
Abstract
Background
Diabetes mellitus is a leading cause of morbidity and mortality worldwide. Studies have shown that Zinc has numerous beneficial effects in both type-1 and type-2 diabetes. We aim to evaluate the literature on the mechanisms and molecular level effects of Zinc on glycaemic control, β-cell function, pathogenesis of diabetes and its complications.
Methods
A review of published studies reporting mechanisms of action of Zinc in diabetes was undertaken in PubMed and SciVerse Scopus medical databases using the following search terms in article title, abstract or keywords; (“Zinc” or “Zn”) and (“mechanism” or “mechanism of action” or “action” or “effect” or “pathogenesis” or “pathology” or “physiology” or “metabolism”) and (“diabetes” or “prediabetes” or “sugar” or “glucose” or “insulin”).
Results
The literature search identified the following number of articles in the two databases; PubMed (n = 1799) and SciVerse Scopus (n = 1879). After removing duplicates the total number of articles included in the present review is 111. Our results show that Zinc plays an important role in β-cell function, insulin action, glucose homeostasis and the pathogenesis of diabetes and its complications.
Conclusion
Numerous in-vitro and in-vivo studies have shown that Zinc has beneficial effects in both type-1 and type-2 diabetes. However further randomized double-blinded placebo-controlled clinical trials conducted for an adequate duration, are required to establish therapeutic safety in humans.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diabetes mellitus is a leading cause of morbidity and mortality worldwide, with an estimated 387 million adults being affected in year 2014, a figure which is expected to increase by nearly 40 % by year 2035 [1]. Ninety to ninety five percent of those with the disease have type-2 diabetes. In a patho-physiologic sense, type-2 diabetes is a multi-organ, multi-factorial condition characterized primarily by insulin resistance, hyper insulinaemia and β-cell dysfunction, which ultimately leads to β-cell failure [2]. Type-1 diabetes has historically been most prevalent in populations of European origin, and the latest edition of the Diabetes Atlas estimates that 490,100 children below the age of 15 years are living with type-1 diabetes [1]. Currently 77 % of those with diabetes live in low- and middle-income countries of the African, Asian, and South American regions [1, 3, 4]. In 2014, diabetes was responsible for 4.9 million deaths worldwide and at least US$ 612 billion in global healthcare expenditures (11 % of the total global healthcare expenditures in adults) [1]. Recent cost estimates, include those for Brazil (US$ 3.9 billion), Argentina (US$ 0.8 billion) and Mexico (US$ 2.0 billion) [5]. Each of these is an annual figure and is rising as diabetes prevalence increases. Overall, direct health care costs of diabetes ranges from 2.5 to 15 % annual health care budgets, depending on local diabetes prevalence and the sophistication of the treatment available [5]. Diabetes is also associated with a host of potentially disabling macro- and micro-vascular complications. Hence, there is also a much larger burden in the form of lost productivity as a result of restricted daily activity. This rapidly increasing prevalence is attributable to population growth, aging, urbanization, unhealthy dietary habits, increasing prevalence of obesity and physical inactivity [6].
Although comprehensive diabetes management guidelines are readily available, even in developed countries like the US 30–50 % adults with diabetes do not meet individualized targets for glycaemic, blood pressure, or lipid control [7]. Reasons for failure to achieve glycaemic control includes the progression of underlying β-cell dysfunction, incomplete adherence to treatment (often due to adverse effects of medication) and reluctance of clinicians to intensify therapy [8]. Anti-diabetic agents currently in use can directly or indirectly enhance the functioning of β-cells. However, reducing the decline and the eventual failure of β-cells is crucial in preventing type-2 diabetes in those at risk and halting disease progression in the affected patients [8]. The increasing worldwide prevalence of type-2 diabetes and the progressive loss of metabolic control in patients are clear demonstrations that the current therapeutic strategies aimed at protecting the β-cells are largely inadequate. Hence there is an urgent need for anti-diabetic agents targeting the intimate mechanisms of β-cell damage and optimizing its function at cellular level.
Insulin, is stored as a hexamer containing two Zinc ions in β-cells of the pancreas and released into the portal venous system at the time of β-cell de-granulation [9]. In-vitro and in-vivo studies in animals and humans have shown that Zinc has numerous beneficial effects in both type-1 and type-2 diabetes [10–14]. A recent meta-analysis confirmed these findings, and concluded that Zinc supplementation in patients with diabetes improves glycaemic control and promotes healthy lipid parameters [15]. Hence, it is evident that Zinc has a promising potential as a novel therapeutic agent in diabetes. Studies have also shown that diabetes is commonly accompanied by hypozincemia and hyperzincuria [16, 17]. Furthermore the high prevalence of Zinc deficiency in developing countries could be contributing towards driving the current diabetes epidemic encountered by them [4, 18]. Numerous research studies have been conducted to clarify the molecular mechanisms underlying the action of Zinc in diabetes. Understanding the molecular mechanisms of action of Zinc will help to further develop targeted therapy and guide future research. The present study aims to systematically evaluate the literature on the mechanisms and molecular level effects of Zinc on glycaemic control, β-cell function, pathogenesis of diabetes and its complications.
Methods
A systematic review of published studies reporting mechanisms of action of Zinc in diabetes was undertaken in accordance with the Preferred Reporting Items for Systematic reviews and Meta-Analyses (PRISMA) statement (Additional file 1).
Search strategy
A comprehensive search of the literature was conducted in the PubMed® (U.S. National Library of Medicine, USA) and SciVerse Scopus® (Elsevier Properties S.A, USA) databases for studies published before 31st July 2015. During the first stage the above databases were searched using the following search terms in article title, abstract or keywords; (“Zinc” or “Zn”) and (“mechanism” or “mechanism of action” or “action” or “effect” or “pathogenesis” or “pathology” or “physiology” or “metabolism”) and (“diabetes” or “prediabetes” or “sugar” or “glucose” or “insulin”).
In the second stage the total hits obtained from searching these three databases were pooled together and duplicates were removed. This was followed by screening of the retrieved articles by reading the article title in the third and abstracts in fourth stage. In the fifth stage individual manuscripts were screened, and those not satisfying inclusion criteria (given below) were excluded. This search process was conducted independently by two reviewers (PR and SP) and the final group of articles to be included in the review was determined after an iterative consensus process.
Inclusion/exclusion criteria and data extraction
The following inclusion criteria were used; a) In-vitro or in-vivo studies reporting effect of Zinc on diabetes/pre-diabetes, evaluating effects of Zinc on sugar/glucose, insulin and/or related metabolic parameters, evaluating the effects of Zinc on pathogenesis and/or complication of diabetes b) Published in English, or with detailed summaries in English and c) Peer-reviewed fully published research papers. Conference proceedings, editorials, commentaries, review articles and book chapters/book reviews were excluded.
Data were extracted from the included studies by one reviewer using a standardized form and checked for accuracy by a second reviewer. The data extracted from each study were: a) study details (lead author, year published/year of survey, type of study—In-vitro/In-vivo), b) methods (study design, sample size, duration) and c) mechanism of action data. Discrepancies in the extracted data were resolved by discussion, with involvement of a third reviewer when necessary.
Results
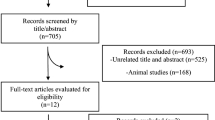
The literature search using the above search criteria identified the following number of articles in the two databases; Medline® (n = 1799) and SciVerse Scopus® (n = 1879). After removing duplicates the total number of articles included in the present review is 111. The search strategy is summarized in Fig. 1.

