
Abstract
Considering the important role of microbiome, many of current investigations have focused on its beneficial aspects. Although, research explores new dimensions of the impact of microbiome and examines the differences in patients and healthy individuals for identifying biomarker patterns, but limited information is available, and investigation in this field seems to be of great value. On the other hand, new therapeutic approaches, called personalized medicine, have opened a new window in medical science, and the association between microbiome and personalized medicine seems to be one of the most interesting aspects of the subsequent research, and has a pivotal perspective on the treatment of diseases such as cancer. Accordingly, given the novelty of the relationship between these two axes, there are very few studies in this regard. The presence of specific strains may have the ability to modulate cancer progression and therapeutics; this increases the likelihood of precision medicine in relation to microbiota, in terms of treatment and prognosis, and therefore, microbiota is a next generation medicine and may develop a novel therapeutic action in this field.
Similar content being viewed by others


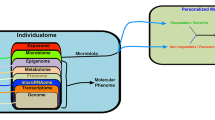
Background
Microbial communities, including bacteria, archaea, fungi, etc., are known as microbiota or microflora, and the genes encoded by them are called microbiome. A healthy microbiome has a series of joint characteristics that can be distinguished from non-healthy individuals, so understanding the microbiome differential properties may contribute in detection and identification of the disease-associated microbiome. The microbiome of healthy people is very diverse with a high number of beneficial microbes that can withstand the changes, occurring during each period of physiological stress; while the disease-associated microbiota is less diverse; the number of beneficial bacteria is lower and leads to the disease in the presence of inflammation [41, 65]. However, the main problem that the researchers are facing is to understand the potential characteristics of microbiome diversity, among individuals [34, 53]. Traditional methods such as cultivation have provided very little information in this regard [20], but today, the approaches such as NGS have been able to introduce an acceptable understanding of this population and their combinations, and identified the archaea, bacteria and viruses in the body [40, 60, 73]. Disturbance in microbial ecology may be associated with many diseases, such as diabetes, inflammatory bowel disease and so on; human microbiome can be used as a primary diagnostic biomarker, and researchers today focus on its therapeutic role [50]. As we know, the microbiota of each human body organ is unique, and its effects on inflammation and cancer are also distinct in each organ, as well as understanding the changes in interpersonal microbiome, and the frequency of microbial population in different positions in organs, leading to information potentially related to the development of diseases such as cancer [27]. These differences may be responsible for the occurrence of cancer in a particular organ, for example, the susceptibility to colorectal cancer is due to the presence of higher microbial density, compared to the small intestine [6, 51]. The microbiome is responsible for various clinical outcomes, and the drug response of individuals can be due to these differences; not all patients show the same response to anticancer therapies. Therefore, given the consideration of each person’s genetic information, and the improvement of drug responses [12], the personalized therapeutics can play a prominent role in the health care program, especially in relation to cancer. Studies have shown that the gut microbiome can also be effective in treating cancer through the regulation of host inflammatory responses [35]; microarray techniques can be helpful in this regard, since they can simultaneously evaluate more than hundreds of cancer-related genes. Modern personalized therapeutic is integrated with each individual genetic structure and disease history before the disease begins, and this is unlike the traditional personalized therapeutic. Each tumor is a specific set of genetic patterns, so that understanding genetic alterations and gene expression profiles in cancer cells can lead to effective therapies. The National Institutes of Health (NIH) has provided a lot of information on the importance of cancer genomics in personalized treatment, and not only genomics, but also proteomics play an important role in personalized treatment [46]. Inflammation, metabolism, and genotoxicity are key mechanisms, in which microbiota can modulate carcinogenesis and can therefore be used to develop anticancer therapies [63]. Today, new therapeutic approaches, called personalized medicine, have opened a new window in medical science, and the link between microbiome and personalized medicine seems to be one of the most interesting aspects of future research and is considered as an important perspective on the treatment of diseases like cancer.
Main text
The role of microbiome metabolites in the development of disease (Fig. 1)
With the advent of sophisticated diseases such as cancer, the association between environmental, microbiome and cancer effects can be very complicated. Changes in cell metabolism and inflammation are a sign of cancer [30]. Even if host-microbiome reactions to cancer are not considered as an essential event, the presence of microbial compounds in some cancers, such as colorectal cancer (CRC), can be indirectly important. In vitro studies have reported a signaling process between bacterial quorum sensing peptides (QSPs) and cancer cells. Bacillus-derived QSPs are synthesized when the bacteria are under stress conditions and have the ability to induce invasive tumor cells in a process called Epithelial mesenchymal like (EMT-Like) (involved in CRC metastasis) [80]. The QSPs participate in both metastatic and angiogenesis behaviors under these conditions [69, 80]. In other types of cancers, microbial activity can reduce the effectiveness of chemotherapy [77] or affect tumor development [35]. Lifestyle and diet are also the ones that play a major role in determining the microbiome. In addition, the production of various metabolites by gut microbiota is effective in cancer-promoting and cancer-protecting induction; however, different determinants are still not fully understood [8].

