expression profiles of long non-coding RNAs located in autoimmune disease-associated regions reveal immune cell-type specificity. Genome Med. 2014;6:88.
Carpenter S, Aiello D, Atianand MK, Ricci EP, Gandhi P, Hall LL, et al. A long noncoding RNA mediates both activation and repression of immune response genes. Science. 2013;341:789–92.
Atianand MK, Fitzgerald KA. Long non-coding RNAs and control of gene expression in the immune system. Trends Mol Med. 2014;20:623–31.
Li Z, Chao T-C, Chang K-Y, Lin N, Patil VS, Shimizu C, et al. The long noncoding RNA THRIL regulates TNFα expression through its interaction with hnRNPL. Proc Natl Acad Sci U S A. 2014;111:1002–7.
Krawczyk M, Emerson BM. p50-associated COX-2 extragenic RNA (PACER) activates COX-2 gene expression by occluding repressive NF-κB complexes. elife. 2014;3:e01776.
IIott NE, Heward JA, Roux B, Tsitsiou E, Fenwick PS, Lenzi L, et al. Long non-coding RNAs and enhancer RNAs regulate the lipopolysaccharide-induced inflammatory response in human monocytes. Nat Commun. 2014;5:3979.
Lu J, Wu X, Hong M, Tobias P, Han J. A potential suppressive effect of natural antisense IL-1β RNA on lipopolysaccharide-induced IL-1β expression. J Immunol. 2013;190:6570–8.
Carpenter S, Fitzgerald KA. Transcription of inflammatory genes: long noncoding RNA and beyond. J Interf Cytokine Res. 2015;35:79–88.
Gomez JA, Wapinski OL, Yang YW, Bureau J-F, Gopinath S, Monack DM, et al. The NeST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-γ locus. Cell. 2013;152:743–54.
Collier SP, Collins PL, Williams CL, Boothby MR, Aune TM. Cutting edge: influence of Tmevpg1, a long intergenic noncoding RNA, on the expression of Ifng by Th1 cells. J Immunol. 2012;189:2084–8.
Rapicavoli NA, Qu K, Zhang J, Mikhail M, Laberge R-M, Chang HY. A mammalian pseudogene lncRNA at the interface of inflammation and anti-inflammatory therapeutics. elife. 2013;2:e00762.
Wang P, Xue Y, Han Y, Lin L, Wu C, Xu S, et al. The STAT3-binding long noncoding RNA lnc-DC controls human dendritic cell differentiation. Science. 2014;344:310–3.
Ryba-Stanisławowska M, Skrzypkowska M, Myśliwiec M, Myśliwska J. Loss of the balance between CD4(+)Foxp3(+) regulatory T cells and CD4(+)IL17A(+) Th17 cells in patients with type 1 diabetes. Hum Immunol. 2013;74:701–7.
Ferraro A, Socci C, Stabilini A, Valle A, Monti P, Piemonti L, et al. Expansion of Th17 cells and functional defects in T regulatory cells are key features of the pancreatic lymph nodes in patients with type 1 diabetes. Diabetes. 2011;60:2903–13.
Huang W, Thomas B, Flynn RA, Gavzy SJ, Wu L, Kim SV, et al. DDX5 and its associated lncRNA Rmrp modulate TH17 cell effector functions. Nature. 2015;528:517–22.
Ricaño-Ponce I, Wijmenga C. Mapping of immune-mediated disease genes. Annu Rev Genomics Hum Genet. 2013;14:325–53.
Schaub MA, Boyle AP, Kundaje A, Batzoglou S, Snyder M. Linking disease associations with regulatory information in the human genome. Genome Res. 2012;22:1748–59.
Hoffman MM, Ernst J, Wilder SP, Kundaje A, Harris RS, Libbrecht M, et al. Integrative annotation of chromatin elements from ENCODE data. Nucleic Acids Res. 2013;41:827–41.
Gong J, Liu W, Zhang J, Miao X, Guo A-Y. lncRNASNP: a database of SNPs in lncRNAs and their potential functions in human and mouse. Nucleic Acids Res. 2015; 43(Database issue):D181–D186.
Kumar V, Westra H-J, Karjalainen J, Zhernakova DV, Esko T, Hrdlickova B, et al. Human disease-associated genetic variation impacts large intergenic non-coding RNA expression. PLoS Genet. 2013;9:e1003201.
Ørom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, et al. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010;143:46–58.
Guttman M, Donaghey J, Carey BW, Garber M, Grenier JK, Munson G, et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature. 2011;477:295–300.
Visel A, Rubin EM, Pennacchio LA. Genomic views of distant-acting enhancers. Nature. 2009;461:199–205.
Sakabe NJ, Savic D, Nobrega MA. Transcriptional enhancers in development and disease. Genome Biol. 2012;13:238.
Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature. 2012;489:109–13.