Summarized search strategy
Anti-oxidant properties
Anti-oxidant properties of Zinc have been evaluated in-vitro, as well as in-vivo animal and human studies. Hypozincaemia and hyperzincuria is known to be present in patients with both type-1 and type-2 diabetes, and Zinc supplementation is known to be helpful in restoring plasma Zinc levels to normal [11, 19–21]. Plasma Thiobarbituric acid reactive substances (TBARS), a marker of oxidative stress is high in both type-1 and type-2 diabetic patients, and significantly decreased by supplementation of Zinc 30 mg/day for 3–6 months [11, 19]. Selenium-dependent glutathione peroxidase (Se-GPx), an anti-oxidant enzyme was also low at baseline in patients with type-1 diabetes, and was normalized after Zinc supplementation [11]. However, no significant difference in antioxidant metallo-enzyme activity was observed in patients with type-2 diabetes [19]. Plasma Zinc levels are known to be negatively correlated with the TBARS levels in both obese and non-obese patients with type-2 diabetes [22].
In animal models of insulin resistance, Zinc supplementation is known to enhance insulin sensitivity and antioxidant status [23, 24]. Furthermore, in diabetes induced animal models anti-oxidant enzymes catalase, GPx and super-oxide dismutase (SOD) are decreased than in normal animals [25–27]. Zinc supplementation in these animals restored the enzyme activity and increased glutathione synthesis [25–27]. Plasma malondialdehyde (MDA) levels, an index of lipid peroxidation was increased in diabetic animals and significantly decreased following Zinc supplementation [26, 28]. When rats were simultaneously treated with a single injection of alloxan and ZnCl2, the increase in blood glucose concentration induced by alloxan was significantly reduced at 24, 48, and 72 h post-treatment with ZnCl2 [29]. ZnCl2 injection also increased the retinal, pancreatic, and liver glutathione and reduced the TBARS content in comparison with the alloxan-treated group [29]. Zinc supplementation or injection is also known to cause a significant induction of anti-oxidant metallothionein (MT) protein synthesis in the pancreatic islets, kidneys, liver and heart of diabetes-induced animals [27, 28, 30–34].
A significant increase in cardiac morphological impairment, fibrosis, and dysfunction is seen in diabetic mice, which is reversed by Zinc [33]. Cultured cardiac cells that were directly exposed to high levels of glucose (HG) and free fatty acid (FFA), treatment that mimics diabetes, resulted in reduced cell survival rate, which was reversed by Zinc [33]. Furthermore, when MT expression was silenced with the use of MT small-interfering RNA, the preventive effect of pretreatment with Zinc was abolished [33]. Hence, the prevention of diabetic cardiomyopathy by Zinc supplementation is predominantly mediated by an increase in cardiac MT and the resultant anti-oxidant effects [33, 35]. Diabetes-induced renal oxidative damage, inflammation and up-regulated expression of pro-fibrosis mediators were also markedly attenuated by Zinc supplementation, mediated via the expression of MT [27, 36]. In-vivo studies in diabetes induced rats have shown that Zinc has a protective effect against diabetes induced peripheral nerve damage by stimulating MT synthesis and reducing oxidative stress [37].
Diabetes induced a significant increase in aortic oxidative damage, inflammation, and remodeling in mice (increased fibrosis and wall thickness) [38]. Zinc treatment of these diabetic mice completely prevented the above pathogenic changes in the aorta, and also significantly up-regulated the expression and function of nuclear factor (erythroid-derived 2)-like 2 (Nrf2), a pivotal regulator of anti-oxidative mechanisms, and the expression of MT [38]. The same up-regulation of Nrf2 by Zinc has been demonstrated in human renal tubule cells in-vitro and mouse kidney in-vivo under the diabetic conditions [39]. In cultured renal tubular epithelial cells (NRK-52E), Zinc supplementation inhibited high glucose induced cell apoptosis by attenuating reactive oxygen species production, facilitated via Nrf2 up-regulation [40]. In postmenopausal women with type-2 diabetes Zinc supplementation increases TNF-α gene expression, suggesting a close interaction between Zinc homeostasis, oxidative stress and inflammation [41].
Effects on carbohydrate and lipid metabolism
Zinc is known to stimulate glycolysis and inhibit gluconeogenesis, an effect that is not overcome by the presence of glucagon [42–44]. In-vitro studies have demonstrated that Zinc increases the activity of glycolytic enzymes, phosphofructokinase (PFK) and pyruvate kinase (PK) in a concentration- and time-dependent manner [45, 46]. Lactate production which reflects the PFK activity, was increased by Zinc [46]. However, the effects of Zinc and insulin were not additive and Zinc pretreatment prevented the stimulation of glycolytic enzymes by insulin. Furthermore, in Zinc-treated cells a progressive activation of ERK2/MAPK1 (Mitogen-activated protein kinase-1) was observed [45]. However, a recent in-vitro study using hepatocytes has shown that at sub-lethal concentrations, ZnO nanoparticles increase both gluconeogenesis and glycogenolysis, contradicting the finding from the earlier studies given above [47].
Zinc is also known to be a concentration dependent reversible inhibitor of α-glucosidase activity in the intestines [48, 49]. Zinc binding to α-glucosidase directly induced tertiary structural changes, however the concentration required to induce structural changes was greater than that required to achieve inhibition [49]. In skeletal muscles Zinc-α2-glycoprotein stimulates the phosphorylation of AMP-activated protein kinase (AMPKα) and increases cellular GLUT4 protein [50]. This increase in the expression of the GLUT4 has also been observed in adipose tissue with resultant increase in glucose uptake [44, 51, 52]. Studies have also shown that whole body as well as adipose tissue specific insulin sensitivity was positively associated with Zinc-α2-glycoprotein expression in subcutaneous adipose tissue and that obesity related decrease in Zinc-α2-glycoprotein is selectively present in subcutaneous but not in visceral adipose tissue [53]. However, two recent studies have indicated that Zinc-α2-glycoprotein functions to inhibit insulin signaling and, in consequence, insulin-induced glucose uptake in adipocytes, whereas, Zinc Finger Protein 407 (ZFP407) is known to stimulate GLUT4 mRNA transcription in adipocytes, facilitating insulin-stimulated glucose uptake via GLUT4 [54, 55].
Zinc also enhanced glucose transport in adipocytes in a dose dependant manner, independent of insulin [43, 56–58]. Zinc was observed to stimulate the phosphorylation of the IR-ß (Insulin Receptor) subunit [52, 56]. Zinc also inhibits Glycogen synthase kinase-3 (GSK-3β), which is a phosphorylating and an inactivating agent of glycogen synthase, with resultant increase in glycogen synthesis [57]. Furthermore, Zinc-α2-glycoproteins increase the expression of the lipolytic enzymes, adipose triglyceride lipase and hormone-sensitive lipase in the white adipose tissue [51]. Incubation of adipocytes with Zinc, augmented lipogenesis and this lipogenic effect was 80 % of maximum insulin stimulation [43, 59]. In isolated rat liver membrane cells, Zinc alone stimulated lipogenesis in a dose-related manner [60].
Treatment of 3 T3-L1 adipocytes with Zinc-chelated vitamin C (ZnC) has been shown to promote adipogenesis, characterized by increased glycerol-3-phosphate dehydrogenase activity and intracellular lipid accumulation in 3 T3-L1 cells, associated with a pronounced up-regulation of the expression of glucose transporter type 4 (GLUT4) [61]. ZnC further increased the expression of peroxisome proliferator-activated receptor gamma (PPARγ) and CCAAT/enhancer-binding protein alpha, the key transcription factors of adipogenesis [61]. A decrease in insulin receptor binding has been observed in Zinc deficient rat adipocytes and addition of Zinc stimulates insulin binding in a dose-dependent manner [60, 62].
Islet cell function
In-vitro environments increasing the extracellular Zinc concentration is known to increase the free insulin concentration in the immediate vicinity of β-cells, mediated by enhanced Zinc-insulin dissociation [63]. However, glucose stimulated insulin secretion in β-cells of the pancreas is inhibited by Zinc, suggesting that co-secreted Zinc acts in an autocrine inhibitory modulator [64, 65]. In pancreatic β-cells of mice and clonal HIT-T15 β-cells, KCl and glucose induced an increase in free cytosolic Zinc levels, which facilitated the processing and/or storage of insulin [66, 67]. This is facilitated by an increase in the expression of cellular Zinc importers (Slc39a6, Slc39a7, and Slc39a8). However, chronic increase in cytosolic Zinc levels following sustained hyperglycemia, as in diabetes may contribute towards β-cell dysfunction and death [66].