Metabolites production by microbiota and role of its on health
Microbiome-derived metabolites have the potential to contribute to cancer development, and this has been recognized [47]. Clearly, the diet is a great source of these metabolites; for example, high-fat and high-protein diets are a feature of the modern Western diet [2, 33], which is one of the risk factors for the occurrence of cancer [5, 22]. On the other hand, the bile acid (BA) is used as a signaling molecule associated with metabolic homeostasis [1]. Specific enzymes convert BA to SBA [54] that can act as a carcinogen [3]. In vitro studies have shown that the exposure of an hour to SBA compounds such as deoxycholic acid (DCA) and lithocholic acid (LCA), leads to extensive damage to DNA that has a dose-dependent behavior [4]. Studies have shown that the African-American population showed more incidence and more deaths than the Native American population relative to CRC. The microbiome combination of these two groups (African–American and Native American) was studied, and the African-American group was abundant in Bacteroides species, while the native group was abundant in Prevotella species [54]. Additionally, the encoded genes for SBA and fecal SBA in the first group had higher levels, whereas short chain fatty acids were higher in Native American, and therefore studies reported [14] that despite the same genetic history, phenotypic and developmental differences of a specific disease are possible, and these differences are mainly due to various diets and microbiome combinations. The consumption of fiber-rich foods induces saccharolytic fermentation, due to different species of gut microbes that produce short chain fatty acids and specifically acetate, propionate and butyrate [32]. For example, Bacteroidetes have high levels of acetate and propionate, while Firmhicute bacteria produce high amounts of butyrate. Some anti-cancer activity is associated with the butyrate. For instance, the butyrate can induce S-phase ablation in colorectal adenocarcinoma cells and result in growth inhibition by inducing apoptosis and expression of cell regulators such as P21 and cyclin B1 [31]. Interestingly, the butyrate effects are in cell-dependent manner; the butyrate in normal cells induces the proliferation as a source of energy, while the butyrate in cell lines inhibits the proliferation and triggers the apoptosis [13].
The relationship between inflammation, cancer, and microbiome (Figs. 2, 3)
Chronic inflammation and inflammatory factors such as reactive oxygen and nitrogen species, cytokines and chemokines can contribute to the growth and metastasis. The microbes in relation to cancer, activating NFκB signaling, are within tumor microenvironments. The NFκB is activated in tumors with high prevalence of Fusobacterium nucleatum (F. nucleatum), which is found to be abundant in colorectal cancer [23]. The NFκB is the regulator of inflammatory responses, also activator of the survival-triggering genes within cancer cells, and inflammatory-inducing genes within the microenvironment [18]. FadA is an adhesin in F. nucleatum. In vitro studies have shown that FadA, by binding to TIGIT inhibitory receptors in NKC, and inhibiting its cytotoxic activity, is helpful in invading immune system in tumor cells [29]. In addition to the innate immune system, microorganisms will also affect the acquired immune system; for example, as soon as a specific bacterium is exposed to the CD4 T cell, it is possible to produce cytokines that induce tumor progression [21]. For instance, IL-23 is one of these cytokines that is produced by tumor-associated myeloid cells, in response to microbial products such as flagellin, which promotes the growth and development of tumor cells and develops tumor IL-17 responses [28].

Diet, environment factors and host influence on microbiota and effect of microbiota on homeostasis and dysbiosis

Effect of B. fragilis and F. nucleatum on inflammation and colorectal cancer
Enterotoxigenic Bacteroides fragilis leads to inflammation in humans and induces colitis, and strongly induces colonic tumor in multiple intestinal neoplasia of mice in vivo. This toxin induces STAT3 signaling via Th17 responses that results in the production of IL-17 and IL-22, and other cytokines linked to human colorectal cancer by activating the STAT3 pathway [36]. On the other hand, the inflammation can be associated with other malignancies and is a risk factor for cancer development; for example, obesity can be a producer of overrepresentation of bacterial species capable of producing pro-carcinogenic metabolites such as SBA. Dysbiosis in obese subjects, changes the intestinal epithelium, causing more permeability to microbial production, [47] which can activate immune cells in the lamina propria and after circulation reach the liver, and lead to the production of pro-inflammatory cytokines such as TNF and IL-6 [19]. Finally, studies have shown that barrier deterioration due to microbial products has been associated with colorectal tumorigenesis [28].