Corradin O, Saiakhova A, Akhtar-Zaidi B, Myeroff L, Willis J, Cowper-Sal lari R, et al. Combinatorial effects of multiple enhancer variants in linkage disequilibrium dictate levels of gene expression to confer susceptibility to common traits. Genome Res. 2014;24:1–13.
Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–8.
Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–12.
Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–83.
Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507:455–61.
Kadauke S, Blobel GA. Chromatin loops in gene regulation. Biochim Biophys Acta. 1789;2009:17–25.
Deng W, Lee J, Wang H, Miller J, Reik A, Gregory PD, et al. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell. 2012;149:1233–44.
Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306–11.
Zhao Z, Tavoosidana G, Sjölinder M, Göndör A, Mariano P, Wang S, et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat Genet. 2006;38:1341–7.
Dostie J, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA, et al. Chromosome conformation capture carbon copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006;16:1299–309.
Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009;462:58–64.
Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–93.
Carter D, Chakalova L, Osborne CS, Dai Y, Fraser P. Long-range chromatin regulatory interactions in vivo. Nat Genet. 2002;32:623–6.
Tolhuis B, Palstra RJ, Splinter E, Grosveld F, de Laat W. Looping and interaction between hypersensitive sites in the active beta-globin locus. Mol Cell. 2002;10:1453–65.
Vernimmen D, De Gobbi M, Sloane-Stanley JA, Wood WG, Higgs DR. Long-range chromosomal interactions regulate the timing of the transition between poised and active gene expression. EMBO J. 2007;26:2041–51.
Spilianakis CG, Flavell RA. Long-range intrachromosomal interactions in the T helper type 2 cytokine locus. Nat Immunol. 2004;5:1017–27.
Kumar PP, Bischof O, Purbey PK, Notani D, Urlaub H, Dejean A, et al. Functional interaction between PML and SATB1 regulates chromatin-loop architecture and transcription of the MHC class I locus. Nat Cell Biol. 2007;9:45–56.
Horike S, Cai S, Miyano M, Cheng J-F, Kohwi-Shigematsu T. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat Genet. 2005;37:31–40.
Murrell A, Heeson S, Reik W. Interaction between differentially methylated regions partitions the imprinted genes Igf2 and H19 into parent-specific chromatin loops. Nat Genet. 2004;36:889–93.
Kurukuti S, Tiwari VK, Tavoosidana G, Pugacheva E, Murrell A, Zhao Z, et al. CTCF binding at the H19 imprinting control region mediates maternally inherited higher-order chromatin conformation to restrict enhancer access to Igf2. Proc Natl Acad Sci U S A. 2006;103:10684–9.
Yoon YS, Jeong S, Rong Q, Park K-Y, Chung JH, Pfeifer K. Analysis of the H19ICR insulator. Mol Cell Biol. 2007;27:3499–510.
Spilianakis CG, Lalioti MD, Town T, Lee GR, Flavell RA. Interchromosomal associations between alternatively expressed loci. Nature. 2005;435:637–45.
Park J-H, Choi Y, Song M-J, Park K, Lee J-J, Kim H-P. Dynamic long-range chromatin interaction controls expression of IL-21 in CD4+ T cells. J Immunol. 2016;196:4378–89.
ENCODE Project Consortium, Birney E, Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816.
Li MJ, Sham PC, Wang J. Genetic variant representation, annotation and prioritization in the post-GWAS era. Cell Res. 2012;22:1505–8.
Raychaudhuri S. VIZ-GRAIL: visualizing functional connections across disease loci. Bioinformatics. 2011;27:1589–90.
Kaur S, Mirza AH, Brorsson CA, Fløyel T, Størling J, Mortensen HB, et al. The genetic and regulatory architecture of ERBB3-type 1 diabetes susceptibility locus. Mol Cell Endocrinol. 2016;419:83–91.
Kim T-K, Hemberg M, Gray JM. Enhancer RNAs: a class of long noncoding RNAs synthesized at enhancers. Cold Spring Harb Perspect Biol. 2015;7:a018622.
Murakami S, Gadad SS, Kraus WL. A PreSTIGEous use of LncRNAs to predict enhancers. Cell Cycle. 2015;14:1619–20.
Ørom UA, Shiekhattar R. Long noncoding RNAs usher in a new era in the biology of enhancers. Cell. 2013;154:1190–3.
Tian D, Sun S, Lee JT. The long noncoding RNA, Jpx, is a molecular switch for X chromosome inactivation. Cell. 2010;143:390–403.
Lai F, Orom UA, Cesaroni M, Beringer M, Taatjes DJ, Blobel GA, et al. Activating RNAs associate with mediator to enhance chromatin architecture and transcription. Nature. 2013;494:497–501.
Pasquali L, Gaulton KJ, Rodríguez-Seguí SA, Mularoni L, Miguel-Escalada I, Akerman I, et al. Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat Genet. 2014;46:136–43.