Human Islet Amyloid Polypeptide (hIAPP) (a polypeptide hormone secreted from pancreatic β-cells in response to glucose) and is cleared by the peptidases in the kidney. hIAPP is known to aggregate in the pancreas to form dense, insoluble extracellular fibrillar deposit, causing β-cell destruction in type-2 diabetes [68]. Zinc, significantly inhibits hIAPP amyloid fibrillogenesis at concentrations similar to those found in-vivo extracellular environments [68]. This probably explains the linkage between the mutations of SLC30A8 zinc transporter (Zinc Transporter 8 [ZnT8]), which transports Zinc into the secretory granules, and type-2 diabetes. In human pancreatic islet cells, ZnT8 is the key protein responsible for both intracellular Zinc accumulation in insulin-containing vesicles and regulation of insulin secretion [69–73]. ZnT8 down regulated cells show reduced insulin content and decreased insulin secretion in response to hyperglycemic stimuli [74]. However, absence of ZnT8 expression did not alter rates of insulin biosynthesis, insulin content and glucose metabolism, but contributed to the packaging efficiency of stored insulin [75–77]. When ZnT8 absent mice were fed a control diet, glucose tolerance and insulin sensitivity were normal. However, after high-fat diet feeding, these mice became glucose intolerant or diabetic, and islets became less responsive to glucose [75]. ZnT8 is down regulated on exposure to metabolic stress associated with diabetic and pre-diabetic states, suggesting that it might further contribute to progression of type-2 diabetes [78, 79]. In β-cell specific SLC30A8 deficiency (ZnT8 knockout mice) a low peripheral blood insulin levels was observed, due to a substantial amount of the insulin being degraded during its first passage through the liver [80]. This is possibly due to the low level of Zinc in the portal circulation co-secreted by β-cells, due to the absence of ZnT8 (reducing uptake of Zinc by β-cells), which leads to augmented hepatic insulin clearance. The ZnT8 is also downregulated in response to exposure of pancreatic β-cells to hypoxia, resulting in lowered cytosolic Zn2+ concentrations [81].
Pancreatic islet cells harvested from rats conditioned under intermittent hypoxia showed a significant reduction in Zinc Influx Transporter 8 (ZIP8) expression in the β-cell membrane, with resultant reduction in cellular Zinc concentration and insulin production [82]. ZIP6 and ZIP7 function as two important zinc influx transporters to regulate cytosolic Zinc concentrations and insulin secretion in β-cells and ZIP-6 is also capable of directly interacting with GLP-1R to facilitate the protective effect of GLP-1 on β-cell survival [83]. Zip4 protein is located in human pancreatic β-cells, is important for the accumulation of Zinc in the cytosol and granules of β-cells [84]. Other Zinc transporters like ZnT3 and ZIP7 might also play a role in insulin secretion and glucose metabolism [85, 86].
L-type voltage-gated Ca2+ channels and TRMP3 (transient receptor potential cation channel subfamily M member 3) are also in part responsible for Zinc transport into β-cells, which is also dependent upon the metabolic status of the cell [87, 88]. Culture of rat pancreatic islets in either low or high vs. intermediate glucose concentrations triggers early mitochondrial oxidative stress and late β-cell apoptosis with loss of glucose stimulated insulin secretion [89]. ZnCl2 reduces mitochondrial oxidative stress and rat β-cell apoptosis under these culture conditions [89]. ZnO nanoparticles at dose of 70 ng/mL improved viability and function of pancreatic islets, by reducing oxidative stress and preventing cells from entering the apoptotic phase [90]. It is well known that Reactive Oxygen Species (ROS) can cause pancreatic β-cell death. This occurs due to the activation of Transient Receptor Potential Melastatin2 (TRPM2) channels by ROS. TRPM2 causes Ca2+ influx into the β-cells causing release of lysosomal Zinc, which results in β-cell death [91]. The tumor suppressor gene ST18 is a neural Zinc finger transcription factor, expressed in pancreatic β-cells and is known to impair insulin secretion, induce β-cell apoptosis and curtail β-cell replication [92].
In glucagon producing α-cells of the pancreas Zinc accumulates under low and high glucose conditions through both Ca2+channels and other Zinc transporting mechanisms, and the intracellular Zinc inhibits glucagon secretion [93]. Furthermore during hypoglycemia the principal signal that initiates glucagon secretion could be the detection by α-cells of a sudden decrease in Zinc paralleling the fall in insulin in the islet peri-portal circulation and this drop in concentration of Zinc, closes α-cell ion channels, promoting entry of calcium which stimulates glucagon secretion [94, 95].
Insulin-mimetic compounds
Zinc ions and its’ complexes, have shown insulin-like action both in-vitro and in-vivo experiments (Table 1) [96–117]. In-vitro, Zinc complexes inhibit the release of FFA from cultured rat adipocytes [96–104, 106, 108, 110–112, 114, 116]. In cultured 3 T3-L1 adipocytes these Zinc complexes activates the insulin signaling cascade through Akt/PKB (protein-kinase B) phosphorylation resulting in GLUT4 translocation to the plasma membrane and enhanced cellular glucose uptake [105, 107, 113]. In-vivo type 2 diabetic KK-Ay mice, these complexes have demonstrated an ability to reduce blood glucose, HbA1c, triglycerides and total cholesterol [98, 100, 102, 104, 110–112, 116, 117]. They are also known to improve the glucose tolerance as demonstrated by Oral Glucose Tolerance Testing (OGTT) [96, 99, 103, 110, 114, 117].
In addition to Zinc containing complexes, Zinc Oxide nanoparticles (ZnO) are known to posses anti-diabetic activity. ZnO nanoparticles induce a significant reduction in blood glucose, elevates serum insulin levels and glucokinase activity, whilst stimulating a higher expression of insulin, insulin receptor, GLUT-2 and glucokinase genes in STZ induced (Type-1) diabetic rats [118]. Blood glucose level is also reduced in Type-2 diabetic rats administered ZnO nanoparticles, with improved glucose tolerance and a 70 % increase in serum insulin levels [119]. In addition a significant lowering of circulating triglycerides and free fatty acids was also observed suggesting a beneficial effect of ZnO on lipid metabolism [119].
Other effects
In STZ-diabetes induced mice, Zinc supplementation has shown to increase the serum leptin concentration [120]. However, this finding has been contradicted by several other studies [100, 106, 121]. Hence, although it is evident that Zinc has an effect on leptin levels, the exact relationship and mechanisms are yet to be determined. In-vitro, ZnCl2 is known to stimulate the trans-differentiation of human heptoma (HpG2) cells into pancreatic-like cells, with increased expression of amylase and insulin mRNAs over 1000 and 10 000 fold respectively [122]. Zinc is also known to activate C-peptide, with resultant increased energy utilization in red cells leading to release of ATP, which in turn stimulates NO production in platelets and endothelium causing reduction in platelet activity [123].
Zinc supplementation has been shown to prevent bone loss in chronic T1DM rats by stimulating expression of the mineralizing phenotype in osteoblasts and reducing expression of the resorptive phenotype in osteoclasts, achieved by osteocalcin up-regulation and RANKL, OPG, COL1A, and MMP-9 protein down-regulation [124]. Furthermore, in-vitro Zinc inhibited advanced glycation end product (AGE)-induced MC3T3-E1 cell (mouse osteoblasts) apoptosis by attenuating the production of reactive oxygen species, inhibiting caspase-3 and caspase-9 activation, and inhibiting the release of cytochrome c from between the mitochondria and the cytosol [125]. Zinc significantly inhibited AGE formation of albumin in-vitro and reduced secondary and tertiary structural modifications of albumin and may have in controlling/preventing AGEs-mediated diabetic pathological conditions in vivo [126, 127].