The role of microbiome in precision diagnosis and personalized treatment
Various evidence suggests that dysregulation of microbiota-host interaction is correlated with different diseases such as IBD, diabetes, cirrhosis and colorectal cancer [45]. Recently, studies have been conducted concerning the reactions between bacteria and cancer treatment drugs [15, 35, 75], and the findings suggest that interactions of the bacteria mediated with the immune system, are necessary for drug efficacy, although little information is available on the effects of human microbiome combinations, and treatment outcomes in cancer patients [37]. Many studies [26, 48, 58] have shown that the patients in accordance with gut microbiome combinations have the potential to respond to or not respond to immunotherapy, and this can be considered in the evaluation of drug interactions. Moreover, the emergence of the role of gut microbiome as a biomarker for disease phenotype, prognosis and response to treatment, is well described in relation to the alteration of microbial population structure in various diseases [79, 78]. Discussions have revealed that the gut microbiome is associated with surgery in CD subjects along with increased mucosa-associated F. prausnitzii in the recurrent disease [25]. Despite many studies in relation to microbiome in IBD, there is no agreement between outcomes, because it is due to geographical differences and the use of antibiotics, diet, and other effective factors that affect the gut microbiome. Therefore, further studies on mucosal bacteria needed in relation to inflammatory diseases such as IBD. In addition, microbiome signatures are associated with many other related gastrointestinal diseases. For example, F. nucleatum is used as a diagnostic marker via FadA adhesin in colorectal cancer [59], or Clostridium difficile (C. difficile) infection is associated with reduced microbial diversity and low production of secondary bile acids [7, 52]. In addition, recently two studies have identified microbiome signatures in the C. difficile infection that can predict the disease [42, 64]. A study found remarkable results in this regard, showing that these patients had 90% clinical improvement after receiving fecal microbiota transplantation (FMT) from stool samples of healthy people [37]. Another example in this regard, was the observation of the spread of Proteobacteria in patients with celiac disease, which has gastrointestinal symptoms, compared to those with the same illness that exhibited extra-intestinal symptoms [76]. Many studies have been interested in describing the gut microbiota signature in the systematic disorders such as rheumatoid arthritis, and confirm that Prevotella is seen in these patients [55, 62]. In another study, these individuals were abundant in Collinsella, Eggrthella and Faecalibacterium [11].
Many microorganisms are associated with treatment responses. For example, it is noteworthy that patients responding to anti-PD1 therapy had a high incidence of Faecalibacterium, while patients who did not respond to treatment showed a high incidence of Bacteroidale. Studies suggested that microbial populations are a source of bacterial immune synergy to respond to anti-PD1 treatment. People with metastatic melanoma who showed a better response to treatment, had a high prevalence of Bifidobacterium longum. The presence of these species in the tumor-bearing rat intestine showed improved treatment for anti-PD-L1 [67]. On the other hand, two species of Ruminococcus obeum and Roseburia intestinalis were observed in people who did not respond to treatment. Routly observed that exposure to antibiotics over the course of cancer therapy can be linked to anti-PD1 treatment responses, and in fact confirms that the destruction of the microbial network and the loss of specific bacteria can interfere with the efficacy of immunity. Comparison of fecal microbiota in those, responding to treatment showed a relative increase in Akkermansia muciniphila, compared to those who did not respond, which also an indication of an optimal outcome for anti-PD1 therapy. The microbiota of the patients, responding to the treatment contained immunoregulatory bacteria, such as Akkermansia, Faecalibacterium and Bifidobacterium, which had a better performance over anti-PD1. In another study [26, 58], it was observed that mice receiving FMT from patients, who responded to treatment, experienced further recovery response to anti-PD1 treatment than the mice receiving FMT from those without treatment response. It was found that this improved response was associated with the frequency of Faecalibacterium in the rat stool. Later in complementary studies [48], it was found that the tumors of mice receiving FMT derived from subjects, responding to the treatment, showed a high CD8 T cell level, compared to the other group. On the other hand, Routly [26] reported that the presence of A. muciniphila in mice, receiving FMT derived from subjects who did not respond to treatment, resulted in an improved antitumor activity of immune cells.
Other interesting observations exhibited that the strains of Akkermansia, Faecalibacterium and Bifidobacterium are associated with anti-inflammatory responses, which is an immune system arm that prevents over-response activation, and leads to the formation and maintenance of homeostasis [10]. For instance, the relative decline of A. muciniphila in the intestine is associated with many diseases, such as IBD, type II diabetes and other diseases [10]. Similarly, F. prausnitzii downregulates intestinal inflammation related to the production of specific metabolites, such as butyrate, salicylic acid derived from host cells or bacteria in the intestines and peripheral blood [49].
All of these studies suggest that the precision medicine strategy, including gut microbiota, can have therapeutic potential. Finally, all these results suggest that efforts are being made to create synthetic microbial communities for the treatment of various diseases such as IBD and CDI. The gut microbiota has the ability to modulate the individual health, through many immune and non-immune cell types such as RNA, DNA, membrane compounds, etc. via the production of a network of metabolites. The interesting point is that in addition to having sporadic microbiota, in the case of patients who respond to treatment, one can assume that better synergy with treatment can be observed in intestinal bacteria in the event of a translocation to the secondary lymphoid organs that create a specific anti-tumor immune response.