Lee GL, Dobi A, Srivastava S. Prostate cancer: diagnostic performance of the PCA3 urine test. Nat Rev Urol. 2011;8:123–4.
Diederichs S. The four dimensions of noncoding RNA conservation. Trends Genet. 2014;30:121–3.
Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–27.
Mercer TR, Mattick JS. Structure and function of long noncoding RNAs in epigenetic regulation. Nat Struct Mol Biol. 2013;20:300–7.
Hon CC, Ramilowski JA, Harshbarger J, Bertin N, Rackham OJ, Gough J, et al. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature. 2017;543:199–204.
Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–89.
Mercer TR, Gerhardt DJ, Dinger ME, Crawford J, Trapnell C, Jeddeloh JA, et al. Targeted RNA sequencing reveals the deep complexity of the human transcriptome. Nat Biotechnol. 2011;30:99–104.
Lagarde J, Uszczynska-Ratajczak B, Santoyo-Lopez J, Gonzalez JM, Tapanari E, Mudge JM, et al. Extension of human lncRNA transcripts by RACE coupled with long-read high-throughput sequencing (RACE-Seq). Nat Commun. 2016;7:12339.
Lagarde J, Uszczynska-Ratajczak B, Carbonell S, Davis C, Gingeras TR, Frankish A. High-throughput annotation of full-length long noncoding RNAs with capture long-read sequencing (CLS). bioRxiv. 2017;105064. doi:https://doi.org/10.1101/105064.
Liu SJ, Horlbeck MA, Cho SW, Birk HS, Malatesta M, He D, et al. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science. 2017;355. doi: 10.1126/science.aah7111.
Nica AC, Ongen H, Irminger JC, Bosco D, Berney T, Antonarakis SE, et al. Cell-type, allelic, and genetic signatures in the human pancreatic beta cell transcriptome. Genome Res. 2013;23:1554–62.
Li W, Notani D, Rosenfeld MG. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat Rev Genet. 2016;17:207–23.
Funding
Not applicable
Availability of data and materials
All data included in this review article are available from published studies and have been properly cited.
Author information
Authors and Affiliations
Contributions
AHM and SK researched the data and drafted and reviewed the manuscript before submission. FP made substantial contribution to discussion of the content and reviewed the manuscript before submission. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Authors’ information
AHM is a postdoctoral fellow and received predoctoral and postdoctoral training in the laboratories of Professor Flemming Pociot (Copenhagen Diabetes Research Center, Copenhagen, Denmark), Professor Jan Gorodkin (Center for non-coding RNA in Technology and Health, Copenhagen, Denmark), and Professor Francis Barany (Sandra and Edward Meyer Cancer Center, Weill Cornell Medicine, NY, USA). AHM obtained his PhD degree in Cellular and Genetic Medicine from the University of Copenhagen, Denmark, under the supervision of Professor Jan Gorodkin and Professor Flemming Pociot. AHM’s research revolves broadly around understanding the role of non-coding RNAs in autoimmune and inflammatory diseases. AHM is currently involved in development of assays and technologies for early detection of tumors using circulating cell-free nucleic acid based genetic and epigenetic biomarkers in collaboration with Professor Francis Barany and Professor Steven Soper (Department of Chemistry, University of Kansas, KS, USA).
SK is a postdoctoral fellow training in the laboratory of Professor Flemming Pociot. SK obtained her PhD degree in Biostatistics and Bioinformatics from University of Copenhagen, Denmark, under the supervision of Professor Flemming Pociot (Copenhagen Diabetes Research Center, Herlev and Gentofte Hospital, Denmark) and Professor Henrik B. Mortensen (Department of Clinical Medicine, University of Copenhagen, Denmark). SK’s research revolves broadly around understanding the role of non-coding RNAs and regulatory elements in β cell function and type 1 diabetes.
FP is a professor of Pediatrics and Clinical Medicine at Herlev and Gentofte Hospital, Denmark and University of Copenhagen, Denmark. FP received his MD from University of Copenhagen and performed his doctoral research at Steno Diabetes Center, Denmark. FP has a strong focus on clinical markers for residual β cell function and to identify early markers for type 1 diabetes risk. FP has been part of the Type 1 Diabetes Genetics Consortium (T1DGC) Steering Group, which published the largest GWAS (genome-wide association study) in T1D. FP has published more than 300 papers in peer reviewed journals accruing >14,000 citations. FP has been ranked no. 21, worldwide by cited papers within the field of Diabetes 1991–2001 and his H-index is 51.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Mirza, A.H., Kaur, S. & Pociot, F. Long non-coding RNAs as novel players in β cell function and type 1 diabetes. Hum Genomics 11, 17 (2017). https://doi.org/10.1186/s40246-017-0113-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-017-0113-7