Discussion
Numerous in-vitro and in-vivo studies have shown that Zinc has beneficial effects in both type-1 and type-2 diabetes. A finding which has been confirmed by a recent meta-analysis, where Zinc supplementation resulted in improved glycaemic control [15]. It is evident from the findings of the present systematic review, that Zinc plays an important role in β-cell function, insulin action, glucose homeostasis and the pathogenesis of diabetes and its complications.
Our results clearly show that Zinc has anti-oxidant properties and that Zinc supplementation reduces oxidative stress. Zinc supplementation enhances the activity and levels of key anti-oxidant enzymes and proteins, whilst significantly reducing lipid peroxidation. Some of these effects are brought on by interactions at nuclear level, by stimulation of nuclear factors like Nrf2 [38]. It is well known that oxidative stress is high in both type-1 and type-2 diabetic patients, as evident by elevated TBARS levels in the plasma [11, 19]. This contributes towards further β-cell dysfunction, with resultant deterioration of glycaemic control. Oxidative stress also plays a pivotal role in the pathogenesis of both micro- and macro-vascular complications of diabetes [128]. The resultant increase in the production of ROSs is responsible for the activation of five major pathways involved in the pathogenesis of diabetes complications [128]. Increased formation of AGEs, and the increased expression of the receptor for AGEs and its activating ligands is one such primary pathway responsible for the pathogenesis of diabetes related complications [128]. Both in-vitro and in-vivo Zinc significantly inhibited the formation of AGEs. Hence, by alleviating the oxidative stress associated with diabetes and by reducing the formation of AGEs, Zinc supplementation could delay the progression of diabetes and also delay/prevent the numerous micro- and macro-vascular complications associated with diabetes.
Furthermore, our results show that Zinc plays an important role in glucose and lipid metabolism. Zinc reduces glucose absorption and synthesis, whilst promoting glucose metabolism and storage. This is primarily via the enhanced activity of key enzymes involved in these metabolic processes, such as α-glucosidase, PFK, PK and glycogen synthase. Its insulinomimetic action possibly mediated via Zinc-α2-glycoporteins increases cellular GLUT4 levels in skeletal muscles and adipose tissue facilitating glucose absorption. Zinc-α2-glycoprotein is gaining increasing recognition as a marker of insulin resistance in type-2 diabetes. Zinc-α2-glycoproteins are also involved in lipid metabolism, affecting the expression of several lipolytic enzymes at hepatic and adipose tissue level. Two recent meta-analyses have shown that Zinc supplementation reduces Fasting Blood Glucose, 2 h Post Prandial Blood Glucose and HbA1c in patients with diabetes, as well as reducing total cholesterol, LDL cholesterol and triglycerides in both patients with and without diabetes [15, 129]. The above molecular/enzymatic level mechanisms probably explain the beneficial effects of Zinc supplementation on glycaemic control and lipids observed in humans.
Zinc also plays an important role in the normal functioning of the islet cells of the pancreas. β-cells and their granules are extremely rich in Zinc. Zinc Transporters (ZnTs) transport zinc from the cytoplasm to extracellular spaces or to intra-cytoplasmic vacuoles, such as secretory granules, while the ZIPs are thought to increase cytoplasmic zinc [130]. Insulin production and efficient packaging into vesicles is closely linked with the transport of Zinc in to the β-cells and subsequent concentration inside vesicles mediated by the Zinc transporter ZnT8, a product of the SLC30A8 gene, specifically expressed in the β-cells of the pancreas [131]. Alterations and/or variations in the activity of ZnT8 is associated with impaired glucose induced insulin response, which promotes progression from glucose intolerance to type-2 diabetes in susceptible individuals [131]. ZnT8 has also been identified as a novel target auto-antigen in patients with type-1 diabetes and hence has diagnostic implications [132]. Auto-antibodies to ZnT8 (ZnT8A) are detected in 50–60 % of Japanese patients with acute-onset and 20 % with slow-onset type-1 diabetes [132]. Hence, it is evident that ZnT8 is a key mediator in the pathogenesis of both type-1 and type-2 diabetes. In addition to ZnT8, altered activity of many other Zinc transporters and Zinc influx proteins (ZIP 6, 7, 8) have been implicated in the pathogenesis of diabetes. Zinc is also an important mediator of α-cell function, as it inhibits glucagon secretion [93]. Identification and characterization of these ZnTs and ZIPs will help in the development of novel therapies for diabetes targeting these molecules and to develop new methods to protect β-cell mass and function, in both type-1 and type-2 diabetes.
Patients’ adherence to present therapeutic regimes for diabetes treatment are poor, resulting in unsatisfactory diabetes control [133]. Regime complexity, hypoglycaemia and other side-effects, lack of confidence in immediate or future benefits and patients’ education/beliefs are among the common reasons identified that limits adherence [134–137]. Inadequacies in current treatments has resulted in 2 to 3.6 million people in USA relying on alternative therapies for management diabetes [138]. In addition current treatment modalities are not very efficacious in preventing and/or delaying the progression of β-cell dysfunction and ultimate β-cell failure in patients with type-2 diabetes. The development of new compounds for treating diabetes is currently important to reduce the need for insulin injection in diabetic patients and to replace the clinically used synthetic therapeutics, which has several severe side effects [139].
Zinc ions and its’ numerous complexes, have shown insulin-like action both in-vitro and in-vivo [71–90]. Zinc complexes activate the insulin signaling cascade via Akt/PKB, which resultant increase in cellular GLUT4 and enhanced cellular glucose uptake. In animal models of type-2 diabetes these complexes have shown a significant ability to reduce blood glucose, HbA1c, serum insulin, triglycerides and total cholesterol, whilst improving glucose tolerance. On the basis of these findings and observations, it is evident that Zinc complexes have promise as a novel therapeutic modality that mimics the action of insulin [139]. However, presently the findings are only from in-vitro and in-vivo animal studies, there is a paucity of data from randomized controlled trials in humans. It is necessary to identify few efficacious compounds, with the least amounts of toxicity and for those complexes to be carefully evaluated in humans. The fruitful outcome of such trials may offer a novel and effective oral medication with better anti-diabetic in place of insulin [140].
Conclusion
Numerous in-vitro and in-vivo studies have shown that Zinc has beneficial effects in both type-1 and type-2 diabetes. It is evident from the findings of the present systematic review, that Zinc plays an important role in β-cell function, insulin action, glucose homeostasis and the pathogenesis of diabetes and its complications. However further randomized double-blinded placebo-controlled clinical trials conducted for an adequate duration, are required to establish therapeutic efficacy and safety in humans.
References
IDF Diabetes Atlas - The Economic Impacts of Diabetes. [http://www.diabetesatlas.com/content/economic-impacts-diabetes]
Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet. 2005;365(9467):1333–46.
Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27(5):1047–53.
WHO Diabetes Fact Sheet. [http://www.who.int/mediacentre/factsheets/fs312/en/index.html]
Diabetes: the cost of diabetes [http://www.who.int/mediacentre/factsheets/fs236/en/]
Stuckler D. Population Causes and Consequences of Leading Chronic Diseases: A Comparative Analysis of Prevailing Explanations. The Milbank Quarterly. 2008;86(2):273–326.
Ali MK, Bullard KM, Saaddine JB, Cowie CC, Imperatore G, Gregg EW. Achievement of goals in U.S. diabetes care 1999–2010. N Engl J Med. 2013;368(17):1613–24.
Khunti K, Davies M. Glycaemic goals in patients with type 2 diabetes: current status, challenges and recent advances. Diabetes Obes Metab. 2010;12(6):474–84.
Dodson G, Steiner D. The role of assembly in insulin's biosynthesis. Curr Opin Struct Biol. 1998;8(2):189–94.
Simon SF, Taylor CG. Dietary zinc supplementation attenuates hyperglycemia in db/db mice. Exp Biol Med (Maywood). 2001;226(1):43–51.
Faure P, Benhamou PY, Perard A, Halimi S, Roussel AM. Lipid peroxidation in insulin-dependent diabetic patients with early retina degenerative lesions: effects of an oral zinc supplementation. Eur J Clin Nutr. 1995;49(4):282–8.