Discussion
The Human Microbiome Project with the mission to supply required resources and expertise to characterize the human microbiome and examine its role in health being was launched by National Institute of Health in 2007. HMP acts as a road map to discover the role microorganisms play in human health, disease, nutrition, and immunity in different parts of the body. It examines the microbes in five different areas of the body: nose, mouth, skin, vagina, and colon. It should be noted that according to the research most communities of the microbes are distinct from each other (e.g. the microbes on the skin are distinct from those in mouth, intestine, and vagina). The microbes do not also appear in mixture, and all major groups, phyla, of the bacteria that may colonize the human body, do not exist in everybody site. Two major strategies so far have been used to analyze microbial communities through NGS: shotgun metagenomics and 16S rDNA sequencing. Shotgun metagenomics is an integral part of sequencing of bacterial DNA isolated from the whole microbial community [72]. 16SrDNA sequencing relies on amplification of the polymerase chain reaction (PCR) in a specific region of the 16S gene [43]. It is assumed that 16S sequencing is a robust, well characterized method that provides adequate information about microbial communities’ composition, starting from a relatively small number of sequences per samples (*200 thousands). However, one of the major limitation of the method is assignment of the taxa based on the sequence of only a single region of the bacterial genome [56]. Shotgun metagenomics, on the other hand, requires a more complex downstream data analysis and higher coverage (10–30 million of reads). Nevertheless, shotgun metagenomics through collecting sequence information about broad genomic regions allows a more accurate definition at the species level and consequently yields a detailed description of bacterial community [66].
Human microbiome can be used to detect biomarker and present research intends to examine its therapeutic role [50]. Given that microbiome is a biomarker of diseased state, examining microbiomics and metagenomics is necessary to find out the processes. Present biomarker will be the future theranostics which could outline the suggested way of diagnostic therapy for the patients and test the new probable medication methods to find the best treatment based on the screening results.
Nevertheless, there are yet many challenges which should be addressed; for example, are we manufacturing antimicrobial drug resistant flora; how the microbiome modifies drugs, what are the side effects and how they can be minimized? [57] Gut microbiome as a tool regarding targeted non-invasive biomarkers has been established by compelling studies for certain diseases or cancers. Microbial metabolites (for example branched chain amino acids) can serve as microbial biomarkers regarding metabolic disorders like prediabetes and type 2 diabetes to prevent or mitigate the disease [74]. More than the well-defined associations of any alterations in the structure of microbial community among different kinds of disease states, gut microbiome recently has been used as a biomarker for disease prognosis, phenotype, and response to treatment [39]. In addition, Inflammatory bowel disease is one of the best examined conditions related to dysbiosis, where microbiome has served as an important marker for response to treatment and disease phenotype. An association also has been reported between gut microbiome signatures and surgical outcomes in CD, where an increase in F. prausnitzii in the ileal mucosa has been associated with a decrease in disease recurrence at 6 months [79, 78]. Although some studies have highlighted the changes in the microbiome in IBD, but there is no consensus in this regard. Accordingly, to overcome the effect of antibiotic use, disease subtype, and other factors affecting the gut microbiome [25], to have large cohorts from different geographic locations is a necessity. Diet in general, and consumption of dietary fiber in particular, seems to affect gut diversity, ecology, and function substantially [71]. The research shows a connection between host health and dietary MAC (Microbiota-Accessible Carbohydrates). Gut microbiota composition is affected by induced alterations in dietary fiber and microbiota composition. Nevertheless, there is large inter-individual variations [16, 68]. According to Kovatcheva-Datchary et al. improved glucose metabolism after dietary fiber supplementation leads to an increase in abundance of Prevotella in gut microbiota [44]. Research also shows that dietary fibers, through modulating the gut microbiota, can prevent high-fat diet induced obesity [45]. Recent studies regarding the impact of protein intake on the gut microbiota suggest that high-protein diets lead to an increase in detrimental metabolites in feces [61]. It is also reported that consumption of omega-3 fatty acids due to modulation of the gut microbiota can reduce chronic inflammation and body weight gain [38]. For example, the composition of gut microbiota in early-life stressed animals changed due to EPA/DHA. According to Degnan et al. cobalamin and related factors shape the composition of human gut microbial communities and their functions [17]. Emulsifiers as a dietary compounds alter gut barrier dysfunction and gut microbiota and have negative impacts on metabolism [9]. In addition, recent studies have identified two microbiome signatures that can be predictor of disease outcomes and allow therapeutic stratification [64]. The patients affected with celiac disease with gastrointestinal symptoms experienced an expansion of proteobacteria in the setting of dysbiotic microbiota in compare with those with extra-intestinal manifestations of celiac disease [76]. In addition to diseases within gastrointestinal tract, gut microbiome signatures is reported in systemic disorders such as rheumatoid arthritis. Expansion of intestinal Prevotella copri is also reported in new onset rheumatoid arthritis (RA) [62]. Enrichment of Faecalibacterium, Eggerthella and Collinsella in patients with RA and a strong correlation between Collinsella and high levels of asparagine and alpha-aminoadipic acid, as well as production of experimental arthritis and alpha-aminoadipic cytokine IL-17A are reported in another recent study [11].
Above mentioned examples, among the others, provide experimental evidence to prove the role of microbiome in human disease and future implications of microbiome based biomarkers for diagnostic and therapeutic purposes. Although much research has been done to identify biomarkers, but validating these signatures in large multicenter cohorts and identifying their causative role needs more combination of in vitro and in vivo models [39].
Conclusion
The development of diagnostic tests, using biomarkers for use in primary diagnosis is one of the key aspects of precision medicine [70]. CRC is one of the cases on which the studies have been conducted, so that researchers evaluated the potential of fecal microbiota for the early detection of CRC, and applied it as a screening tool among various clinical groups of healthy people, and those with adenoma and carcinoma [81]. These limited studies have confirmed the role of microbiome in human diseases, and that the microbiome population may be used as a diagnostic and a therapeutic biomarker in the near future. Although these studies are preliminary, and there is definitely a need for in vitro and in vivo studies with more confirmatory tests for each disease, in order to achieve a suitable microbiome signature.