Shidfar F, Aghasi M, Vafa M, Heydari I, Hosseini S, Shidfar S. Effects of combination of zinc and vitamin A supplementation on serum fasting blood sugar, insulin, apoprotein B and apoprotein A-I in patients with type i diabetes. Int J Food Sci Nutr. 2010;61(2):182–91.
Afkhami-Ardekani M, Karimi M, Mohammadi SM, Nourani F. Effect of zinc sulfate supplementation on lipid and glucose in type 2 diabetic patients. Pak J Nutr. 2008;7(4):550–3.
Al-Maroof RA, Al-Sharbatti SS. Serum zinc levels in diabetic patients and effect of zinc supplementation on glycemic control of type 2 diabetics. Saudi Med J. 2006;27(3):344–50.
Jayawardena R, Ranasinghe P, Galappatthy P, Malkanthi R, Constantine G, Katulanda P. Effects of zinc supplementation on diabetes mellitus: a systematic review and meta-analysis. Diabetol Metab Syndr. 2012;4(1):13.
Garg VK, Gupta R, Goyal RK. Hypozincemia in diabetes mellitus. J Assoc Physicians India. 1994;42(9):720–1.
Pidduck HG, Wren PJ, Evans DA. Hyperzincuria of diabetes mellitus and possible genetical implications of this observation. Diabetes. 1970;19(4):240–7.
Black RE. Zinc deficiency, infectious disease and mortality in the developing world. J Nutr. 2003;133(5 Suppl 1):1485S–9S.
Anderson RA, Roussel AM, Zouari N, Mahjoub S, Matheau JM, Kerkeni A. Potential antioxidant effects of zinc and chromium supplementation in people with type 2 diabetes mellitus. J Am Coll Nutr. 2001;20(3):212–8.
Blostein-Fujii A, DiSilvestro RA, Frid D, Katz C, Malarkey W. Short-term zinc supplementation in women with non-insulin-dependent diabetes mellitus: effects on plasma 5'-nucleotidase activities, insulin-like growth factor I concentrations, and lipoprotein oxidation rates in vitro. Am J Clin Nutr. 1997;66(3):639–42.
Pathak A, Sharma V, Kumar S, Dhawan DK. Supplementation of zinc mitigates the altered uptake and turnover of 65Zn in liver and whole body of diabetic rats. Biometals. 2011;24(6):1027–34.
Konukoglu D, Turhan MS, Ercan M, Serin O. Relationship between plasma leptin and zinc levels and the effect of insulin and oxidative stress on leptin levels in obese diabetic patients. J Nutr Biochem. 2004;15(12):757–60.
Faure P, Barclay D, Joyeux-Faure M, Halimi S. Comparison of the effects of zinc alone and zinc associated with selenium and vitamin E on insulin sensitivity and oxidative stress in high-fructose-fed rats. J Trace Elem Med Biol. 2007;21(2):113–9.
Vijayaraghavan K, Iyyampillai S, Subramanian SP. Antioxidant potential of zinc-flavonol complex studied in streptozotocin-diabetic rats. J Diabetes. 2013;5(2):149–56.
Bădescu M, Păduraru I, Colev V, Saramet A, Bohotin C, Bădescu L. The relation zinc-lipidic peroxidation in experimental diabetes mellitus. Rom J Physiol. 1993;30(3–4):167–71.
Duzguner V, Kaya S. Effect of zinc on the lipid peroxidation and the antioxidant defense systems of the alloxan-induced diabetic rabbits. Free Radic Biol Med. 2007;42(10):1481–6.
Tang Y, Yang Q, Lu J, Zhang X, Suen D, Tan Y, et al. Zinc supplementation partially prevents renal pathological changes in diabetic rats. J Nutr Biochem. 2010;21(3):237–46.
Wang X, Li H, Fan Z, Liu Y. Effect of zinc supplementation on type 2 diabetes parameters and liver metallothionein expressions in Wistar rats. J Physiol Biochem. 2012;68(4):563–72.
Moustafa SA. Zinc might protect oxidative changes in the retina and pancreas at the early stage of diabetic rats. Toxicol Appl Pharmacol. 2004;201(2):149–55.
Ohly P, Dohle C, Abel J, Seissler J, Gleichmann H. Zinc sulphate induces metallothionein in pancreatic islets of mice and protects against diabetes induced by multiple low doses of streptozotocin. Diabetologia. 2000;43(8):1020–30.
Özcelik D, Nazıroglu M, Tunçdemir M, Çelik Ö, Öztürk M, Flores-Arce MF. Zinc supplementation attenuates metallothionein and oxidative stress changes in kidney of streptozotocin-induced diabetic rats. Biol Trace Elem Res. 2012;150(1–3):342–9.
Zimny S, Gogolin F, Abel J, Gleichmann H. Metallothionein in isolated pancreatic islets of mice: induction by zinc and streptozotocin, a naturally occurring diabetogen. Arch Toxicol. 1993;67(1):61–5.
Wang J, Song Y, Elsherif L, Song Z, Zhou G, Prabhu SD, et al. Cardiac metallothionein induction plays the major role in the prevention of diabetic cardiomyopathy by zinc supplementation. Circulation. 2006;113(4):544–54.
Liang T, Zhang Q, Sun W, Xin Y, Zhang Z, Tan Y, et al. Zinc treatment prevents type 1 diabetes-induced hepatic oxidative damage, endoplasmic reticulum stress, and cell death, and even prevents possible steatohepatitis in the OVE26 mouse model: Important role of metallothionein. Toxicol Lett. 2015;233(2):114–24.
Lu Y, Liu Y, Li H, Wang X, Wu W, Gao L. Effect and mechanisms of zinc supplementation in protecting against diabetic cardiomyopathy in a rat model of type 2 diabetes. Bosn J Basic Med Sci. 2015;15(1):14–20.
Zhang X, Liang D, Chi ZH, Chu Q, Zhao C, Ma RZ, et al. Effect of zinc on high glucose-induced epithelial-to-mesenchymal transition in renal tubular epithelial cells. Int J Mol Med. 2015;35(6):1747–54.
Liu F, Ma F, Kong G, Wu K, Deng Z, Wang H. Zinc supplementation alleviates diabetic peripheral neuropathy by inhibiting oxidative stress and upregulating metallothionein in peripheral nerves of diabetic rats. Biol Trace Elem Res. 2014;158(2):211–8.
Miao X, Wang Y, Sun J, Sun W, Tan Y, Cai L, et al. Zinc protects against diabetes-induced pathogenic changes in the aorta: Roles of metallothionein and nuclear factor (erythroid-derived 2)-like 2. Cardiovasc Diabetol. 2013;12:54.
Li B, Cui W, Tan Y, Luo P, Chen Q, Zhang C, et al. Zinc is essential for the transcription function of Nrf2 in human renal tubule cells in vitro and mouse kidney in vivo under the diabetic condition. J Cell Mol Med. 2014;18(5):895–906.
Zhang X, Zhao Y, Chu Q, Wang ZY, Li H, Chi ZH. Zinc modulates high glucose-induced apoptosis by suppressing oxidative stress in renal tubular epithelial cells. Biol Trace Elem Res. 2014;158(2):259–67.
Chu A, Foster M, Hancock D, Bell-Anderson K, Petocz P, Samman S. TNF-alpha gene expression is increased following zinc supplementation in type 2 diabetes mellitus. Genes & nutrition. 2015;10(1):440.
Brand IA, Kleineke J. Intracellular zinc movement and its effect on the carbohydrate metabolism of isolated rat hepatocytes. J Biol Chem. 1996;271(4):1941–9.
Shisheva A, Gefel D, Shechter Y. Insulinlike effects of zinc ion in vitro and in vivo. Preferential effects on desensitized adipocytes and induction of normoglycemia in streptozocin-induced rats. Diabetes. 1992;41(8):982–8.
May JM, Contoreggi CS. The mechanism of the insulin-like effects of ionic zinc. J Biol Chem. 1982;257(8):4362–8.
Canesi L, Betti M, Ciacci C, Gallo G. Insulin-like effect of zinc in mytilus digestive gland cells: modulation of tyrosine kinase-mediated cell signaling. Gen Comp Endocrinol. 2001;122(1):60–6.
Tamaki N, Ikeda T, Funatsuka A. Zinc as activating cation for muscle glycolysis. J Nutr Sci Vitaminol. 1983;29(6):655–62.
Filippi C, Pryde A, Cowan P, Lee T, Hayes P, Donaldson K, et al. Toxicology of ZnO and TiO2 nanoparticles on hepatocytes: impact on metabolism and bioenergetics. Nanotoxicology. 2015;9(1):126–34.
Yoshikawa Y, Hirata R, Yasui H, Sakurai H. Alpha-glucosidase inhibitory effect of anti-diabetic metal ions and their complexes. Biochimie. 2009;91(10):1339–41.
Zeng YF, Lee J, Si YX, Yan L, Kim TR, Qian GY, et al. Inhibitory effect of Zn2+ on α-glucosidase: Inhibition kinetics and molecular dynamics simulation. Process Biochem. 2012;47(12):2510–7.
Eckardt K, Schober A, Platzbecker B, Mracek T, Bing C, Trayhurn P, et al. The adipokine zinc-α2-glycoprotein activates AMP kinase in human primary skeletal muscle cells. Arch Physiol Biochem. 2011;117(2):88–93.
Russell ST, Tisdale MJ. Studies on the anti-obesity activity of zinc-α 2-glycoprotein in the rat. Int J Obes (Lond). 2011;35(5):658–65.
Miranda ER, Dey CS. Effect of chromium and zinc on insulin signaling in skeletal muscle cells. Biol Trace Elem Res. 2004;101(1):19–36.
Balaz M, Vician M, Janakova Z, Kurdiova T, Surova M, Imrich R, et al. Subcutaneous adipose tissue zinc-alpha2-glycoprotein is associated with adipose tissue and whole-body insulin sensitivity. Obesity (Silver Spring, Md). 2014;22(8):1821–9.
Ceperuelo-Mallafre V, Ejarque M, Duran X, Pachon G, Vazquez-Carballo A, Roche K, et al. Zinc-alpha2-glycoprotein modulates AKT-dependent insulin signaling in human adipocytes by activation of the PP2A phosphatase. PLoS One. 2015;10(6):e0129644.
Buchner DA, Charrier A, Srinivasan E, Wang L, Paulsen MT, Ljungman M, et al. Zinc finger protein 407 (ZFP407) regulates insulin-stimulated glucose uptake and glucose transporter 4 (Glut4) mRNA. J Biol Chem. 2015;290(10):6376–86.
Tang X, Shay NF. Zinc has an insulin-like effect on glucose transport mediated by phosphoinositol-3-kinase and Akt in 3 T3-L1 fibroblasts and adipocytes. J Nutr. 2001;131(5):1414–20.
Ilouz R, Kaidanovich O, Gurwitz D, Eldar-Finkelman H. Inhibition of glycogen synthase kinase-3beta by bivalent zinc ions: insight into the insulin-mimetic action of zinc. Biochem Biophys Res Commun. 2002;295(1):102–6.
Ezaki O. IIb group metal ions (Zn2+, Cd2+, Hg2+) stimulate glucose transport activity by post-insulin receptor kinase mechanism in rat adipocytes. J Biol Chem. 1989;264(27):16118–22.
Coulston L, Dandona P. Insulin-like effect of zinc on adipocytes. Diabetes. 1980;29(8):665–7.
Herington AC. Effect of zinc on insulin binding to rat adipocytes and hepatic membranes and to human placental membranes and IM-9 lymphocytes. Horm Metab Res. 1985;17(7):328–32.
Ghosh C, Yang SH, Kim JG, Jeon TI, Yoon BH, Lee JY, et al. Zinc-chelated vitamin C stimulates adipogenesis of 3 T3-L1 cells. Asian-Australas J Anim Sci. 2013;26(8):1189–96.
Gomot MJ, Faure P, Roussel AM, Coudray C, Osman M, Favier A. Effect of acute zinc deficiency on insulin receptor binding in rat adipocytes. Biol Trace Elem Res. 1992;32:331–5.
Aspinwall CA, Brooks SA, Kennedy RT, Lakey JR. Effects of intravesicular H+ and extracellular H+ and Zn2+ on insulin secretion in pancreatic beta cells. J Biol Chem. 1997;272(50):31308–14.
Slepchenko KG, James CB, Li YV. Inhibitory effect of zinc on glucose-stimulated zinc/insulin secretion in an insulin-secreting beta-cell line. Exp Physiol. 2013;98(8):1301–11.
Slepchenko KG, Daniels NA, Guo A, Li YV. Autocrine effect of Zn on the glucose-stimulated insulin secretion. Endocrine. 2015;50:110–22.
Bellomo EA, Meur G, Rutter GA. Glucose regulates free cytosolic Zn2+ concentration, Slc39 (ZiP), and metallothionein gene expression in primary pancreatic islet β-cells. J Biol Chem. 2011;286(29):25778–89.
Slepchenko KG, Li YV. Rising intracellular zinc by membrane depolarization and glucose in insulin-secreting clonal HIT-T15 beta cells. Exp Diabetes Res. 2012;2012:190309–9.
Brender JR, Hartman K, Nanga RPR, Popovych N, De La Salud BR, Vivekanandan S, et al. Role of zinc in human islet amyloid polypeptide aggregation. J Am Chem Soc. 2010;132(26):8973–83.
Chimienti F, Devergnas S, Pattou F, Schult F, Garcia-Cuenca R, Vandewalle B, et al. In vivo expression and functional characterization of the zinc transporter ZnT8 in glucose-induced insulin secretion. J Cell Sci. 2006;119(20):4199–206.
Chimienti F, Devergnas S, Favier A, Seve M. Identification and cloning of a beta-cell-specific zinc transporter, ZnT-8, localized into insulin secretory granules. Diabetes. 2004;53(9):2330–7.
Chimienti F, Favier A, Seve M. ZnT-8, a pancreatic beta-cell-specific zinc transporter. Biometals. 2005;18(4):313–7.
Wijesekara N, Dai FF, Hardy AB, Giglou PR, Bhattacharjee A, Koshkin V, et al. Beta cell-specific Znt8 deletion in mice causes marked defects in insulin processing, crystallisation and secretion. Diabetologia. 2010;53(8):1656–68.
Nicolson TJ, Bellomo EA, Wijesekara N, Loder MK, Baldwin JM, Gyulkhandanyan AV, et al. Insulin storage and glucose homeostasis in mice null for the granule zinc transporter ZnT8 and studies of the type 2 diabetes-associated variants. Diabetes. 2009;58(9):2070–83.
Fu Y, Tian W, Pratt EB, Dirling LB, Shyng S-L, Meshul CK, et al. Down-regulation of ZnT8 expression in INS-1 rat pancreatic beta cells reduces insulin content and glucose-inducible insulin secretion. PLoS One. 2009;4(5):e5679–9.
Lemaire K, Ravier MA, Schraenen A, Creemers JWM, Van de Plas R, Granvik M, et al. Insulin crystallization depends on zinc transporter ZnT8 expression, but is not required for normal glucose homeostasis in mice. Proc Natl Acad Sci U S A. 2009;106(35):14872–7.
Pound LD, Sarkar SA, Benninger RKP, Wang Y, Suwanichkul A, Shadoan MK, et al. Deletion of the mouse Slc30a8 gene encoding zinc transporter-8 results in impaired insulin secretion. Biochem J. 2009;421(3):371–6.
Pound LD, Sarkar SA, Ustione A, Dadi PK, Shadoan MK, Lee CE, et al. The physiological effects of deleting the mouse SLC30A8 gene encoding zinc transporter-8 are influenced by gender and genetic background. PLoS One. 2012;7(7):e40972–2.