Further studies are needed to understand how bacteria affect the immune system and tumor microenvironments, and on the other hand, the association between microbial populations and antitumor therapy response is complicated. In fact, selective reduction and bacterial taxa by means such as exposure to antibiotics or other stressors, may result in reduced immunotherapy responses. Additionally, the presence of specific microorganisms on other sites may lead to interference with treatment [24]. For example, E. coli, by metabolizing and deactivating the active form of the drug, reduces the effects of chemotherapy, which can have a negative interaction with tumor responses [24], so the presence of specific strains may have the ability to modulate cancer progression and therapeutics. This increases the likelihood of precision medicine in relation to microbiota, in terms of treatment and prognosis, and therefore, microbiota is a next generation medicine and may develop a novel therapeutic role in this field.
Abbreviations
- NGS:
-
next generation sequencing
- NIH:
-
National Institute of Health
- CRC:
-
colorectal cancer
- QSPs:
-
quroum sensing peptides
- EMT-Like:
-
epithelial mesenchymal like
- BA:
-
bile acid
- DCA:
-
deoxycholic acid
- LCA:
-
lithocholic acid
- NFkB:
-
nuclear factor kB
- STAT3:
-
Signal Transducer and Activator of Transcription 3
- IBD:
-
inflammatory bowel disease
- FTM:
-
fecal microbiota transplanation
References
de Aguiar Vallim TQ, Tarling EJ, Edwards PA (2013) Pleiotropic roles of bile acids in metabolism. Cell Metab 17:657–669
Albenberg LG, Wu GD (2014) Diet and the intestinal microbiome: associations, functions, and implications for health and disease. Gastroenterology 146:1564–1572
Bernstein H, Bernstein C, Payne CM, Dvorakova K, Garewal H (2005) Bile acids as carcinogens in human gastrointestinal cancers. Mutat Res 589:47–65
Booth LA, Gilmore IT, Bilton RF (1997) Secondary bile acid induced DNA damage in HT29 cells: are free radicals involved? Free Rad Res 26:135–144
Bouvard V, Loomis D, Guyton KZ, Grosse Y, Ghissassi FE, Benbrahim-Tallaa L, Guha N, Mattock H, Straif K, International Agency for Research on Cancer Monograph Working, G (2015) Carcinogenicity of consumption of red and processed meat. Lancet Oncol 16:1599–1600
Breitbart M, Haynes M, Kelley S, Angly F, Edwards RA, Felts B, Mahaffy JM, Mueller J, Nulton J, Rayhawk S, Rodriguez-Brito B, Salamon P, Rohwer F (2008) Viral diversity and dynamics in an infant gut. Res Microbiol 159:367–373
Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, Littmann E, van den Brink MR, Jenq RR, Taur Y, Sander C, Cross JR, Toussaint NC, Xavier JB, Pamer EG (2015) Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517:205–208
Bultman SJ, Jobin C (2014) Microbial-derived butyrate: an oncometabolite or tumor-suppressive metabolite? Cell Host Microbe 16:143–145
Cani PD, Everard A (2015) Keeping gut lining at bay: impact of emulsifiers. Trends Endocrinol Metab 26(6):273–274
Cani PD, de Vos WM (2017) Next-generation beneficial microbes: the case of Akkermansia muciniphila. Front Microbiol 8:1765
Chen J, Wright K, Davis JM, Jeraldo P, Marietta EV, Murray J, Nelson H, Matteson EL, Taneja V (2016) An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Med 8:43
Cho SH, Jeon J, Kim SI (2012) Personalized medicine in breast cancer: a systematic review. J Breast Cancer 15:265–272
Comalada M, Bailon E, de Haro O, Lara-Villoslada F, Xaus J, Zarzuelo A, Galvez J (2006) The effects of short-chain fatty acids on colon epithelial proliferation and survival depend on the cellular phenotype. J Cancer Res Clin Oncol 132:487–497
Contreras AV, Cocom-Chan B, Hernandez-Montes G, Portillo-Bobadilla T, Resendis-Antonio O (2016) Host-microbiome interaction and cancer: potential application in precision medicine. Front Physiol 7:606
Daillere R, Vetizou M, Waldschmitt N, Yamazaki T, Isnard C, Poirier-Colame V, Duong CPM, Flament C, Lepage P, Roberti MP, Routy B, Jacquelot N, Apetoh L, Becharef S, Rusakiewicz S, Langella P, Sokol H, Kroemer G, Enot D, Roux A, Eggermont A, Tartour E, Johannes L, Woerther PL, Chachaty E, Soria JC, Golden E, Formenti S, Plebanski M, Madondo M, Rosenstiel P, Raoult D, Cattoir V, Boneca IG, Chamaillard M, Zitvogel L (2016) Enterococcus hirae and Barnesiella intestinihominis facilitate cyclophosphamide-induced therapeutic immunomodulatory effects. Immunity 45:931–943
Deehan EC, Walter J (2016) The fiber gap and the disappearing gut microbiome: implications for human nutrition. Trends Endocrinol Metab 27(5):239–242