Lefebvre B, Vandewalle B, Balavoine AS, Queniat G, Moerman E, Vantyghem MC, et al. Regulation and functional effects of ZNT8 in human pancreatic islets. J Endocrinol. 2012;214(2):225–32.
Liu B-Y, Jiang Y, Lu Z, Li S, Lu D, Chen B. Down-regulation of zinc transporter 8 in the pancreas of db/db mice is rescued by Exendin-4 administration. Mol Med Rep. 2011;4(1):47–52.
Tamaki M, Fujitani Y, Hara A, Uchida T, Tamura Y, Takeno K, et al. The diabetes-susceptible gene SLC30A8/ZnT8 regulates hepatic insulin clearance. J Clin Invest. 2013;123(10):4513–24.
Gerber PA, Bellomo EA, Hodson DJ, Meur G, Solomou A, Mitchell RK, et al. Hypoxia lowers SLC30A8/ZnT8 expression and free cytosolic Zn2+ in pancreatic beta cells. Diabetologia. 2014;57(8):1635–44.
Pae EK, Kim G. Insulin production hampered by intermittent hypoxia via impaired zinc homeostasis. PLoS One. 2014;9(2):e90192.
Liu Y, Batchuluun B, Ho L, Zhu D, Prentice KJ, Bhattacharjee A, et al. Characterization of zinc influx transporters (ZIPS) in pancreatic beta cells: roles in regulating cytosolic zinc homeostasis and insulin secretion. J Biol Chem. 2015;290(30):18757–69.
Hardy AB, Prentice KJ, Froese S, Liu Y, Andrews GK, Wheeler MB. Zip4 mediated zinc influx stimulates insulin secretion in pancreatic beta cells. PLoS One. 2015;10(3):e0119136.
Smidt K, Jessen N, Petersen AB, Larsen A, Magnusson N, Jeppesen JB, et al. SLC30A3 responds to glucose- and zinc variations in beta-cells and is critical for insulin production and in vivo glucose-metabolism during beta-cell stress. PLoS One. 2009;4(5):e5684–4.
Myers SA, Nield A, Chew GS, Myers MA. The zinc transporter, Slc39a7 (Zip7) is implicated in glycaemic control in skeletal muscle cells. PLoS One. 2013;8(11):e79316.
Gyulkhandanyan AV, Lee SC, Bikopoulos G, Dai F, Wheeler MB. The Zn2 + −transporting pathways in pancreatic β-cells: A role for the L-type voltage-gated Ca2+ channel. J Biol Chem. 2006;281(14):9361–72.
Wagner TFJ, Drews A, Loch S, Mohr F, Philipp SE, Lambert S, et al. TRPM3 channels provide a regulated influx pathway for zinc in pancreatic beta cells. Pflugers Arch. 2010;460(4):755–65.
Duprez J, Roma LP, Close A-F, Jonas J-C. Protective antioxidant and antiapoptotic effects of ZnCl2 in rat pancreatic islets cultured in low and high glucose concentrations. PLoS One. 2012;7(10):e46831–1.
Shoae-Hagh P, Rahimifard M, Navaei-Nigjeh M, Baeeri M, Gholami M, Mohammadirad A, et al. Zinc oxide nanoparticles reduce apoptosis and oxidative stress values in isolated rat pancreatic islets. Biol Trace Elem Res. 2014;162(1–3):262–9.
Manna PT, Munsey TS, Abuarab N, Li F, Asipu A, Howell G, et al. TRPM2-mediated intracellular Zn2+ release triggers pancreatic beta-cell death. Biochem J. 2015;466(3):537–46.
Henry C, Close AF, Buteau J. A critical role for the neural zinc factor ST18 in pancreatic beta-cell apoptosis. J Biol Chem. 2014;289(12):8413–9.
Gyulkhandanyan AV, Lu H, Lee SC, Bhattacharjee A, Wijesekara N, Fox JEM, et al. Investigation of transport mechanisms and regulation of intracellular Zn2+ in pancreatic alpha-cells. J Biol Chem. 2008;283(15):10184–97.
Zhou H, Zhang T, Harmon JS, Bryan J, Robertson RP. Zinc, not insulin, regulates the rat alpha-cell response to hypoglycemia in vivo. Diabetes. 2007;56(4):1107–12.
Slucca M, Harmon JS, Oseid EA, Bryan J, Robertson RP. ATP-sensitive K+ channel mediates the zinc switch-off signal for glucagon response during glucose deprivation. Diabetes. 2010;59(1):128–34.
Yoshikawa Y, Ueda E, Miyake H, Sakurai H, Kojima Y. Insulinomimetic bis(maltolato)zinc(II) complex: blood glucose normalizing effect in KK-A(y) mice with type 2 diabetes mellitus. Biochem Biophys Res Commun. 2001;281(5):1190–3.
Yoshikawa Y, Ueda E, Suzuki Y, Yanagihara N, Sakurai H, Kojima Y. New insulinomimetic zinc(II) complexes of alpha-amino acids and their derivatives with Zn(N2O2) coordination mode. Chem Pharm Bull. 2001;49(5):652–4.
Ueda E, Yoshikawa Y, Ishino Y, Sakurai H, Kojima Y. Potential insulinominetic agents of zinc(II) complexes with picolinamide derivatives: preparations of complexes, in vitro and in vivo studies. Chem Pharm Bull. 2002;50(3):337–40.
Yoshikawa Y, Ueda E, Sakurai H, Kojima Y. Anti-diabetes effect of Zn(II)/carnitine complex by oral administration. Chem Pharm Bull. 2003;51(2):230–1.
Adachi Y, Yoshida J, Kodera Y, Kato A, Yoshikawa Y, Kojima Y, et al. A new insulin-mimetic bis(allixinato)zinc(II) complex: structure-activity relationship of zinc(II) complexes. J Biol Inorg Chem. 2004;9(7):885–93.
Yoshikawa Y, Ueda E, Kojima Y, Sakurai H. The action mechanism of zinc(II) complexes with insulinomimetic activity in rat adipocytes. Life Sci. 2004;75(6):741–51.
Kojima Y, Yoshikawa Y, Ueda E, Kishimoto N, Tadokoro M, Sakurai H. Synthesis, structure, and in vitro and in vivo insulinomimetic activities of the zinc(II)-6-ethylpicolinate complex. Bull Chem Soc Jpn. 2005;78(3):451–5.
Yoshikawa Y, Kondo M, Sakurai H, Kojima Y. A family of insulinomimetic zinc(II) complexes of amino ligands with Zn(Nn) (n = 3 and 4) coordination modes. J Inorg Biochem. 2005;99(7):1497–503.
Adachi Y, Yoshikawa Y, Sakurai H. Antidiabetic zinc(II)-N-acetyl-L-cysteine complex: evaluations of in vitro insulinomimetic and in vivo blood glucose-lowering activities. Biofactors (Oxford, England). 2007;29(4):213–23.
Basuki W, Hiromura M, Sakurai H. Insulinomimetic Zn complex (Zn(opt)2) enhances insulin signaling pathway in 3 T3-L1 adipocytes. J Inorg Biochem. 2007;101(4):692–9.
Yoshikawa Y, Adachi Y, Sakurai H. A new type of orally active anti-diabetic Zn(II)-dithiocarbamate complex. Life Sci. 2007;80(8):759–66.
Nakayama A, Hiromura M, Adachi Y, Sakurai H. Molecular mechanism of Antidiabetic zinc-allixin complexes: regulations of glucose utilization and lipid metabolism. J Biol Inorg Chem. 2008;13(5):675–84.
Nishide M, Yoshikawa Y, Yoshikawa EU, Matsumoto K, Sakurai H, Kajiwara NM. Insulinomimetic Zn(II) complexes as evaluated by both glucose-uptake activity and inhibition of free fatty acids release in isolated rat adipocytes. Chem Pharm Bull. 2008;56(8):1181–3.
Rasheed K, Tariq MI, Munir C, Hussain I, Siddiqui HL. Synthesis, characterization and hypoglycemic activity of Zn(II), Cd(II) and Hg(II) complexes with glibenclamide. Chem Pharm Bull. 2008;56(2):168–72.
Karmaker S, Saha TK, Yoshikawa Y, Sakurai H. A zinc(II)/poly(γ-glutamic acid) complex as an oral therapeutic for the treatment of type-2 diabetic KKAy mice. Macromol Biosci. 2009;9(3):279–86.
Matsumoto K, Motoyasu N, Sera K, Fujii T, Yoshikawa Y, Yasui H, et al. Effects of Zn(II) complex with vitamins C and U, and carnitine on metabolic syndrome model rats. Metallomics. 2011;3(7):683–5.
Moniz T, Amorim MJ, Ferreira R, Nunes A, Silva A, Queirós C, et al. Investigation of the insulin-like properties of zinc(II) complexes of 3-hydroxy-4-pyridinones: identification of a compound with glucose lowering effect in STZ-induced type I diabetic animals. J Inorg Biochem. 2011;105(12):1675–82.
Naito Y, Yoshikawa Y, Yasui H. Cellular mechanism of zinchinokitiol complexes in diabetes mellitus. Bull Chem Soc Jpn. 2011;84(3):298–305.
Yoshikawa Y, Adachi Y, Yasui H, Hattori M, Sakurai H. Oral administration of Bis(aspirinato)zinc(II) complex ameliorates hyperglycemia and metabolic syndrome-like disorders in spontaneously diabetic KK-A(y) mice: structure-activity relationship on zinc-salicylate complexes. Chem Pharm Bull. 2011;59(8):972–7.
Vijayaraghavan K, Iyyam Pillai S, Subramanian SP. Design, synthesis and characterization of zinc-3 hydroxy flavone, a novel zinc metallo complex for the treatment of experimental diabetes in rats. Eur J Pharmacol. 2012;680(1–3):122–9.
Fujimoto S, Yasui H, Yoshikawa Y. Development of a novel antidiabetic zinc complex with an organoselenium ligand at the lowest dosage in KK-A(y) mice. J Inorg Biochem. 2013;121:10–5.
Kadowaki S, Munekane M, Kitamura Y, Hiromura M, Kamino S, Yoshikawa Y, et al. Development of new zinc dithiosemicarbazone complex for use as oral antidiabetic agent. Biol Trace Elem Res. 2013;154(1):111–9.
Alkaladi A, Abdelazim AM, Afifi M. Antidiabetic activity of zinc oxide and silver nanoparticles on streptozotocin-induced diabetic rats. Int J Mol Sci. 2014;15(2):2015–23.
Umrani RD, Paknikar KM. Zinc oxide nanoparticles show antidiabetic activity in streptozotocin-induced Type 1 and 2 diabetic rats. Nanomedicine (Lond). 2014;9(1):89–104.
Chen MD, Song YM, Lin PY. Zinc effects on hyperglycemia and hypoleptinemia in streptozotocin-induced diabetic mice. Horm Metab Res. 2000;32(3):107–9.
Liu MJ, Bao S, Bolin ER, Burris DL, Xu X, Sun Q, et al. Zinc deficiency augments leptin production and exacerbates macrophage infiltration into adipose tissue in mice fed a high-fat diet. J Nutr. 2013;143(7):1036–45.
Kanoh Y, Tomotsune D, Shirasawa S, Yoshie S, Ichikawa H, Yokoyama T, et al. In vitro transdifferentiation of HepG2 cells to pancreatic-like cells by CCl4, D-galactosamine, and ZnCl2. Pancreas. 2011;40(8):1245–52.
Meyer JA, Subasinghe W, Sima AAF, Keltner Z, Reid GE, Daleke D, et al. Zinc-activated C-peptide resistance to the type 2 diabetic erythrocyte is associated with hyperglycemia-induced phosphatidylserine externalization and reversed by metformin. Mol Biosyst. 2009;5(10):1157–62.
Bortolin RH, da Graca Azevedo Abreu BJ, Abbott Galvao Ururahy M, de Souza KS C, Bezerra JF, Loureiro MB, et al. Protection against T1DM-induced bone loss by zinc supplementation: biomechanical, histomorphometric, and molecular analyses in STZ-induced diabetic rats. PLoS One. 2015;10(5):e0125349.
Xiong M, Liu L, Liu Z, Gao H. Inhibitory effect of zinc on the advanced glycation end product-induced apoptosis of mouse osteoblastic cells. Molecular medicine reports 2015. 10.3892/mmr.2015.4088.
Tupe R, Kulkarni A, Adeshara K, Sankhe N, Shaikh S, Dalal S, et al. Zinc inhibits glycation induced structural, functional modifications in albumin and protects erythrocytes from glycated albumin toxicity. Int J Biol Macromol. 2015;79:601–10.
Baraka-Vidot J, Navarra G, Leone M, Bourdon E, Militello V, Rondeau P. Deciphering metal-induced oxidative damages on glycated albumin structure and function. Biochim Biophys Acta. 2014;1840(6):1712–24.
Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107(9):1058–70.
Ranasinghe P, Wathurapatha WS, Ishara MH, Jayawardana R, Galappatthy P, Katulanda P, et al. Effects of Zinc supplementation on serum lipids: a systematic review and meta-analysis. Nutr Metab. 2015;12:26.
Rungby J. Zinc, zinc transporters and diabetes. Diabetologia. 2010;53(8):1549–51.
Chistiakov DA, Voronova NV. Zn2 + −transporter-8: A dual role in diabetes. Biofactors. 2009;35(4):356–63.
Kawasaki E. ZnT8 and type 1 diabetes. Endocr J. 2012;59(7):531–7.
Cramer JA. A systematic review of adherence with medications for diabetes. Diabetes Care. 2004;27(5):1218–24.
Alvarez Guisasola F, Tofe Povedano S, Krishnarajah G, Lyu R, Mavros P, Yin D. Hypoglycaemic symptoms, treatment satisfaction, adherence and their associations with glycaemic goal in patients with type 2 diabetes mellitus: findings from the Real-Life Effectiveness and Care Patterns of Diabetes Management (RECAP-DM) Study. Diabetes Obes Metab. 2008;10 Suppl 1:25–32.
Odegard PS, Capoccia K. Medication taking and diabetes: a systematic review of the literature. Diabetes Educ. 2007;33(6):1014–29. discussion 1030–1011.
Grant RW, Devita NG, Singer DE, Meigs JB. Polypharmacy and medication adherence in patients with type 2 diabetes. Diabetes Care. 2003;26(5):1408–12.
Bailey CJ, Kodack M. Patient adherence to medication requirements for therapy of type 2 diabetes. Int J Clin Pract. 2011;65(3):314–22.
Dham S, Shah V, Hirsch S, Banerji MA. The role of complementary and alternative medicine in diabetes. Curr Diab Rep. 2006;6(3):251–8.
Sakurai H, Adachi Y. The pharmacology of the insulinomimetic effect of zinc complexes. Biometals. 2005;18(4):319–23.
Sakurai H, Katoh A, Kiss T, Jakusch T, Hattori M. Metallo-allixinate complexes with anti-diabetic and anti-metabolic syndrome activities. Metallomics. 2010;2(10):670–82.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
The authors declare that they have no competing interests.
Authors’ contributions
PR and SP substantially contributed to the general idea and design of the study. PR and SP were involved in data collection. PR, PK, PG and GRC planned data analysis. PR and SP drafted the manuscript. All authors have read and consented to the manuscript.
Additional file
Additional file 1:
PRISMA 2009 Checklist. (DOC 64 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ranasinghe, P., Pigera, S., Galappatthy, P. et al. Zinc and diabetes mellitus: understanding molecular mechanisms and clinical implications. DARU J Pharm Sci 23, 44 (2015). https://doi.org/10.1186/s40199-015-0127-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40199-015-0127-4