Degnan PH, Taga ME, Goodman AL (2014) Vitamin B 12 as a modulator of gut microbial ecology. Cell Metab 20(5):769–778
DiDonato JA, Mercurio F, Karin M (2012) NF-kappaB and the link between inflammation and cancer. Immunol Rev 246:379–400
Font-Burgada J, Sun B, Karin M (2016) Obesity and cancer: the oil that feeds the flame. Cell Metab 23:48–62
Fraher MH, O’Toole PW, Quigley EM (2012) Techniques used to characterize the gut microbiota: a guide for the clinician. Nat Rev 9:312–322
Gagliani N, Hu B, Huber S, Elinav E, Flavell RA (2014) The fire within: microbes inflame tumors. Cell 157:776–783
Gallagher EJ, LeRoith D (2015) Obesity and diabetes: the increased risk of cancer and cancer-related mortality. Physiol Rev 95:727–748
Garrett WS (2015) Cancer and the microbiota. Science (New York, NY) 348:80–86
Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, Nejman D, Gavert N, Zwang Y, Cooper ZA, Shee K, Thaiss CA, Reuben A, Livny J, Avraham R, Frederick DT, Ligorio M, Chatman K, Johnston SE, Mosher CM, Brandis A, Fuks G, Gurbatri C, Gopalakrishnan V, Kim M, Hurd MW, Katz M, Fleming J, Maitra A, Smith DA, Skalak M, Bu J, Michaud M, Trauger SA, Barshack I, Golan T, Sandbank J, Flaherty KT, Mandinova A, Garrett WS, Thayer SP, Ferrone CR, Huttenhower C, Bhatia SN, Gevers D, Wargo JA, Golub TR, Straussman R (2017) Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science (New York, NY) 357:1156–1160
Gevers D, Kugathasan S, Denson LA, Vazquez-Baeza Y, Van Treuren W, Ren B, Schwager E, Knights D, Song SJ, Yassour M, Morgan XC, Kostic AD, Luo C, Gonzalez A, McDonald D, Haberman Y, Walters T, Baker S, Rosh J, Stephens M, Heyman M, Markowitz J, Baldassano R, Griffiths A, Sylvester F, Mack D, Kim S, Crandall W, Hyams J, Huttenhower C, Knight R, Xavier RJ (2014) The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 15:382–392
Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, Prieto PA, Vicente D, Hoffman K, Wei SC, Cogdill AP, Zhao L, Hudgens CW, Hutchinson DS, Manzo T, Petaccia de Macedo M, Cotechini T, Kumar T, Chen WS, Reddy SM, Szczepaniak Sloane R, Galloway-Pena J, Jiang H, Chen PL, Shpall EJ, Rezvani K, Alousi AM, Chemaly RF, Shelburne S, Vence LM, Okhuysen PC, Jensen VB, Swennes AG, McAllister F, Sanchez MR et al (2018) Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science (New York, NY) 359:97–103
Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, Program NCS, Bouffard GG, Blakesley RW, Murray PR, Green ED, Turner ML, Segre JA (2009) Topographical and temporal diversity of the human skin microbiome. Science (New York, NY) 324:1190–1192
Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, Taniguchi K, Yu GY, Osterreicher CH, Hung KE, Datz C, Feng Y, Fearon ER, Oukka M, Tessarollo L, Coppola V, Yarovinsky F, Cheroutre H, Eckmann L, Trinchieri G, Karin M (2012) Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 491:254–258
Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, Enk J, Bar-On Y, Stanietsky-Kaynan N, Coppenhagen-Glazer S, Shussman N, Almogy G, Cuapio A, Hofer E, Mevorach D, Tabib A, Ortenberg R, Markel G, Miklic K, Jonjic S, Brennan CA, Garrett WS, Bachrach G, Mandelboim O (2015) Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity 42:344–355
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Hinnebusch BF, Meng S, Wu JT, Archer SY, Hodin RA (2002) The effects of short-chain fatty acids on human colon cancer cell phenotype are associated with histone hyperacetylation. The Journal of nutrition 132:1012–1017
Holmes E, Li JV, Marchesi JR, Nicholson JK (2012) Gut microbiota composition and activity in relation to host metabolic phenotype and disease risk. Cell Metab 16:559–564
Hughes R, Magee EA, Bingham S (2000) Protein degradation in the large intestine: relevance to colorectal cancer. Curr Issues Intest Microbiol 1:51–58
Human Microbiome Project (2012) Structure, function and diversity of the healthy human microbiome. Nature 486:207–214
Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, Molina DA, Salcedo R, Back T, Cramer S, Dai RM, Kiu H, Cardone M, Naik S, Patri AK, Wang E, Marincola FM, Frank KM, Belkaid Y, Trinchieri G, Goldszmid RS (2013) Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science (New York, NY) 342:967–970
Jiang R, Wang H, Deng L, Hou J, Shi R, Yao M, Gao Y, Yao A, Wang X, Yu L, Sun B (2013) IL-22 is related to development of human colon cancer by activation of STAT3. BMC Cancer 13:59
Jobin C (2018) Precision medicine using microbiota. Science (New York, NY) 359:32–34
Kaliannan K, Wang B, Li XY, Bhan AK, Kang JX (2016) Omega-3 fatty acids prevent early-life antibiotic exposure-induced gut microbiota dysbiosis and later-life obesity. Int J Obes. 40(6):1039
Kashyap PC, Chia N, Nelson H, Segal E, Elinav E (2017) Microbiome at the Frontier of personalized medicine. Mayo Clin Proc 92(12):1855–1864
Kau AL, Ahern PP, Griffin NW, Goodman AL, Gordon JI (2011) Human nutrition, the gut microbiome and the immune system. Nature 474:327–336
Kelly BJ, Imai I, Bittinger K, Laughlin A, Fuchs BD, Bushman FD, Collman RG (2016) Composition and dynamics of the respiratory tract microbiome in intubated patients. Microbiome 4:7
Khanna S, Montassier E, Schmidt B, Patel R, Knights D, Pardi DS, Kashyap P (2016) Gut microbiome predictors of treatment response and recurrence in primary Clostridium difficile infection. Aliment Pharmacol Ther 44:715–727
Klindworth A, Pruesse E, Schweer T et al (2013) Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res 41:e1
Kovatcheva-Datchary P, Nilsson A, Akrami R, Lee YS, De Vadder F, Arora T, Bäckhed F (2015) Dietary fiber-induced improvement in glucose metabolism is associated with increased abundance of prevotella. Cell Metab 22(6):971–982
Li X, Guo J, Ji K, Zhang P (2016) Bamboo shoot fiber prevents obesity in mice by modulating the gut microbiota. Scientific Rep 6:32953
Ling Z, Kong J, Liu F, Zhu H, Chen X, Wang Y, Li L, Nelson KE, Xia Y, Xiang C (2010) Molecular analysis of the diversity of vaginal microbiota associated with bacterial vaginosis. BMC Genomics 11:488
Louis P, Hold GL, Flint HJ (2014) The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev 12:661–672
Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, Luke JJ, Gajewski TF (2018) The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science (New York, NY) 359:104–108
Miquel S, Leclerc M, Martin R, Chain F, Lenoir M, Raguideau S, Hudault S, Bridonneau C, Northen T, Bowen B, Bermudez-Humaran LG, Sokol H, Thomas M, Langella P (2015) Identification of metabolic signatures linked to anti-inflammatory effects of Faecalibacterium prausnitzii. mBio 6:1
Morgan XC, Huttenhower C (2012) Chapter 12: human microbiome analysis. PLoS Comput Biol 8:e1002808
O’Hara AM, Shanahan F (2006) The gut flora as a forgotten organ. EMBO Rep 7:688–693
Pasolli E, Truong DT, Malik F, Waldron L, Segata N (2016) Machine learning meta-analysis of large metagenomic datasets: tools and biological insights. PLoS Comput Biol 12:e1004977
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto JM, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Li S, Jian M, Zhou Y, Li Y, Zhang X, Li S, Qin N, Yang H, Wang J, Brunak S, Dore J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, Meta HITC, Bork P, Ehrlich SD, Wang J (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464:59–65
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glockner FO (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–596
Rajpoot M, Sharma AK, Sharma A, Gupta GK (2018) Understanding the microbiome: emerging biomarkers for exploiting the microbiota for personalized medicine against cancer. Semin Cancer Biol 52(Pt 1):1–8
Rintala A, Pietila S, Munukka E et al (2017) Gut microbiota analysis results are highly dependent on the 16S rRNA gene target region, whereas the impact of DNA extraction is minor. J Biomol Tech 28:19–30
Rintala A, Riikonen I, Toivonen A, Pietila S, Munukka E, Pursiheimo JP et al (2018) Early fecal microbiota composition in children who later develop celiac disease and associated autoimmunity. Scand J Gastroenterol 53:403–409. https://doi.org/10.1080/00365521.2018.1444788
Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, Fluckiger A, Messaoudene M, Rauber C, Roberti MP, Fidelle M, Flament C, Poirier-Colame V, Opolon P, Klein C, Iribarren K, Mondragon L, Jacquelot N, Qu B, Ferrere G, Clemenson C, Mezquita L, Masip JR, Naltet C, Brosseau S, Kaderbhai C, Richard C, Rizvi H, Levenez F, Galleron N, Quinquis B, Pons N, Ryffel B, Minard-Colin V, Gonin P, Soria JC, Deutsch E, Loriot Y, Ghiringhelli F, Zalcman G, Goldwasser F, Escudier B, Hellmann MD, Eggermont A, Raoult D, Albiges L, Kroemer G, Zitvogel L (2018) Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science (New York, NY) 359:91–97
Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW (2013) Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell Host Microbe 14:195–206
Russo E, Taddei A, Ringressi MN, Ricci F, Amedei A (2016) The interplay between the microbiome and the adaptive immune response in cancer development. Therap Adv Gastroenterol 9:594–605
Salonen A, de Vos WM (2014) Impact of diet on human intestinal microbiota and health. Ann Rev Food Sci Technol 5:239–262
Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, Rostron T, Cerundolo V, Pamer EG, Abramson SB, Huttenhower C, Littman DR (2013) Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. eLife 2:e01202
Schwabe RF, Jobin C (2013) The microbiome and cancer. Nat Rev Cancer 13:800–812
Seekatz AM, Rao K, Santhosh K, Young VB (2016) Dynamics of the fecal microbiome in patients with recurrent and nonrecurrent Clostridium difficile infection. Genome Med 8:47
Sekirov I, Russell SL, Antunes LC, Finlay BB (2010) Gut microbiota in health and disease. Physiol Rev 90:859–904
Shah N, Tang H, Doak TG, Ye Y (2010) Comparing bacterial communities inferred from 16 s rRNA gene sequencing and shotgun metagenomics. Pac Symp Biocomput 2011:165–176
Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Lei YM, Jabri B, Alegre ML, Chang EB, Gajewski TF (2015) Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science (New York, NY) 350:1084–1089
Sonnenburg ED, Sonnenburg JL (2014) Starving our microbial self: the deleterious consequences of a diet deficient in microbiota-accessible carbohydrates. Cell Metab 20(5):779–786
De Spiegeleer B, Verbeke F, D’Hondt M, Hendrix A, Van De Wiele C, Burvenich C, Peremans K, De Wever O, Bracke M, Wynendaele E (2015) The quorum sensing peptides PhrG, CSP and EDF promote angiogenesis and invasion of breast cancer cells in vitro. PLoS ONE 10:e0119471
Stafford P, Cichacz Z, Woodbury NW, Johnston SA (2014) Immunosignature system for diagnosis of cancer. Proc Natl Acad Sci USA 111:E3072–3080
Tan J, McKenzie C, Vuillermin PJ, Goverse G, Vinuesa CG, Mebius RE, Mackay CR (2016) Dietary fiber and bacterial SCFA enhance oral tolerance and protect against food allergy through diverse cellular pathways. Cell Rep 15(12):2809–2824
Truong DT, Franzosa EA, Tickle TL et al (2015) MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods 12:902–903
Tyler AD, Smith MI, Silverberg MS (2014) Analyzing the human microbiome: a “how to” guide for physicians. Am J Gastroenterol 109:983–993
Versalovic J, Dore J, Guarner F, Luna RA, Ringel Y (2017) Microbiome-based diagnostics: ready for applications in laboratory Medicine? Clin Chem 63(11):1674–1679
Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillere R, Hannani D, Enot DP, Pfirschke C, Engblom C, Pittet MJ, Schlitzer A, Ginhoux F, Apetoh L, Chachaty E, Woerther PL, Eberl G, Berard M, Ecobichon C, Clermont D, Bizet C, Gaboriau-Routhiau V, Cerf-Bensussan N, Opolon P, Yessaad N, Vivier E, Ryffel B, Elson CO, Dore J, Kroemer G, Lepage P, Boneca IG, Ghiringhelli F, Zitvogel L (2013) The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science (New York, NY) 342:971–976
Wacklin P, Kaukinen K, Tuovinen E, Collin P, Lindfors K, Partanen J, Maki M, Matto J (2013) The duodenal microbiota composition of adult celiac disease patients is associated with the clinical manifestation of the disease. Inflamm Bowel Dis 19:934–941
Wallace BD, Wang H, Lane KT, Scott JE, Orans J, Koo JS, Venkatesh M, Jobin C, Yeh LA, Mani S, Redinbo MR (2010) Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science (New York, NY) 330:831–835
Willing BP, Dicksved J, Halfvarson J, Andersson AF, Lucio M, Zheng Z, Jarnerot G, Tysk C, Jansson JK, Engstrand L (2010) A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology 139(1844–1854):e1841
Willing B, Halfvarson J, Dicksved J, Rosenquist M, Jarnerot G, Engstrand L, Tysk C, Jansson JK (2009) Twin studies reveal specific imbalances in the mucosa-associated microbiota of patients with ileal Crohn’s disease. Inflamm Bowel Dis 15:653–660
Wynendaele E, Verbeke F, D’Hondt M, Hendrix A, Van De Wiele C, Burvenich C, Peremans K, De Wever O, Bracke M, De Spiegeleer B (2015) Crosstalk between the microbiome and cancer cells by quorum sensing peptides. Peptides 64:40–48
Zackular JP, Rogers MA, Ruffin MT, Schloss PD (2014) The human gut microbiome as a screening tool for colorectal cancer. Cancer Prev Res (Philadelphia, Pa) 7:1112–1121
Authors’ contributions
AB prepare collection of main data. SS edit the main manuscript text. All authors read and approved the final manuscript.
Acknowledgements
We thank all the colleagues in the Department of Mycobacteriology and Pulmonary Research.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This study was financially supported by Pasteur Institute of Iran (Grant No. 1006).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Behrouzi, A., Nafari, A.H. & Siadat, S.D. The significance of microbiome in personalized medicine. Clin Trans Med 8, 16 (2019). https://doi.org/10.1186/s40169-019-0232-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40169-019-0232-y